

Abstract
Background and purpose:
In vitro assays that determine activities of drug candidates with isolated targets have only limited predictive value for activities in cellular assays. Poor membrane permeability and off-target binding are major reasons for such discrepancies. However, it still difficult to directly analyse off-target binding at the same time as target binding, on a subcellular level. Here, we present a combination of fluorescence correlation spectroscopy (FCS) and fluorescence cross-correlation spectroscopy (FCCS) as a solution to this problem.
Experimental approach:
The well-established dihydrofolate reductase inhibitor methotrexate and the kinase inhibitors PD173956 and purvalanol B were conjugated via polyethylene glycol linkers with the fluorophore Cy5. The cellular uptake and subcellular distribution of these compounds in single human cancer-derived cells were investigated by confocal laser scanning microscopy. In addition, molecular interactions inside the cell with the respective target proteins and off-target binding were detected simultaneously in the nanomolar range by FCCS and FCS, respectively, using cells expressing green fluorescent protein fusion proteins of dihydrofolate reductase and Abelson kinase 1.
Key results:
Large differences in the interaction patterns were found for these compounds. For methotrexate-Cy5, drug–target interactions could be detected and dissociation constants determined. In contrast, PD173956-Cy5 showed strong interactions with intracellular high-molecular weight structures, other than its target.
Conclusions and implications:
The combination of FCS and FCCS provides a powerful means to assess subcellular pharmacokinetics and dynamics of drug candidates at nanomolar concentrations.
Keywords: fluorescence correlation spectroscopy, fluorescence cross-correlation spectroscopy, anti-cancer drug, cellular pharmacology, drug-target interaction, drug development
Introduction
Cellular assays are gaining significance in drug development (Perlman et al., 2004; Lang et al., 2006; Korn and Krausz, 2007). While providing less information on the interaction of a drug candidate towards a specific molecular target, such assays reveal off-target effects and cytotoxicity. In addition, many compounds fail to show any biological effect, even if activity was detected towards an isolated target molecule in an in vitro screen.
For compounds that exert their activity inside the cell, such failure is typically attributed to poor cellular uptake. However, this assumption rather reflects a lack of knowledge of the intracellular pharmacology of a drug, than being based on direct evidence. This deficit is due to an absence of methods describing the interaction of compounds on the subcellular level. Next to the failure to cross the plasma membrane, lack of activity may also be due to interactions with membranes, sequestration in vesicular compartments, binding to DNA in the cell nucleus and binding to cytoskeletal structures and other proteins. For example, numerous cationic amphiphilic drugs are enriched in the lysosomal compartment (Bareford and Swaan, 2007; Almela et al., 2009).
In the past decades, numerous techniques have been developed to investigate the subcellular localization of small molecule drugs. The molecular localization of proteins, peptides and small molecules in tissues can be determined by mass spectrometry imaging with a resolution down to approximately 4 µm (Chaurand et al., 2005; Signor et al., 2007). Infrared microscopy and Raman scanning techniques provide information about the localization and molecular nature of macromolecular assemblies, but the detection is rather non-specific for particular molecules and the spatial resolution (>1 µm) is limited in comparison to high-resolution microscopy (Lasch and Naumann, 2006). Microscopic autoradiography enables high spatial resolution in the subcellular localization of drugs but requires the fixation of cells and the synthesis and manipulation of radioactively labelled drug molecules (Stumpf, 2005).
Generally, high-resolution fluorescence microscopy has become the method of choice for studying molecular dynamics on a subcellular level in living cells. For small molecule drugs, detection is either based on intrinsic fluorescence – one such example is doxorubicin (de Lange et al., 1992; Lankelma et al., 1999) – or molecules are conjugated to fluorophores for visualization. The dihydrofolate reductase (DHFR) inhibitor methotrexate (MTX) is a prominent example that illustrates the potential of fluorescence microscopy for an analysis of cellular transport and localization of a drug molecule (Breen et al., 2004).
All the methods described above, however, do not report on molecular interactions. Here, fluorescence correlation spectroscopy (FCS) and in particular fluorescence cross-correlation spectroscopy (FCCS) are two highly powerful techniques.
In FCS, information on the mobility and concentration of molecules is obtained from an analysis of fluorescence fluctuations, caused by diffusion of molecules through a subfemtoliter confocal detection volume (Rigler et al., 1993). With a sensitivity in the subnanomolar range, FCS has become an important tool to address molecular dynamics and interactions in cells and subcellular compartments (Brock et al., 1999; Schwille et al., 1999; Hink et al., 2000; Wachsmuth et al., 2000; Koopman et al., 2007). Binding of a low-molecular weight drug candidate to a target protein slows down diffusion. Binding equilibria can be derived from the contributions of a slow- and fast-moving fraction to the total autocorrelation function (Meseth et al., 1999). In the context of determining drug activity inside a cell, FCS has been employed to assess the activity of a small molecule on protein–DNA interactions (Michelman-Ribeiro et al., 2009). However, for measurements in complex mixtures of targets, FCS lacks specificity for reporting on a particular interaction. This information is provided by FCCS. In FCCS, concerted fluorescence fluctuations, caused by co-diffusion of two spectrally well-separated fluorophores, are detected (Schwille et al., 1997b). For the detection of drug–target interactions, two main characteristics set this method apart from fluorescence resonance energy transfer (FRET) (Clegg, 1995): First, there is no dependence of the signal on the distance of the fluorophores. Therefore, there are no constraints on the use of linker molecules between drug and fluorophore. Second, especially for interacting molecules that are homogeneously distributed throughout a cell and at low concentrations, FCCS has a much higher sensitivity for the detection of interactions. Also intracellular applications have been presented for FCCS (Bacia et al., 2006), including the determination of binding constants of fluorescent proteins (Sudhaharan et al., 2009).
Given these powerful characteristics for the detection of molecular interactions, we here employed a combination of confocal laser scanning microscopy, FCS and FCCS to simultaneously investigate target and off-target binding of Cy5-labelled anti-cancer drugs. Cy5 has an excitation maximum around 650 nm and is spectrally well separated from green and yellow fluorophores with emission below 600 nm, enabling the simultaneous detection with proteins fused to green fluorescent protein (GFP). MTX, purvalanol B and PD173956 were selected for this purpose and coupled with Cy5 to yield MTX-Cy5, Pur-Cy5 and PD-Cy5, respectively. Previous studies had shown that these compounds show bioactivity against cancer cells and cancer-associated molecular targets in the lower nanomolar range (Washtien, 1982; Gray et al., 1998; Kraker et al., 2000). Hydrophobicity is a major determinant for the pharmacological properties of a drug (Lipinski et al., 2001). Therefore, the drugs were selected to cover a broad range of hydrophobicity. Whereas the anti-metabolite MTX is hydrophilic, having a logarithmic octanol/water partition coefficient (logP) of −1.8, the kinase inhibitor purvalanol B has a logP of 3.8 and the kinase inhibitor PD173956 a logP of approximately 5, according to the PubChem database (National Center for Biotechnology Information, Bethesda, USA).
The intracellular localization of MTX-Cy5, Pur-Cy5 and PD-Cy5 was determined in living human cervical cancer HeLa cells and the human embryonic kidney cell line Hek293. For cells expressing GFP-tagged target proteins of MTX and PD 173956, FCS and FCCS were employed for detecting interactions of these two molecules inside the cell. Whereas FCCS specifically detected interactions with GFP-tagged target proteins, FCS yielded information on the total fraction of bound molecules. In our experiments, PD-Cy5 showed a much stronger off-target binding to intracellular structures than MTX-Cy5. The different characteristics of these drugs also confirmed that the Cy5 label did not impair the analysis of subcellular pharmacology.
Methods
Synthesis of labelled compounds
Compounds carrying a 5-polyethylene glycol (PEG) linker with a terminal amino group were synthesized at GPC Biotech (Munich, Germany). Five hundred nmol of these conjugates were dissolved in 100 µL dimethyl sulphoxide and 0.7 µL N,N-diisopropylethylamine. The solution was added to one aliquot of Cy5 monofunctional reactive dye and incubated in the dark at room temperature for 20 h. Subsequently, purification was performed by preparative reversed-phase high-performance liquid chromatography (Gilson, Middleton, Wisconsin, USA) using a linear gradient of water (0.1% trifluoroacetic acid) – acetonitrile (0.1% trifluoroacetic acid). The identity of the Cy5-labelled compounds was confirmed by mass spectrometry (see Figure 1).
