

Abstract
Background and purpose:
Tetracyclines were recently found to induce tumour cell death, but the early processes involved in this cytotoxic effect remain unclear.
Experimental approach:
Viability of human and mouse melanoma cells was determined by MTT assay and flow cytometry. Kinase/protein/caspase activation was measured by Western blotting and mitochondrial membrane potential (ΔΨm) was analyzed by fluorescence microscopy and flow cytometry.
Key results:
Human and mouse melanoma cells were treated with doxycycline or minocycline but only doxycycline was cytotoxic. This cell death (apoptosis) in A2058 cells involved activation of caspase-3, -7 and -9 and contributed to inhibition, by doxycycline, of matrix metalloproteinase (MMP) activity and migration of these cells. Doxycycline induced intra-cellular reactive oxygen species (ROS) production, apoptosis signal-regulated kinase 1 (ASK1), c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) activation at an early stage of treatment and induced mitochondrial cytochrome c release into cytosol and ΔΨm change during apoptosis. The JNK inhibitor/small interference RNA inhibited doxycycline-induced JNK activation, ΔΨm change and apoptosis, but did not affect ASK1 activation, suggesting a role of ASK1 for JNK activation in melanoma cell apoptosis. Two ROS scavengers reduced doxycycline-induced JNK and caspase activation, and apoptosis. Taken together, the results suggest the involvement of a ROS-ASK1-JNK pathway in doxycycline-induced melanoma cell apoptosis.
Conclusions and implications:
We have shown a promising cytotoxic effect of doxycycline on melanoma cells, have identified ROS and ASK1 as the possible initiators and have demonstrated that JNK activation is necessary for doxycycline-induced melanoma cell apoptosis.
Keywords: apoptosis, ASK, caspase, doxycycline, free radical, JNK, melanoma, mitochondria, ROS, tetracycline
Introduction
Tetracyclines, including tetracycline, doxycycline and minocycline, are widely used antibiotics. However, recent studies have shown that tetracyclines possess other potentially therapeutic effects that are distinct from their antimicrobial actions (Kroon et al., 1984; Ryan et al., 1996). For example, doxycycline inhibits the activity of matrix metalloproteinases (MMPs), including collagenase, gelatinase and stromelysin in vitro (Gilbertson-Beadling et al., 1995). In several in vitro studies, doxycycline and minocycline were found to decrease human endothelial cell proliferation and tube formation, tumour cell migration, inducible nitric oxide synthetase expression and induce macrophage apoptosis (Bettany and Wolowacz, 1998; Bettany et al., 2000).
Melanoma, with a rapidly rising incidence, is the deadliest form of skin cancer. In the United States, melanoma is the fifth and sixth most common cancer in men and women respectively with an estimated lifetime average risk of 1 in 75 (Jemal et al., 2004). Although melanoma accounts for only 4% of all dermatological cancers, it is responsible for 80% of deaths from skin cancer; only 14% of patients with metastatic melanoma survive for 5 years (Miller and Mihm, 2006). In its advanced stages, melanoma is highly resistant to medical treatment. Dacarbazine remains the most efficacious therapy for metastatic melanoma. The other two drugs approved for metastatic melanoma are high-dose interleukin-2 (IL-2) and hydroxyurea, but their response rates do not approach that of dacarbazine (Ibrahim and Haluska, 2009).
A previous study reported that doxycycline and chemically modified tetracyclines (CMTs) could inhibit melanoma cell invasion and metastasis (Seftor et al., 1998). The authors suggested that melanoma cells treated with doxycycline and CMT-1, -3 and -6 were less invasive in vitro, which were coincident with their ability to differentially suppress extracellular levels of gelatinase activity. However, it was recently shown that tetracycline analogues (TCNAs) can also act as apoptotic inducers and the activity appears to be independent of their anti-MMP activity (Fife et al., 1997; D'Agostino et al., 2003). For example, doxycycline induces apoptosis in human pancreatic cancer cells (Mouratidis et al., 2007). Doxycycline and CMT-3 induce apoptosis in human prostate and colon cancer cells (Lokeshwar et al., 2002; Onoda et al., 2006). Although caspase-dependent and/or -independent pathways are suggested to be involved in tetracycline-induced cancer cell death, the early process(es) responsible for the cytotoxicity remains unclear.