Figure 1.
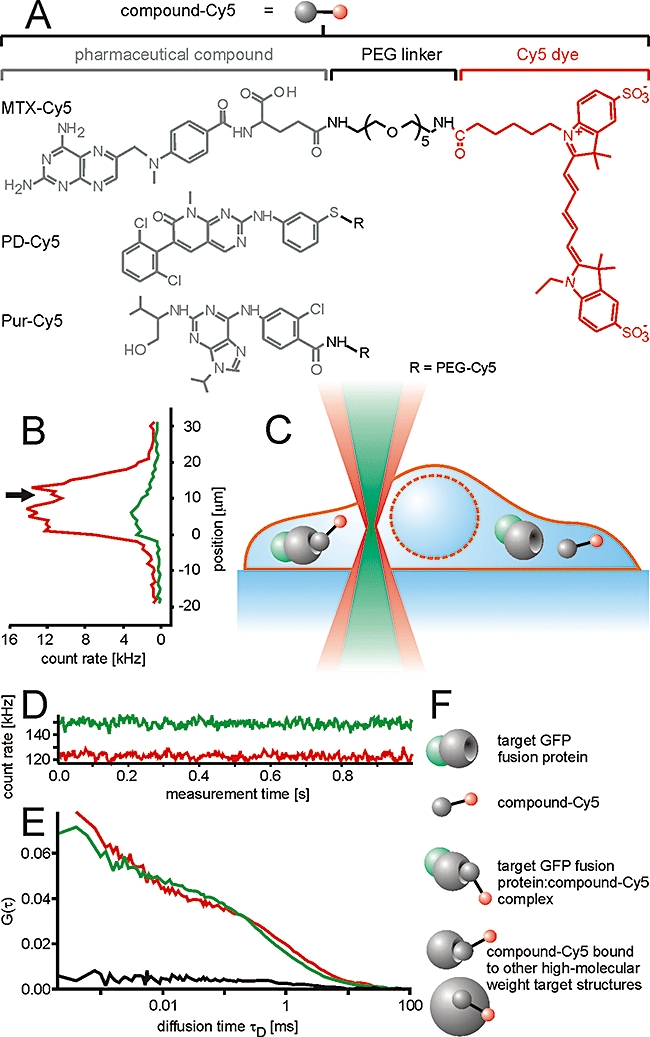
Determination of the intracellular pharmacological properties of small molecule drugs by a combination of fluorescence correlation spectroscopy and fluorescence cross-correlation spectroscopy measurements in living cells. (A) The drug molecules were conjugated to the fluorescent dye Cy5 via a 5-polyethylene glycol (PEG) linker. (B) Cells expressing green fluorescent protein (GFP)-tagged target proteins were loaded with the Cy5-labelled compounds and observed by confocal laser scanning microscopy. Subsequently, the laser beam was displaced vertically in order to identify a measurement position of the detection volume inside the cytoplasm along the optical axis (z-position). Alternatively, the z-position was identified from a confocal section in x-z-position. Measurements of interactions inside the cell (C) resulted in (D) a fluorescent count trace over time from which (E) autocorrelation functions G(τ) for both channels (red and green) and a cross-correlation function GCC(τ) (black) were calculated. (F) From the auto- and cross-correlation functions, concentrations of the various fluorescent species could be derived such as free GFP-tagged target protein, unbound compound-Cy5, compound-Cy5 : target protein–GFP complexes and compound-Cy5 bound to high-molecular weight structures. MTX, methotrexate.
Tissue culture
Hek293 and HeLa wild-type cell lines were grown in Roswell Park Memorial Institute (RPMI) 1640 medium containing 5% fetal calf serum at 37°C and 5% CO2. The cell line T-REx-293 transfected with the pcDNA4/TO-based expression plasmids was grown in RPMI1640 medium containing 10% fetal calf serum and the selection factors zeocin (100 µg·mL−1) and blasticidin (5 mg·mL−1) at 37°C and 5% CO2.
Transient transfection of HeLa cells with the DHFR-GFP expression plasmid
1 × 106 HeLa cells were detached by trypsin/EDTA, washed with medium, and resuspended in 200 µL medium containing 1 µM of a pcDNA4/TO-based DHFR-GFP expression plasmid for electroporation (single pulse, 130 V, 500 µF, in 2 mm cuvettes with a Fischer Electroporator, Heidelberg, Germany). The electroporated cells were seeded into dishes and incubated at 37°C and 5% CO2 overnight before they were trypsinized again for the electroporation of compounds.
Generation of T-REx-293 cell lines stably expressing Abelson kinase 1-GFP and DHFR-GFP
5 × 106 T-REx-293 cells were transfected with 2 µg of the respective plasmids encoding GFP-tagged target proteins and a zeocin resistance cassette using effectene transfection reagent. Transfected cells were selected using zeocin at a concentration of 0.1 mg·mL−1 over a period of several weeks, eliminating all cells that did not express the zeocin resistance gene at sufficient amounts. The polyclonal T-REx-293 strain was kept under selective pressure of 100 µg·mL−1 zeocin and 5 mg·mL−1 blasticidin throughout the experiment.
Cellular import of drug molecules
T-REx-293 cells expressing low amounts of Abelson kinase 1 (ABL1)-GFP were detached by trypsin/EDTA and washed in medium. Subsequently, different concentrations of PD-Cy5 were electroporated into the cells (single pulse, 130 V, 500 µF, Fischer Electroporator). For direct uptake of PD-Cy5 without electroporation, T-REx-293 cells expressing ABL1-GFP were grown in 8-well chambered coverglass slides (Nunc, Rochester, New York, USA) and incubated with different concentrations of PD-Cy5 for 3 h at 37°C.
For analysing the interaction of MTX-Cy5 with DHFR-GFP, T-REx-293 cells stably transfected with DHFR-GFP were detached by trypsin/EDTA, washed in medium and resuspended in 100 µL Nucleofector Solution R containing 4 µM MTX-Cy5. MTX-Cy5 was imported by electroporation (Nucleofector, Lonza Cologne, Cologne, Germany) using the electroporation method I-013 according to the recommendations of the manufacturer. Subsequently, 500 µL RPMI medium was added carefully and the cells were incubated for 10 min at 37°C. Subsequently, the cell suspension was washed in medium twice by centrifugation. The cells were seeded in 8-well chambered coverglass slides.
HeLa cells expressing low amounts of DHFR-GFP were detached by trypsin/EDTA and washed in medium. Cells were electroporated in a solution of 2 µM MTX-Cy5 in RPMI medium (single pulse, 130 V, 500 µF, Fischer Electroporator). The cells were washed three times with medium and finally seeded in an 8-well chambered coverglass slide.
Intracellular FCCS measurements
FCCS measurements were performed with a TCS SP5 confocal microscope/dual channel FCS unit equipped with an HCX PL APO 63x N.A. 1.2 water immersion lens (Leica Microsystems, Mannheim, Germany) and with a ConfoCor2 FCS unit connected to an Axiovert 100M equipped with a C-Apochromat 40x N.A. 1.2 water immersion lens (Carl Zeiss, Jena, Germany). For the TCS SP5, fluorescence was split by a BS 625 beam splitter. Fluorescence in the green channel was detected using a BP 500–550 bandpass filter, fluorescence in the red channel with a BP 647–703 bandpass filter. For the ConfoCor2, fluorescence was split by an NFT 610 beam splitter, green fluorescence was detected with a BP 500–550 IR bandpass filter, red fluorescence with an LP 650 long pass filter. The GFP fusion proteins were excited with the 488 nm laser line of an argon-ion laser, Cy5 with a 633 helium-neon laser. In order to avoid high background signals resulting from autofluorescence and minimize bleaching, the power of both lasers was adjusted to a low intensity. The argon-ion laser was adjusted to 20 µW and 11 µW for the TCS SP5 and the ConfoCor2, respectively, the helium-neon laser to 19 µW and 8 µW, measured with an X1-1 laser power meter equipped with an LSM-9901 Luminous Flux detector head (Gigahetz-Optik, Puchheim, Germany) at the exit aperture of the lens.