At a molecular level, abnormal activation of the mitogen-activated protein kinase (MAPK) signalling pathway, especially extracellular signal-regulated kinase (ERK), stimulates growth in melanoma cells (Lin et al., 1998; Brunet et al., 1999; Welsh et al., 2001). Activation of ERK is the result of somatic mutations of N-Ras, which are associated with about 15% of melanomas, or BRAF, which are associated with about 50% of melanomas (Miller and Mihm, 2006). The MAPK family, including ERK, c-Jun N-terminal kinase (JNK) and p38 MAPK, are also activated in human epidermal keratinocytes following exposure to UV irradiation (Peus et al., 1999; 2000;) and activation of p38 MAPK correlates with skin cell death in response to UV (Assefa et al., 2000), suggesting a differential role of MAPKs in controlling skin cell behaviours under physiological and pathological conditions. It was reported that apoptosis signal-regulating kinase 1 [ASK1, also known as MAPK kinase kinase (MAPKKK)] is a serine/threonine protein kinase, which can activate both the SEK1-JNK and MKK3/6-p38 MAPK pathways and constitute a pivotal signalling pathway in cytokine- and stress-induced apoptosis (Matsuzawa and Ichijo, 2001).
Given that tetracyclines are widely used in dermatology, in this study, we have investigated the in vitro cytotoxic activity of two commonly used tetracyclines, doxycycline and minocycline. We found that doxycycline exhibited a stronger cytotoxic effect than minocycline on melanoma cells. Moreover, the cytotoxicity contributed to the inhibitory effect of doxycycline on melanoma cell MMP activity and migration. Caspase(s) activation, mitochondrial cytochrome c release and change in mitochondrial membrane potential (ΔΨm) occurred during doxycycline-induced melanoma cell apoptosis. JNK and p38 MAPK were activated at the early stage of doxycycline treatment, but only JNK activation was necessary for doxycycline-induced ΔΨm change and melanoma cell death. Moreover, we found that reactive oxygen species (ROS) and ASK1 were increased and activated respectively. Two ROS scavengers reduced doxycycline-induced caspase activation and melanoma cell death. Based on our findings, we suggest that ROS, ASK1 and JNK are involved in the early stages of doxycycline-induced melanoma cell apoptosis.
Methods
Cell cultures
A2058 human metastatic melanoma cell line, A375 malignant melanoma cell line and B16F10 murine melanoma cells were purchased from Food Industry Research and Development Institute (Hsinchu, Taiwan). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), penicillin (100 units·mL−1), streptomycin (100 µg·mL−1) and fungizone (250 ng·mL−1) (defined as ‘complete medium’) (Invitrogen Life Technologies, Carlsbad, CA, USA). A549 cells, a human pulmonary epithelial carcinoma cell line with type II alveolar epithelial cell differentiation, were cultured in DMEM/Ham's F-12 nutrient mixture containing 10% FBS, penicillin (100 units·mL−1), streptomycin (100 µg·mL−1) and fungizone (250 ng·mL−1). Except when otherwise indicated, human A2058 melanoma cells were used in the experiments. For some experiments, rat aortic smooth muscle cells were used and prepared as previously described (Lo et al., 2007).
Cell growth/viability assay (MTT assay)
Cells growing in complete medium and reaching 80–90% of confluency were treated with tetracyclines for the indicated times. After tetracycline treatment, cells were incubated with 0.5 mg·mL−1 MTT for 2 h at 37°C. Formazan crystals resulting from MTT reduction were dissolved by adding DMSO and gently agitating for 20 min. The absorbance of the supernatant was then measured spectrophotometrically in an ELISA reader at 550 nm.
DNA fragmentation assay
A procedure for DNA extraction from apoptotic cells has been previously described with a minor modification (Wu et al., 2001). Doxycycline or PBS-treated cancer cells were collected by centrifugation, re-suspended in 1 mL Hank's buffered salt solution (HBSS) (Invitrogen Life Technologies, Carlsbad, CA, USA), and fixed by adding 10 mL pre-chilled 70% ethanol and kept at −20°C for 24 h. After centrifugation, the cell pellets were re-suspended in 40 µL phosphate-citrate buffer (pH 7.8) and incubated at room temperature for 30 min to extract DNA. The mixture was analyzed by 1% agarose gel electrophoresis. DNA ladders were visualized by ethidium bromide staining.