Laser power is an important factor for FCS measurements, especially if high- and low-mobility fractions of molecules are to be analysed in parallel. For rapidly diffusing molecules, high laser powers are required in order to enable, within the short residence time of the molecule in the detection volume, the detection of a sufficient number of photons to accurately determine the diffusional autocorrelation time of the molecule. However, at such laser powers, molecules moving slowly through the laser focus may already be bleached. As a result, the fraction of slowly moving molecules, for example, target-bound drugs, may be underestimated. The laser powers that we selected covered the full dynamic range of diffusional autocorrelation times required for our analytical question.
For each set-up, these parameters led to comparable molecular brightness (counts per molecule – cpm) in both channels. Solution measurements were carried out with a sample volume of 20 µL in a 384-well plate (175 µm, low-base design, MMI, Eching, Germany). The short wavelength channel was calibrated with a 10 nM sodium fluorescein solution. For measurements of GFP fusion proteins, the structure parameter corresponding to the ratio of the radii of the detection volume along the optical axis and in the focal plane S = z0/ω0 was fixed to the value determined by fitting the autocorrelation function for the fluorescein measurement (Supporting Information Table S1). The structure parameter S for the long wavelength channel was determined by an autocorrelation measurement of a 10 nM S0387 solution. The spectral overlap of the detection volumes was determined by a cross-correlation measurement of a doubly labelled DNA standard, terminally labelled with Rhodamine Green and Cy5, similar to a standard described previously (Baudendistel et al., 2005). Cells with low expression of GFP fusion proteins were selected by confocal imaging. The detection volume for the FCCS measurements was placed inside the cytosol near the nucleus. Fluorescence of immobile molecules was removed by a pre-bleaching pulse of a few seconds at the same laser power employed for the subsequent measurements, followed by recording of either 5–60 autocorrelation functions of 1 s each for the ConfoCor2 or 5–10 measurements of 5 s each for the TCS SP5. Autocorrelation functions not affected by strong fluctuations of fluorescence that are caused by diffusion of vesicles were averaged for each cell for further analysis.
Autocorrelation functions of Cy5-labelled compounds were fitted with a 3D Gaussian algorithm containing two diffusing components and a triplet term (Equation 1)
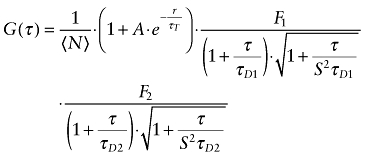 |
(1) |
in which A is the amplitude of the contribution of transitions into the triplet state to the total autocorrelation function, S is the structure parameter, N is the average number of particles in the detection volume, τT the relaxation time of the triplet state and τD1 and τD2 are the diffusional autocorrelation times of the rapid and slow component and F1 and F2 are the fractional contributions of both components with F1 + F2 = 1.
Either the diffusional autocorrelation times τD and fractions of both components were variable or the τD of the fast component was fixed to the value determined for the respective Cy5-labelled compound in solution multiplied by a factor of four in order to account for the higher viscosity of the cytoplasm (Berland et al., 1995; Schwille et al., 1999; Ellis, 2001). The number of molecules N and the fractional contributions A, F1 and F2 were variable in all cases.
The triplet times of GFP and Cy5 and the structure parameters in both channels were fixed to values derived from measurements in solution. For the cross-correlation curve, no triplet term was included in the fitting algorithm. The autocorrelation amplitudes of the GFP channel Ggreen(0) and the Cy5 channel Gred(0) and the cross-correlation amplitude GCC(0) were determined. The ratio CC(measured) between Ggreen(0) and GCC(0) represented the fraction of the Cy5-labelled compounds that formed a complex with GFP fusion proteins (Schwille et al., 1997a). The results were normalized to the value CC(max) determined for the doubly labelled DNA standard in the same experiment.
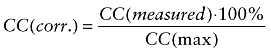 |
(2) |
Here, the device-dependent values were between 41% and 45 %. Due to the wavelength dependence of diffraction, this value can maximally amount to 54% in a perfectly adjusted microscope (Weidemann et al., 2002).
The particle number N derived from the autocorrelation functions was corrected for non-correlating autofluorescence background (Koppel, 1974),
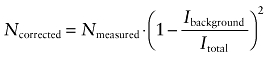 |
(3) |
as was the cpm.
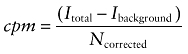 |
(4) |
Deviations of the cpm from values measured in solution would indicate the presence of fluorescence energy transfer (FRET) and quenching (Weidemann et al., 2002; Foldes-Papp, 2005) or poor overlap of the detection volume with the cell. The number of Cy5-labelled drug molecules bound to their respective GFP target proteins Nred-bound was calculated according to
| (5) |
With knowledge of the size of the detection volumes, which were approximately 0.3 fl for the detection volume of the green and 0.6 fl for the detection volume of the red channel, the concentrations of Cy5-labelled compounds and GFP-tagged target protein could be calculated. The determination of detection volumes was based on experimentally determined point-spread functions using TetraSpeck microspheres of 0.1 µm (Supporting Information Figure S1).
Assuming thermodynamic equilibrium for the interaction of compound and target, the dissociation constant Kd was determined by
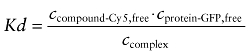 |
(6) |
Here, the concentration of unbound compound-Cy5 (Ccompound-Cy5, free) was deduced from the fraction of free molecules obtained from two-component fits of the autocorrelation functions. The concentration of the compound-Cy5 : target protein-GFP complex (Ccomplex) was determined by cross-correlation measurements, and the concentration of unbound GFP fusion protein (Cprotein-GFP, free) was calculated by the subtraction of Ccomplex from the total concentration of the GFP fusion protein.
Materials
The human cervical carcinoma cell line HeLa was obtained from the American Type Culture Collection (Manassas, Virginia, USA). T-REx-293 cells, the associated vector pcDNA4/TO, zeocin, blasticidin and TetraSpeck microspheres were purchased from Invitrogen (Carlsbad, California, USA). Effectene transfection reagent kits were obtained from QIAGEN (Hagen, Germany). Tissue culture media and fetal calf serum were purchased from PAN biotech (Aidenbach, Germany), the Cy5 monofunctional reactive dye from GE Healthcare (Little Chalfont, UK). The electroporation kits Cell Line Nucleofector Kit V and Cell Line Nucleofector Kit R were obtained from Lonza Cologne. The dye S0387, with Cy5-like spectral characteristics (Mader et al., 2004), was purchased from FEW Chemicals (Bitterfeld-Wolfen, Germany), the dye sodium fluorescein was obtained from Fluka (Deisenhofen, Germany). Drug/molecular target nomenclature follows Alexander et al. (2009).
Results
Synthesis of Cy5-labelled drug molecules
The pentamethine indocyanine dye Cy5 was selected as a fluorophore for the labelling of the drug molecules. With an emission maximum of around 670 nm, the fluorescence of this molecule shows little spectral overlap with the emission of the GFP (Southwick et al., 1990). For conjugation, functional groups of the drug molecules had to be selected that are not part of the pharmacophore. MTX was labelled at the free carboxyl group of the glutamate residue as described before (Gapski et al., 1975). For the compounds PD173956 and purvalanol B, the position for labelling was based on earlier SAR studies that verified that the conjugated compounds maintained their specific activity (Becker et al., 2004; Caligiuri et al., 2005).
In order to further minimize interference of the fluorophore with the binding of the compounds and to prevent FRET, the fluorophore was coupled via a PEG spacer. Because of its hydrophilicity, the PEG spacer should promote the solubility of the compounds and minimize hydrophobic interactions with membranes and proteins. Cy5 was employed as a commercially available, pre-activated succinimidyl active ester (Figure 1A).
Intracellular distribution of the Cy5-labelled compounds
First, we explored the intracellular distribution of the compounds. When using fluorescently labelled analogues, an influence of the fluorophore on the distribution of the molecule of interest is an important concern. HeLa and Hek293 cells were incubated with 5 µM of the respective compounds for 90 min. In comparison to previous studies, the incubation time with MTX was rather short (Kaufman et al., 1978). This short incubation time was mainly chosen to prevent unintended effects on cell viability.
The hydrophobic compounds PD-Cy5 and Pur-Cy5 were taken up efficiently into HeLa and Hek293 cells. Both conjugates were primarily localized in membranes and vesicular structures. In contrast, the uptake efficiency of the hydrophilic drug MTX-Cy5 was lower and its localization was more diffuse. For Cy5 alone, no uptake was observed (Figure 2).
Figure 2.
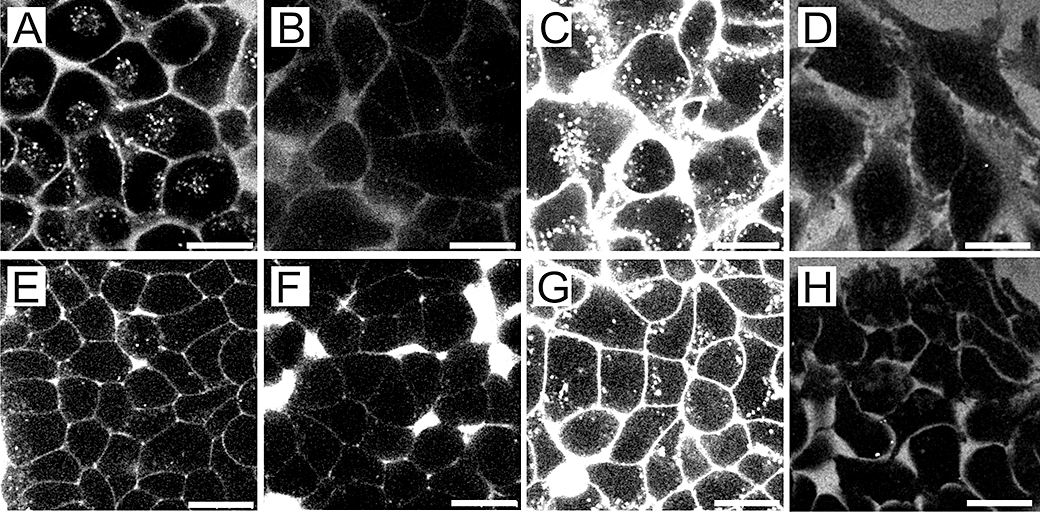
Intracellular localization of Cy5-labelled compounds and of Cy5. (A–D) HeLa cells and (E–H) Hek293 cells were treated with 5 µM of (A and E) PD-Cy5, (B and F) methotrexate-Cy5 and (C and G) Pur-Cy5 or free Cy5 (D–H) for 90 min at 37°C. The scale bars denote 20 µm.
While these results do not fully exclude an influence of the fluorophore on the characteristics of the compounds, they clearly show that the compound is decisive for the subcellular pharmacokinetics of the conjugates. As we had cells expressing GFP-tagged DHFR and the ABL1 kinase domain in our hands, the labelled compounds MTX-Cy5 and PD-Cy5 were selected for further experiments to address intracellular interactions by FCS and FCCS.
Intracellular interactions of MTX-Cy5 in DHFR-GFP-expressing HeLa cells
Confocal microscopy is very instructive to study the subcellular distribution of molecules (Pygall et al., 2007). However, the method provides little information on the possible association of compounds with homogenously distributed intracellular structures. Moreover, information on concentrations is very difficult to obtain. Fluorescence recovery after photobleaching (Meyvis et al., 1999), a method frequently used to study association and mobility of molecules inside cells, does not provide sufficient temporal resolution to analyse the mobility of small molecules and/or very low concentrations of fluorescent molecules (Lippincott-Schwartz et al., 2001). Therefore, for the analysis of the association of small molecules with subcellular structures at low concentrations, FCS and FCCS are the methods of choice.
When incubating cells with low micromolar concentrations of Cy5-MTX for 90 min, we failed to acquire intracellular autocorrelation functions. It has been shown previously, that the cellular uptake of fluorescein-labelled MTX is slow, requiring several hours to reach equilibrium (Kaufman et al., 1978). In order to achieve rapid cytoplasmic delivery, we explored the use of electroporation for the cytoplasmic import of the conjugate. Electroporation achieves a cytoplasmic import of solutes by application of short high-voltage pulses that induce transient pore formation in the plasma membrane (Gehl, 2003).
Subsequently, FCCS measurements were performed with a combined FCCS/confocal microscope while the cells were still not entirely flattened and attached to the bottom. Confocal imaging enabled a precise placement of the confocal measurement volume for FCCS measurements in three dimensions (Figure 1B-F). HeLa cells transiently expressing DHFR-GFP were electroporated with a 2 µM solution of MTX-Cy5. The intracellular DHFR-GFP concentration was in the range of 313 ± 187 nM in cells with low expression levels. The electroporation of compound yielded typical intracellular MTX-Cy5 concentrations of approximately 40–70 nM, thus being in a concentration range in which standard microscopy techniques fail to provide suitable results. The concentrations were derived from the amplitudes of the autocorrelation functions. In FCS, the amplitude is inversely correlated to the average number of molecules in the detection volume. Using experimentally determined detection volumes for both channels, these molecule numbers were converted into concentrations.
Dividing the fluorescence by the number of molecules yields the cpm. For cellular FCS measurements, large variations of this value between individual measurements indicate inhomogeneities in the sample or cell movement. Moreover, the cpm may vary depending on the molecular environment of a fluorophore.
The molecular brightness cpm of GFP and MTX-Cy5 were highly constant within the samples with 3.9 ± 1.2 kHz for GFP, and 0.8 ± 0.1 kHz and 1.2 ± 0.9 kHz for MTX-Cy5 outside and inside the cells, respectively. Furthermore, the intracellular molecular brightness of MTX-Cy5 was about the same as the extracellular cpm indicating that no significant quenching occurred in the cytosol.
In most cases, the first two measurements of a consecutive series of measurements in a single cell were affected by bleaching of immobilized MTX-Cy5 and therefore were omitted from the further analysis. The autocorrelation functions for MTX-Cy5 were fitted with two diffusing components and a triplet term (Figure 3A). The diffusional autocorrelation time τD reflects the average time that a fluorophore needs to diffuse through the detection volume. This time is directly correlated with the radius of the molecule. Therefore, the larger the molecule is, the bigger the τD will be. The ability to describe the autocorrelation functions with two components does not imply that MTX-Cy5 was only present in two molecular forms, as, for example, free drug molecule and in a molecular complex of a defined molecular weight. The molecular radius correlates with the cubic root of the molecular weight of a molecule, which means that it is much less dependent on the molecular weight. The high molecular weight form may especially represent many different complexes.
Figure 3.
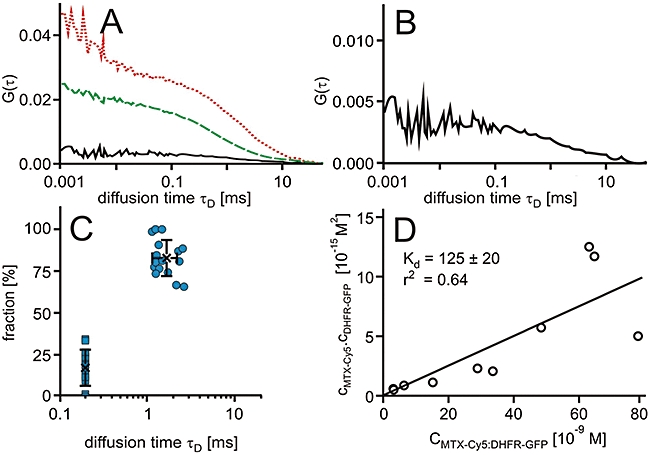
Interaction of methotrexate (MTX)-Cy5 in HeLa cells expressing green-fluorescent protein fusion proteins of dihydrofolate reductase (DHFR-GFP). (A) Characteristic autocorrelation functions for DHFR-GFP (dashed line, green) and MTX-Cy5 (dotted line, red) and the cross-correlation function for the DHFR-GFP : MTX-Cy5 complex (solid line, black). (B) Characteristic cross-correlation function at a larger scale. (C) Scatter plots of relative contributions to the autocorrelation function versus diffusional autocorrelation times derived from fits to the autocorrelation functions with an algorithm comprising two diffusing components in which the diffusion time τD of the low-molecular weight fraction was fixed to 200 µs. Averages (cross) and standard deviations are indicated. (D) Plot of the concentrations of the free molecules against the concentration of the complex and a linear regression.