DNA content analysis
DNA content was analyzed by flow cytometry. Cancer cells with or without tetracycline treatment were collected by centrifugation and adjusted to 3 × 106 cells·mL−1, pre-chilled absolute methanol was added to 0.5 mL of the cells and incubated at 4°C overnight. Methanol was then removed by centrifugation and DNA of the cells was stained with PI staining solution (100 µg·mL−1 PI, 0.1% Triton-X, 1 mM EDTA in PBS) in the presence of equal volume of DNase-free RNase (200 µg·mL−1) and analyzed immediately by a Partec CyFlow ML cytometer (Partech GmBH, Munster, Germany). Debris and clumps were excluded by monitoring DNA area (FL3-A) and peak (i.e. signal height, FL3-W). Fluorescence signals from 10 000-gated cells were collected.
Analysis of early and late apoptosis of melanoma cells
Melanoma cells undergoing early/late apoptosis or necrosis were analyzed by annexin V-FITC and PI staining according to the protocol supplied by the manufacturer. Cells were left untreated or incubated with doxycycline for the indicated times at 37°C. Cells were collected, washed with phosphate-buffered saline, and re-suspended in 100 µL of binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2). Cells were then incubated with 5 µL of annexin V-FITC for 10 min at the room temperature in the dark, followed by the addition of PI to binding buffer and analyzed immediately by the Partec CyFlow ML cytometer.
Migration assay and gelatin zymography
Migration assay with melanoma cells was performed using a modified Boyden chamber model (Transwell apparatus, 8.0-µm pore size, Costar) according to the previously described protocol with a minor modification (Lo et al., 2007). Briefly, the lower face of polycarbonate filter (Transwell insert) was coated with collagen (10 µg·mL−1) for 30 min in the laminar flow hood. The lower chamber was filled with serum (10%)-containing medium. Melanoma cells (2.5 × 105 cells·mL−1) were plated to the upper chamber in the presence of doxycycline. For some experiments, cells were pre-treated with DMSO or pan-caspase inhibitors for 30 min. After 16 h of incubation, all non-migrant cells were removed from the upper face of the Transwell membrane with a cotton swab and migrant cells were fixed and stained with 0.5% toluidine blue in 4% paraformaldehyde. Migration was quantified by counting the number of stained cells per × 100 field (high power field, HPF) under the phase-contrast microscope (Leica DMIL®) and photographed.
The zymography was performed using a previously described method (Hung et al., 2005). Samples were separated on an 8.5% polyacrylamide gel containing 0.1% gelatin. After electrophoresis, gels were washed and then developed in the buffer (50 mM Tris-HCl, pH 7.6, containing 5 mM CaCl2, 1 mM ZnCl2, 1% Triton X-100 and 0.02% NaN3) at 37°C for 24 h. The gel was stained with 0.1% Coomassie Brilliant Blue R250 (Sigma Co., USA), and the location of gelatinolytic activity was detected as clear bands.
Protein preparation and Western blotting
For total protein preparation, cells were washed with pre-chilled PBS and lysed in radioimmunoprecipitation assay buffer (RIPA buffer). After sonication, the lysate was centrifuged (14 000 ×g for 15 min at 4°C), and supernatant was transferred to a tube. The protein content was quantified by the Pierce protein assay kit (Pierce, Rockford, IL). Total proteins were separated by electrophoresis, electroblotted onto PVDF membranes, and then probed using primary mAbs. Immunoblots were detected by enhanced chemiluminescence (Perkin-Elmer, Waltham, MA, USA).
Preparation of cytosolic protein fractions without mitochondria was performed as previously described (Wang et al., 2006). Briefly, cells were added to buffer A (20 mM HEPES, pH 7.5, 250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM EDTA, 0.5 mM EGTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride) and placed on ice for 15 min. After centrifugation at 4°C for 10 min (600 × g), the resultant supernatant was centrifuged a second time under the same conditions. This supernatant was centrifuged at 4°C for 15 min at 10 000 × g. The pellet was redissolved in buffer A and was the mitochondrial fraction. The supernatant was further centrifuged at 4°C for 10 min at 10 000 × g. This last supernatant comprised the cytosolic protein fraction.
Measurement of mitochondrial membrane potential (ΔΨm)
The values of ΔΨm during melanoma cell death was determined by flow cytometry and fluorescence microscopy using Cayman's JC-1 mitochondrial membrane potential assay kit according to the manufacturer's protocol. Briefly, 75 µL of JC-1 staining solution in 1 mL culture medium was added to each well of plate and cells were incubated in a CO2 incubator at 37°C for 30 min. After a brief wash with serum-free medium, cells were directly analyzed by the Leica DMIL® microscope or flowcytometer.