The τD of the fast component was fixed to 200 µs. This value was based on the τD of MTX-Cy5 in phosphate-buffered saline of approximately 50 µs, considering the higher viscosity of the cytoplasm. In order to represent the variation between individual measurements, a graphical presentation according to Brock et al. (1999) was chosen (Figure 3C). Here, for each fraction, the relative contribution to the autocorrelation amplitude is plotted versus the diffusional autocorrelation time τD.
The bound MTX-Cy5 fraction accounted for approximately 83 ± 11% and had an average diffusional autocorrelation time τD of 1.7 ± 0.5 ms. The diffusional autocorrelation time τD of the 48 kDa DHFR-GFP was 1.3 ± 0.6 ms. Assuming identical mobilities for the DHFR-GFP and the MTX-Cy5, the MTX-Cy5 should have a diffusional autocorrelation time of about 2.2 ms. This estimate is based on the larger dimensions of the detection volume due to the wavelength dependence of diffraction (Weidemann et al., 2002) and the proportionality of τD ∼ ωx2.
In addition to the autocorrelation functions of MTX-Cy5 and DHFR-GFP, the cross-correlation functions were calculated. Cross-correlation amplitudes of 19.0 ± 6.4% in comparison to the autocorrelation amplitude of the DHFR-GFP channel were obtained (Figure 3A,B). By relating this value to the maximum attainable cross-correlation amplitude of 45% if all molecules were in a complex, these data showed that on average, approximately 42% of the MTX-Cy5 was bound to the specific target protein DHFR-GFP. This in turn means that on average, about 51% of the MTX-Cy5 molecules that were bound to high-molecular weight targets were in complex with DHFR-GFP. Based on these numbers, the dissociation constant Kd for the interaction of MTX-Cy5 with DHFR-GFP was calculated. The dissociation constant of MTX-Cy5 : DHFR-GFP determined in 10 HeLa cells was 125 ± 54 nM. As a means to validate that the concentration of drug–target complex derived from the cross-correlation amplitudes related to the concentration of interactors according to mass–action law, the concentrations of free interactors were plotted against the concentration of complex (Sudhaharan et al., 2009). Particularly in the lower concentration range, all points fell on a line, the slope of which corresponds to the Kd value, which was 125 ± 20 nM in this case (Figure 3D).
Intracellular interactions of MTX-Cy5 in DHFR-GFP-expressing HEK293 cells
In order to investigate to which degree this interaction pattern was a characteristic of the specific cell type, we next performed a corresponding set of measurements in HEK293 cells, expressing the DHFR-GFP fusion protein under control of the tetracycline promoter.
In the absence of tetracycline, an intracellular DHFR-GFP concentration of 27 ± 14 nM was obtained, indicating a leakiness of the promoter. As expected, the concentrations were considerably lower than in the HeLa cells in which DHFR-GFP was expressed transiently.
Again, the molecular brightness of GFP was highly constant within the samples. The cpm of the MTX-Cy5 varied between independent measurements (1.5 ± 1.0 kHz). This variation was likely due to the interactions of the compound with high-molecular weight structures that partially perturbed the autocorrelation measurements.
For the analysis of the autocorrelation functions (Figure 4A), first, the τD of the fast component was fixed again to 200 µs. In this case, the fast fraction accounted for 62% of the autocorrelation amplitude; the slow component accounted for 38% of MTX-Cy5 with a τD of 5.8 ± 2.6 ms. The τD of the slow component had a much stronger variation than for the HeLa cells (Figure 4C). Therefore, the autocorrelation functions were also fitted with both diffusional autocorrelation times τD kept variable. In that case, the fast fraction had an average τD of 437 ± 193 µs representing 65 ± 20% of the total amount of MTX-Cy5, the slow-diffusing fraction a τD of 132 ± 165 ms (35% of the total amount; Figure 4D). The comparability of results obtained with both fitting procedures strongly indicates that the rapidly diffusing component corresponds to free compound and that in this case, more than half of the molecules diffused freely in the cytoplasm.
Figure 4.
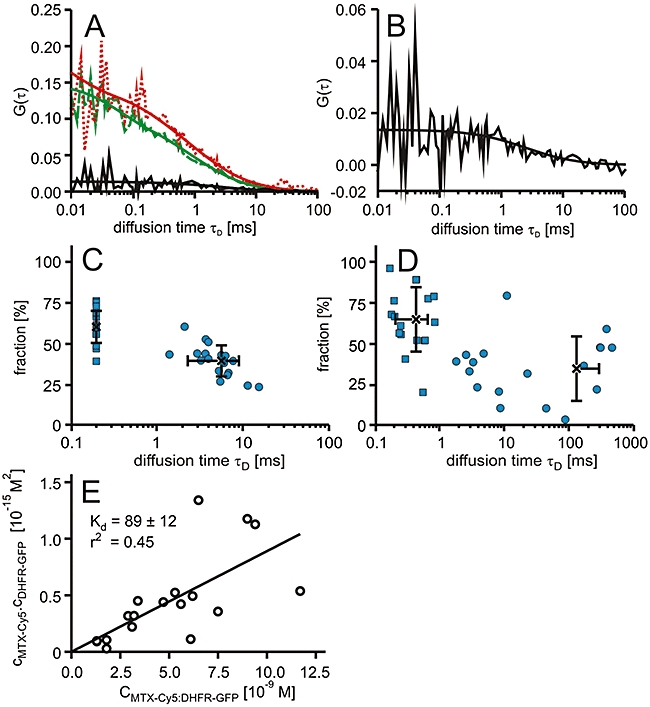
Interactions of methotrexate (MTX)-Cy5 in T-REx-293 cells expressing green-fluorescent protein fusion proteins of dihydrofolate reductase (DHFR-GFP). (A) Characteristic autocorrelation functions for DHFR-GFP (dashed line, green), MTX-Cy5 (dotted line, red) and the respective cross-correlation function (solid line) and the corresponding fits. (B) Characteristic cross-correlation function at a larger scale. (C,D) Dot plots of the relative fractions of each component versus the diffusional autocorrelation τD of MTX-Cy5 derived from two-component fits with averages (cross) and standard deviations, in which the fast component was fixed to 200 µs (C) or both diffusional autocorrelation times were allowed to vary (D). (E) Plot of the concentrations of the free molecules against the concentration of the complex and a linear regression.
The diffusional autocorrelation time τD of the DHFR-GFP was 972 ± 294 µs and hence slightly shorter than the one measured for HeLa cells. Thus, DHFR-GFP-bound MTX-Cy5 should have a diffusional autocorrelation time of about 1.6 ms. However, the bound compound showed a clearly longer diffusion time than expected for MTX-Cy5 bound to its target protein DHFR-GFP.
The cross-correlation functions had amplitudes in the range of 7.2 ± 1.9% of the one of the DHFR-GFP channel. These data indicate that on average, a fraction of approximately 16% of the MTX-Cy5 molecules were bound to the specific target protein DHFR-GFP. Given the total fraction of bound molecules of 38%, this result showed that on average, about 42% of the MTX-Cy5 molecules bound to high-molecular weight targets was bound to DHFR-GFP, considerably less than for the HeLa cells. The average dissociation constant determined from measurements in 18 cells was 86 ± 45 nM. Again, the plot of the concentrations of interactors against the concentration of complex demonstrated that for this group of cells, the concentrations of drug target complex followed the same binding equilibrium (Figure 4E).
From the linear regression analysis, a dissociation constant of 89 ± 12 nM was obtained.
The Kd value determined in these experiments was measured in the presence of intrinsic dihydrofolate (DHF) which competes with MTX-Cy5 for the binding sites on DHFR-GFP; thus, the calculated binding constants are regarded as apparent Kd values, that may differ from values determined in biochemical assays.
Intracellular binding of PD-Cy5 to high-molecular weight target structures in T-REx-293 cells
Next, we were interested to investigate the intracellular interaction pattern for the hydrophobic compound PD-Cy5. T-REx-293 cells expressing low amounts of ABL1-GFP in the range of 50–130 nM were electroporated with 0.2 µM PD-Cy5. This gave rise to intracellular PD-Cy5 concentrations of 320 ± 165 nM. These concentrations were significantly higher than the concentrations determined for MTX-Cy5. Also, in measurements in which cells were directly incubated with the compound, much higher intracellular concentrations were reached. These results indicate that PD-Cy5 enters cells much more efficiently than MTX-Cy5, and that also, during the electroporation procedure, for this hydrophobic compound, a direct crossing of the plasma membrane greatly contributed to cell entry.
Again, the autocorrelation functions were analysed with two-component fits in which the diffusion time τD of the low-molecular weight component was fixed to 200 µs (Figure 5). The determined diffusional autocorrelation time τD of the high-molecular weight fraction was high, mostly >10 ms, indicating an interaction between the compound and cellular structures that were significantly larger in molecular weight than the 60 kDa ABL1-GFP molecule. The diffusion time τD of the ABL1-GFP was 541 ± 112 µs, and therefore shorter than the one for the DHFR-GFP. The high-molecular weight fraction accounted for 70 ± 10% of the total PD-Cy5 fraction. Additionally, a large fraction of PD-Cy5 was bleached within the first seconds of the measurements indicating complete immobilization or trapping in vesicular structures. This strong bleaching of immobile fluorescence is therefore consistent with the distribution of fluorescence seen by confocal microscopy.
Figure 5.
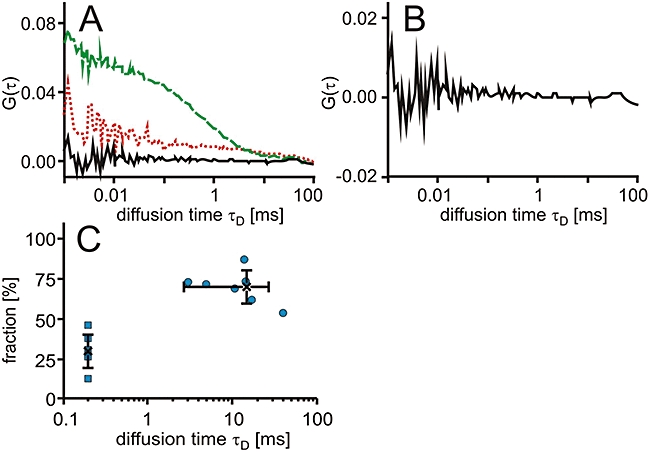
Interaction of PD-Cy5 in cells. T-REx-293 cells expressing ABL1-GFP were electroporated with PD-Cy5. (A) Characteristic autocorrelation functions for ABL1-GFP (dashed line, green) and PD-Cy5 (dotted line, red) and the cross-correlation function (solid line). (B) Characteristic cross-correlation function at a larger scale. (C) Dot plot of the relative contribution to the autocorrelation amplitude versus diffusional autocorrelation time τD for PD-Cy5 with the fast component fixed to 200 µs with averages (cross) and standard deviations.
Interestingly, no cross-correlation could be observed in the cells, although in vitro titrations had shown a high affinity of binding in cell lysate with Kd values in the lower nanomolar range. Varying the concentration of PD-Cy5 used for electroporation did not improve the result. The measurements with MTX-Cy5 had demonstrated the possibility of determining dissociation constants inside cells at a similar total degree of drug binding, as determined by FCS. Apparently, the fraction of PD-Cy5 molecules binding to their target was below the detection limit. We therefore conclude that off-target binding to other proteins and/or membranes inside the cell plays a much stronger role for PD-Cy5 than for MTX-Cy5.
Specificity of drug–target interactions
Finally, we intended to further confirm the inertness of Cy5 with respect to interactions inside the cell and with respect to drug–target interactions. As the live cell microscopy had shown that Cy5 by itself does not enter cells, intracellular autocorrelation functions were recorded for T-REx293 cells expressing DHFR-GFP electroporated with free Cy5 at a concentration of 2 µM (Figure 6). Autocorrelation functions were fitted with two diffusional components with either both components freely variable or the fast component fixed to four times the diffusional autocorrelation time of Cy5 in solution, that is, 128 µs. In both cases, about 90% of the total autocorrelation amplitude corresponded to the fast component, demonstrating that inside the cell, Cy5 engages in very little non-specific interactions with cellular components. For the free fit, a diffusional autocorrelation time of 185 ± 91 µs was obtained for the fast component, demonstrating that the diffusional hindrance was somewhat bigger than the previously reported reference value. Assuming an equivalent behaviour of Cy5 and MTX-Cy5, a fixed fast fraction of 300 µs had to be considered. However, comparison of fits in which both components were allowed to vary or the fast component fixed demonstrated that variations of the diffusional autocorrelation time by a factor of two was without influence on the analysis of binding equilibria.
Figure 6.
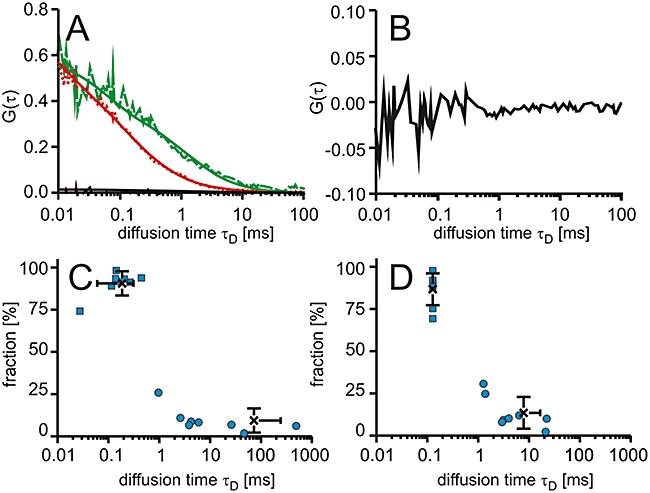
Intracellular diffusion of free Cy5. T-REx-293 cells expressing green-fluorescent protein fusion proteins of dihydrofolate reductase (DHFR-GFP) were electroporated with Cy5. (A) Characteristic autocorrelation functions for DHFR-GFP (dashed line, green) and Cy5 (dotted line, red) and the cross-correlation function (solid line). The fits of the autocorrelation functions are included. Cy5 was fitted with a model containing two diffusing components. (B) Characteristic cross-correlation function at a larger scale. (C,D) Dot plots of the relative contribution to the autocorrelation amplitude versus diffusional autocorrelation time τD for Cy5 with (C) the fast component variable and (D) the fast component fixed to 128 µs with averages (cross) and standard deviations.
Discussion
At present, detailed molecular analyses of the biological activity of drugs and drug candidates are restricted to in vitro tests. Cellular assays typically report on the activity of a compound in a very complex molecular context. Molecular interactions with the drug target and off-target binding are not addressed explicitly. In this work, we achieved such an analysis for small molecule anti-cancer drugs using laser scanning microscopy and a combination of FCS and FCCS. For this purpose, Cy5-labelled molecules were synthesized. To our knowledge, Cy5 has not been used for the labelling of small molecule drugs and their application in intracellular studies before.
Confocal microscopy revealed that Pur-Cy5 and PD-Cy5 were taken up into HeLa and Hek293 cells efficiently, but localized in vesicular structures, whereas MTX-Cy5 entered the cells poorly but showed a homogenous intracellular distribution. The localization and poor uptake for such short incubation times were consistent with results reported earlier for fluorescein-MTX (Kaufman et al., 1978). The subcellular distribution of PD-Cy5 and Pur-Cy5 demonstrated that sequestration in membranes and vesicular structures is a limiting factor for the intracellular bioavailability of these compounds. Indeed, FCS revealed that a considerable fraction of PD-Cy5 was bound to high-molecular weight structures. Cross-correlations reflecting an interaction of PD-Cy5 with the ABL1-GFP target molecule could not be detected. If such complexes were present, their fraction in comparison to the non-interacting molecules was too low to give rise to a cross-correlation amplitude. A too high number of non-interacting molecules limits the detectability of molecular interactions in FCCS as these lead to a decrease in the amplitude of the cross-correlation function. The ability of PD-Cy5 to interact with the target was confirmed in lysates of cells strongly overexpressing the fusion protein. However, in these cell lysates, a significant fraction of membranes had been removed by centrifugation prior to the FCCS measurements.