Detection of intra-cellular reactive oxygen species (ROS)
Intra-cellular ROS were assayed by fluorescence microscopy. Melanoma cells grown on 24-well culture plates were loaded with 5-(and-6)-chloromethyl-2′7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA, 2.5 µg·mL−1) (Invitrogen) and were incubated at 37°C for 10 min. After a brief wash, cells were treated with H2O2 or doxycycline at 37°C for 10 min and immediately analyzed by the fluorescence microscope (Leica DMIL®) and photographed by a digital camera. Alternatively, cells were washed by PBS and lysed by RIPA buffer and ROS production was measured by the fluorescence plate reader (Wallac Victor 31420 multilabel counter, Perkin Elmer, Turku, Finland) using excitation and emission wavelength at 485 and 535 nm.
Small interference RNA transfection
As JNK1 (∼46 kDa) was activated by doxycycline, small interference RNA (siRNA) targeting JNK1 was selected and synthesized. Dharmacon ON-TARGET plus SMARTpool JNK1 (gene id: 5599) and negative control siRNA (Thermo Scientific, Dharmacon RNAi Technologies) were synthesized. Transfection was performed according to the manufacturer's protocol with some modifications. The A2058 melanoma cells (5 × 104 per well) were seeded in a 24-well plate for overnight in complete medium. siRNA (2 µM, 25 µL) was added to serum-free medium (25 µL). After incubation with DharmaFECT transfection reagent (2 µL) and medium for 20 min, they were diluted with antibiotic-free complete medium (0.4 mL) and were added to cell cultures for 72 h. The JNK1 expression and activation were analyzed by Western blotting and cell viability after doxycycline treatment for an additional 24 h was determined by MTT assay.
Statistical analysis
Data are expressed as mean ± standard error mean (SEM). Comparison of means of two groups of data was made by using the unpaired and two-tailed Student t test.
Materials
Doxycycline hyclate (MW = 512.94), minocycline hydrochloride (MW = 493.94), bovine type I collagen, propidium iodide (PI), protease inhibitors for Western blotting, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (+/−)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) and vitamin C were purchased from Sigma Chemical Co. (St Louis, MO, USA). Caspase inhibitors were from R&D systems, Inc. (MN, USA). JC-1 kit was purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). The antibodies raised against ASK1 and phospho-ERK1/2 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies raised against phospho-p38, phospho-JNK, phospho-ASK1 (Thr845), caspase-3, -7, -9, poly(ADP-ribose) polymerase (PARP), and cytochrome c were from New England Biolabs, Inc. (Beverly, MA, USA). The antibodies for total p38 and ERK1/2 were from R&D systems, Inc. (MN, USA). Annexin-V-fluorescein isothiocyanate (Annexin-V-FITC) conjugate was from Biosource International, Inc. (Camarillo, CA, USA). The antibody for α-tubulin was purchased from Calbiochem EMD Bioscience Inc. (San Diego, CA, USA). Doxycycline was freshly prepared for each experiment and 20 µg·mL−1 of doxycycline is about 39 µM.
Results
Effect of doxycycline and minocycline on growth of melanoma and lung epithelial carcinoma cells
The effects of two tetracyclines, doxycycline and minocycline, on growth of human and mouse melanoma and lung epithelial carcinoma cells were examined by MTT assay. Figure 1A shows that doxycycline caused an inhibition on growth of melanoma and lung epithelial carcinoma cells. Doxycycline at the tested concentrations induced much more growth inhibition on melanoma than on lung epithelial carcinoma cells. Among these melanoma cells, the concentration-dependent effect of doxycycline on growth inhibition was found to be marked in mouse B16F10 melanoma cells than in human A2058 and A375 melanoma cells. The IC50 of doxycycline on A2058, A375 and B16F10 melanoma cell growth were estimated to be 15.7, 14.8 and 5.9 µg·mL−1 respectively. On the other hand, minocycline, a tetracycline with a similar structure to doxycycline, only slightly affected growth in melanoma and lung epithelial carcinoma cells (Figure 1B). Comparison of their effect at an equal concentration (40 µM) demonstrated their differential cytotoxic effect on melanoma cells (Figure 1B, inset). We next examined the time-dependent effect of doxycycline on growth of human 2058 melanoma cells. Figure 1C shows that doxycycline slightly affected melanoma cell growth at 8 h but markedly reduced cell growth at incubation times greater than 16 h. In parallel, we tested whether doxycycline affected normal cell growth and found that growth of rat aortic vascular smooth muscle cells (SMCs) was not affected even at 50 µg·mL−1 for 24 h (data not shown). As human A2058 melanoma cell growth was reduced to about 60% by doxycycline at 20 µg·mL−1, this melanoma cell line and concentration were used in the subsequent experiments in this study.