In contrast to PD-Cy5, the interaction of the hydrophilic drug MTX, as MTX-Cy5, with its specific molecular target DHFR could be detected in living cells. The Kd values determined in the T-REx-293 and HeLa cells were highly comparable, even though expression levels of the target protein varied by one order of magnitude and the overall interaction pattern was also different. In the T-REx-293 cells, about two-thirds of the molecules were freely diffusive, in contrast to only 17% in HeLa cells. Moreover, in T-REx-293 cells, a lower fraction of MTX-Cy5 specifically bound to DHFR-GFP. This reduced binding was accompanied by increased off-target binding to high-molecular weight intracellular structures, as indicated by the longer and more heterogeneous τD values.
It is a particular strength of FCCS that it can directly provide information on all molecular species in equilibrium. Therefore, a dissociation constant can be derived from one single measurement. In this case, in which off-target binding also occurred, the combination of FCS and FCCS proved to be highly powerful. Both measurement modalities provided complementary information on the fraction of bound molecules. With FCS, total binding of the compound was determined. FCCS selectively reported on those MTX-Cy5 molecules specifically bound to DHFR-GFP.
The dissociation constants were about two orders of magnitude higher than the values determined in vitro for purified DHFR (Mayer et al., 1986; Rajagopalan et al., 2002). A major reason for this difference will be the presence of endogenous DHF. Whereas in resting cells, the intracellular DHF concentration is in the lower nanomolar range, it can increase over several orders of magnitudes in proliferating cells (Assaraf, 2007). Both Hek293 and HeLa cells are highly proliferating cell lines, so it is reasonable to assume a fairly high DHF concentration.
Apart from the fusion protein, endogenous DHFR is also present inside the cells. However, since the binding of MTX-Cy5 to DHFR-GFP was measured in equilibrium, this endogenous protein will not affect the apparent Kd values measured for DHFR-GFP : MTX-Cy5 by FCCS. Instead, we may assume that endogenous DHFR may account at least for a fraction of the off-target binding, derived from the difference of bound fractions determined with FCS and FCCS.
In addition to a fraction with low mobility, in most cases, a considerable loss of fluorescence due to bleaching of immobile Cy5-labeled drug molecules occurred. This loss in fluorescence may result from compound associated with vesicular or cytoskeletal structures. The fraction of drug molecules available for binding to their respective targets will therefore be even lower than indicated by the combined FCS/FCCS measurements. Due to the heterogenous distribution of this immobile fraction, it is not possible to draw conclusions concerning the relative amounts of immobilized molecules and those available for target binding. However, in thermodynamic equilibrium, this immobilized fraction will not affect the dissociation constant for the drug–target interaction derived from the FCCS measurements.
In summary, this study demonstrates the potential of a combination of FCS and FCCS to simultaneously define target and off-target binding of drugs inside cells. Exploitation of the leakiness of inducible promoters turned out as an important technical detail to achieve reliably low expression levels of fusion proteins in only transiently transfected cells.
Acknowledgments
This work was supported by the BioChance Plus program of the German Ministry of Education and Research (FKZ 0313666). R. B. gratefully acknowledges financial support from the Volkswagen-Foundation (Nachwuchsgruppen an Universitäten, I/77 472).
Glossary
Abbreviations:
- Abl 1
Abelson kinase 1
- cpm
counts per molecule
- DHFR
dihydrofolate reductase
- FCS
fluorescence correlation spectroscopy
- FCCS
fluorescence cross- correlation spectroscopy
- FRET
fluorescence resonance energy transfer
- Hek
human embryonic kidney
- MTX
methotrexate
- RP-HPLC
reversed-phase high- performance liquid chromatography
- PBS
phosphate-buffered saline
- PD
Park Davis
- PEG
polyethylene glycol
- Pur
purvalanol
- RT
room temperature
Conflicts of interest
Intana Biosciences offers services in FCCS-based analyses of drug–target interactions.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Experimental determination of point spread functions (PSF). Confocal images of the same field of sub-resolution fluorescent beads imaged with (A) 488 nm and (B) 633 nm laser excitation (scale bar corresponds to 5 µm) and detection of fluorescence using (A) a BP 500–550 nm bandpass filter and (B) a BP 647–703 nm bandpass filter. Fluorescence between both channels was separated with a 625 nm beamsplitter. The avalanche photodetectors (APD) of the FCS module were used for imaging. (C) Intensity distributions along x and y were acquired along the axes of the crosses. (E) Intensity distributions along the optical axis were detected for the centres of the circles shown. The determination of intensity distributions was assisted by functions of the Leica instrument software. (E, F) Averaged intensity distributions of multiple objects in one image fit with a Gaussian function for (E) the x- and y-axis, and (F) the z-axis. The full widths of half maximum (FWHW) were taken as the radii of the detection volume. The effective FCS detection volume Veff was then calculated by
where ωxy is the radius for xy-axis and ωz for z-axis of detection volume. The effective FCS detection volumes for the green and red channel were 0.31 ± 0.05 fl and 0.61 ± 0.05 fl, respectively.
Table S1 Parameters obtained from fits to autocorrelation functions of the fluorescent standards1.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almela MJ, Torres PA, Lozano S, Herreros E. Characterization of the phospholipidogenic potential of 4(1H)-pyridone antimalarial derivatives. Toxicol In Vitro. 2009;23:1528–1534. doi: 10.1016/j.tiv.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Assaraf YG. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007;26:153–181. doi: 10.1007/s10555-007-9049-z. [DOI] [PubMed] [Google Scholar]
- Bacia K, Kim SA, Schwille P. Fluorescence cross-correlation spectroscopy in living cells. Nat Methods. 2006;3:83–89. doi: 10.1038/nmeth822. [DOI] [PubMed] [Google Scholar]
- Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59:748–758. doi: 10.1016/j.addr.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudendistel N, Muller G, Waldeck W, Angel P, Langowski J. Two-hybrid fluorescence cross-correlation spectroscopy detects protein–protein interactions in vivo. Chemphyschem. 2005;6:984–990. doi: 10.1002/cphc.200400639. [DOI] [PubMed] [Google Scholar]
- Becker F, Murthi K, Smith C, Come J, Costa-Roldan N, Kaufmann C, et al. A three-hybrid approach to scanning the proteome for targets of small molecule kinase inhibitors. Chem Biol. 2004;11:211–223. doi: 10.1016/j.chembiol.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Berland KM, So PTC, Gratton E. Two-photon fluorescence correlation spectroscopy: method and application to the intracellular environment. Biophys J. 1995;68:694–701. doi: 10.1016/S0006-3495(95)80230-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen CM, Sykes DB, Baehr C, Fricker G, Miller DS. Fluorescein-methotrexate transport in rat choroid plexus analyzed using confocal microscopy. Am J Physiol Renal Physiol. 2004;287:F562–F569. doi: 10.1152/ajprenal.00045.2003. [DOI] [PubMed] [Google Scholar]
- Brock R, Vàmosi G, Vereb G, Jovin TM. Rapid characterization of GFP fusion proteins by intracellular fluorescence correlation microscopy (FCM) Proc Natl Acad Sci U S A. 1999;96:10123–10128. doi: 10.1073/pnas.96.18.10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri M, Becker F, Murthi K, Kaplan F, Dedier S, Kaufmann C, et al. A proteome-wide CDK/CRK-specific kinase inhibitor promotes tumor cell death in the absence of cell cycle progression. Chem Biol. 2005;12:1103–1115. doi: 10.1016/j.chembiol.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Chaurand P, Schwartz SA, Reyzer ML, Caprioli RM. Imaging mass spectrometry: principles and potentials. Toxicol Pathol. 2005;33:92–101. doi: 10.1080/01926230590881862. [DOI] [PubMed] [Google Scholar]