Figure 1.
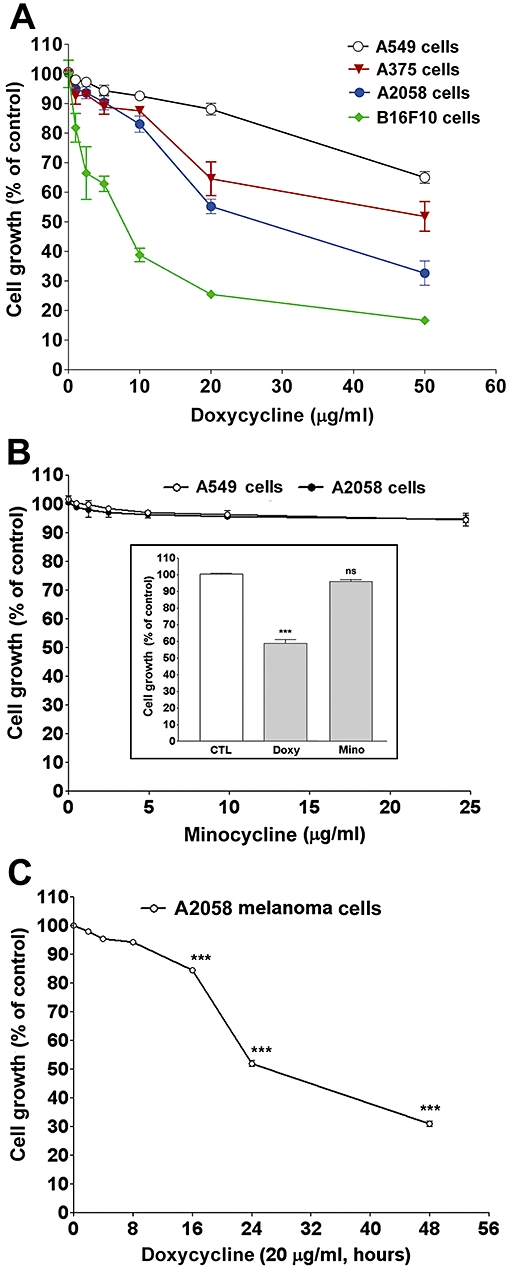
Effects of doxycycline (doxy) and minocycline (mino) on growth of melanoma and lung epithelial cancer cells. (A) The cells (A549 human lung carcinoma; A375 human melanoma; A2058 human melanoma; B16F10 mouse melanoma) were treated with PBS or doxycycline for 24 h. (B) Human lung cancer and melanoma cells were treated with minocycline or tetracyclines (inset) for 24 h. Inset: Comparison of doxycycline and minocycline effect on human A2058 melanoma cell growth, at the same concentration (40 µM) (C) Time-dependent effect of doxycycline on human A2058 melanoma cell growth. Cell growth was measured by MTT assay. Data were presented as percentage of control and were mean ± SEM (n = 3–4). ***P < 0.001 versus control.
Doxycycline induces apoptosis in melanoma cells
We have examined inhibitory effects of doxycycline on cell growth/viability by MTT assay (Figure 1). We also observed some floating cells in melanoma cell culture medium upon doxycycline treatment (data not shown), suggesting that interference of cell-extracellular matrix and/or cell death were caused by doxycycline in this process. To determine whether doxycycline caused melanoma cell apoptosis, a DNA content analysis was performed. As shown in Figure 2A, exposure of human A2058 melanoma cells to doxycycline resulted in a significant increase in the proportion of subG0/G1 phase cells, that is, the appearance of cells with a fractional DNA content. Doxycycline at 10 and 20 µg·mL−1 induced a marked increase in melanoma cell apoptosis to 26.8 and 33.6% respectively. In contrast, doxycycline at 10 and 20 µg·mL−1 only induced 2.4 and 6.7% of apoptosis in A549 lung epithelial carcinoma cells respectively. The differential effects of doxycycline in causing apoptosis of these two cancer cells were confirmed by DNA fragmentation analysis, in which doxycycline induced clearly fragmented DNA ladders in melanoma but only slightly fragmented DNA ladders in lung epithelial carcinoma cells (Figure 2B).