- Clegg RM. Fluorescence resonance energy transfer. Curr Opin Biotechnol. 1995;6:103–110. doi: 10.1016/0958-1669(95)80016-6. [DOI] [PubMed] [Google Scholar]
- de Lange JH, Schipper NW, Schuurhuis GJ, ten Kate TK, van Heijningen TH, Pinedo HM, et al. Quantification by laser scan microscopy of intracellular doxorubicin distribution. Cytometry. 1992;13:571–576. doi: 10.1002/cyto.990130604. [DOI] [PubMed] [Google Scholar]
- Ellis RJ. Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr Opin Struct Biol. 2001;11:114–119. doi: 10.1016/s0959-440x(00)00172-x. [DOI] [PubMed] [Google Scholar]
- Foldes-Papp Z. How the molecule number is correctly quantified in two-color fluorescence cross-correlation spectroscopy: corrections for cross-talk and quenching in experiments. Curr Pharm Biotechnol. 2005;6:437–444. doi: 10.2174/138920105775159296. [DOI] [PubMed] [Google Scholar]
- Gapski GR, Whiteley JM, Rader JI, Cramer PL, Henderson GB, Neef V, et al. Synthesis of a fluorescent derivative of amethopterin. J Med Chem. 1975;18:526–528. doi: 10.1021/jm00239a020. [DOI] [PubMed] [Google Scholar]
- Gehl J. Electroporation: theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol Scand. 2003;177:437–447. doi: 10.1046/j.1365-201X.2003.01093.x. [DOI] [PubMed] [Google Scholar]
- Gray NS, Wodicka L, Thunnissen AM, Norman TC, Kwon S, Espinoza FH, et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science. 1998;281:533–538. doi: 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]
- Hink MA, Griep RA, Borst JW, van Hoek A, Eppink MH, Schots A, et al. Structural dynamics of green fluorescent protein alone and fused with a single chain Fv protein. J Biol Chem. 2000;275:17556–17560. doi: 10.1074/jbc.M001348200. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ, Bertino JR, Schimke RT. Quantitation of dihydrofolate reductase in individual parental and methotrexate-resistant murine cells. Use of a fluorescence activated cell sorter. J Biol Chem. 1978;253:5852–5860. [PubMed] [Google Scholar]
- Koopman WJ, Hink MA, Verkaart S, Visch HJ, Smeitink JA, Willems PH. Partial complex I inhibition decreases mitochondrial motility and increases matrix protein diffusion as revealed by fluorescence correlation spectroscopy. Biochim Biophys Acta. 2007;1767:940–947. doi: 10.1016/j.bbabio.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Koppel DE. Statistical accuracy in fluorescence correlation spectroscopy. Phys Rev A. 1974;10:1938–1945. [Google Scholar]
- Korn K, Krausz E. Cell-based high-content screening of small-molecule libraries. Curr Opin Chem Biol. 2007;11:503–510. doi: 10.1016/j.cbpa.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Kraker AJ, Hartl BG, Amar AM, Barvian MR, Showalter HD, Moore CW. Biochemical and cellular effects of c-Src kinase-selective pyrido[2, 3-d]pyrimidine tyrosine kinase inhibitors. Biochem Pharmacol. 2000;60:885–898. doi: 10.1016/s0006-2952(00)00405-6. [DOI] [PubMed] [Google Scholar]
- Lang P, Yeow K, Nichols A, Scheer A. Cellular imaging in drug discovery. Nat Rev Drug Discov. 2006;5:343–356. doi: 10.1038/nrd2008. [DOI] [PubMed] [Google Scholar]
- Lankelma J, Dekker H, Luque FR, Luykx S, Hoekman K, van der Valk P, et al. Doxorubicin gradients in human breast cancer. Clin Cancer Res. 1999;5:1703–1707. [PubMed] [Google Scholar]
- Lasch P, Naumann D. Spatial resolution in infrared microspectroscopic imaging of tissues. Biochim Biophys Acta. 2006;1758:814–829. doi: 10.1016/j.bbamem.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Snapp E, Kenworthy A. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol. 2001;2:445–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- Mader O, Reiner K, Egelhaaf H-J, Fischer R, Brock R. Structure property analysis of pentamethine indocynanine dyes: identification of a new dye for life science applications. Bioconjug Chem. 2004;15:70–78. doi: 10.1021/bc034191h. [DOI] [PubMed] [Google Scholar]
- Mayer RJ, Chen JT, Taira K, Fierke CA, Benkovic SJ. Importance of a hydrophobic residue in binding and catalysis by dihydrofolate reductase. Proc Natl Acad Sci U S A. 1986;83:7718–7720. doi: 10.1073/pnas.83.20.7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meseth U, Wohland T, Rigler R, Vogel H. Resolution of fluorescence correlation measurements. Biophys J. 1999;76:1619–1631. doi: 10.1016/S0006-3495(99)77321-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyvis TKL, De Smedt SC, Van Oostveldt P, Demeester J. Fluorescence recovery after photobleaching: a versatile tool for mobility and interaction measurements in pharmaceutical research. Pharm Res. 1999;16:1153–1162. doi: 10.1023/a:1011924909138. [DOI] [PubMed] [Google Scholar]
- Michelman-Ribeiro A, Mazza D, Rosales T, Stasevich TJ, Boukari H, Rishi V, et al. Direct measurement of association and dissociation rates of DNA binding in live cells by fluorescence correlation spectroscopy. Biophys J. 2009;97:337–346. doi: 10.1016/j.bpj.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman ZE, Slack MD, Feng Y, Mitchison TJ, Wu LF, Altschuler SJ. Multidimensional drug profiling by automated microscopy. Science. 2004;306:1194–1198. doi: 10.1126/science.1100709. [DOI] [PubMed] [Google Scholar]
- Pygall SR, Whetstone J, Timmins P, Melia CD. Pharmaceutical applications of confocal laser scanning microscopy: the physical characterisation of pharmaceutical systems. Adv Drug Deliv Rev. 2007;59:1434–1452. doi: 10.1016/j.addr.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Rajagopalan PT, Zhang Z, McCourt L, Dwyer M, Benkovic SJ, Hammes GG. Interaction of dihydrofolate reductase with methotrexate: ensemble and single-molecule kinetics. Proc Natl Acad Sci U S A. 2002;99:13481–13486. doi: 10.1073/pnas.172501499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigler R, Mets Ü, Widengren J, Kask P. Fluorescence correlation spectroscopy with high count rate and low background: analysis of translational diffusion. Eur Biophys J. 1993;22:169–175. [Google Scholar]
- Schwille P, Bieschke J, Oehlenschläger F. Kinetic investigations by fluorescence correlation spectroscopy: the analytical and diagnostic potential of diffusion studies. Biophys Chem. 1997a;66:211–228. doi: 10.1016/s0301-4622(97)00061-6. [DOI] [PubMed] [Google Scholar]
- Schwille P, Meyer-Almes F-J, Rigler R. Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys J. 1997b;72:1878–1886. doi: 10.1016/S0006-3495(97)78833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille P, Haupts U, Maiti S, Webb WW. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys J. 1999;77:2251–2265. doi: 10.1016/S0006-3495(99)77065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signor L, Varesio E, Staack RF, Starke V, Richter WF, Hopfgartner G. Analysis of erlotinib and its metabolites in rat tissue sections by MALDI quadrupole time-of-flight mass spectrometry. J Mass Spectrom. 2007;42:900–909. doi: 10.1002/jms.1225. [DOI] [PubMed] [Google Scholar]
- Southwick PL, Ernst LA, Tauriello EW, Parker SR, Mujumdar RB, Mujumdar SR, et al. Cyanine dye labeling reagents – Carboxymethylindocyanine succinimidyl esters. Cytometry. 1990;11:418–430. doi: 10.1002/cyto.990110313. [DOI] [PubMed] [Google Scholar]
- Stumpf WE. Drug localization and targeting with receptor microscopic autoradiography. J Pharmacol Toxicol Methods. 2005;51:25–40. doi: 10.1016/j.vascn.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Sudhaharan T, Liu P, Foo YH, Bu W, Lim KB, Wohland T, et al. Determination of in vivo dissociation constant, KD, of Cdc42-effector complexes in live mammalian cells using single wavelength fluorescence cross-correlation spectroscopy. J Biol Chem. 2009;284:13602–13609. doi: 10.1074/jbc.M900894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsmuth M, Waldeck W, Langowski J. Anomalous diffusion of fluorescent probes inside living cell nuclei investigated by spatially-resolved fluorescence correlation spectroscopy. J Mol Biol. 2000;298:677–689. doi: 10.1006/jmbi.2000.3692. [DOI] [PubMed] [Google Scholar]
- Washtien WL. Thymidylate synthetase levels as a factor in 5-fluorodeoxyuridine and methotrexate cytotoxicity in gastrointestinal tumor cells. Mol Pharmacol. 1982;21:723–728. [PubMed] [Google Scholar]
- Weidemann T, Wachsmuth M, Tewes M, Rippe K, Langowski J. Analysis of ligand binding by two-color fluorescence cross-correlation spectroscopy. Single Mol. 2002;3:49–61. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.