Figure 2.
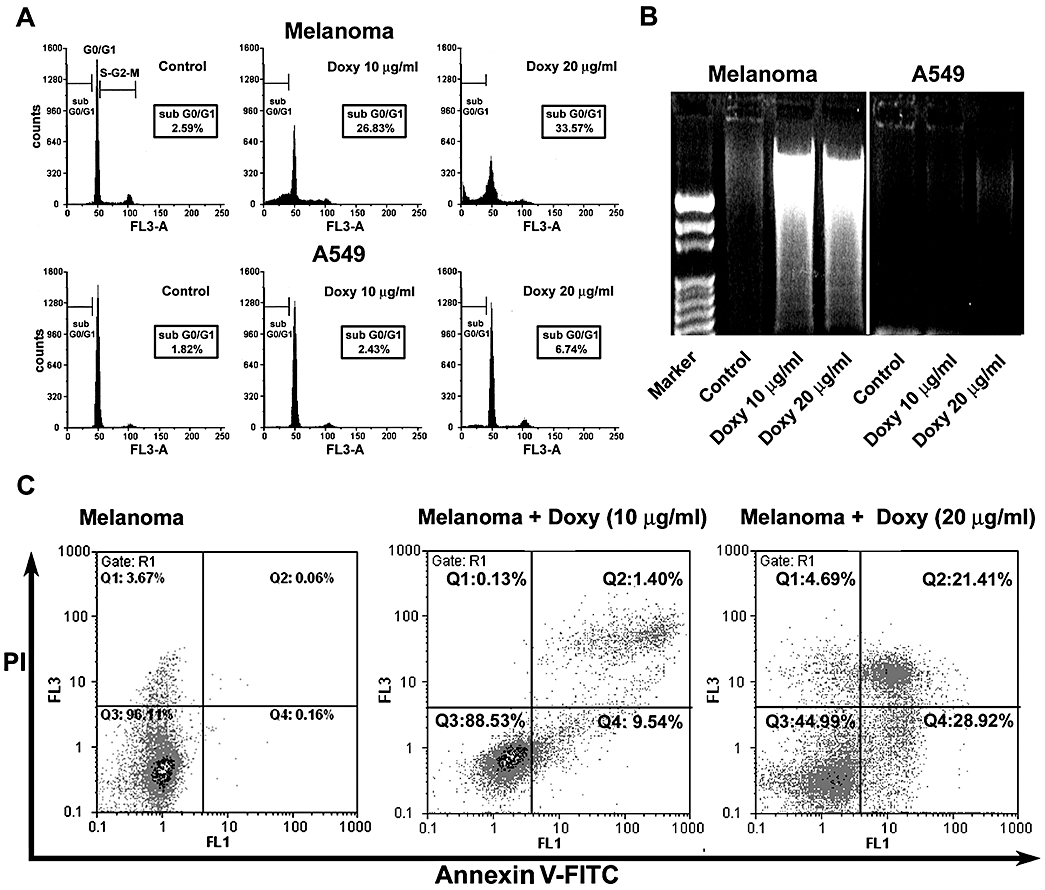
Doxycycline (doxy) induces melanoma cell apoptosis. Human melanoma or A549 lung cancer cells were treated with PBS or doxycycline for 24 h. (A and C) Cells were collected, stained with (A) propidium iodide (PI) or (C) PI + annexin V-FITC and followed by analysis with flow cytometry. (B) Cells were collected and DNA from apoptotic cells were extracted and analyzed as described in Methods. Marker, a 100-bp DNA ladder marker. Each panel is representative of three to four experiments.
To further characterize doxycycline-induced cell death, we performed flow cytometric analysis of DNA content using annexin-V-FITC and PI, which has been used to determine early apoptosis and necrosis/late apoptosis. Figure 2C shows that the percentage of early apoptosis (FL1 positive/FL3 negative) and necrosis/late apoptosis (FL1 positive/FL3 positive) was increased in melanoma cells challenged with 10 and 20 µg·mL−1 of doxycycline.
Caspases are partially involved in doxycycline-induced melanoma apoptosis
Next, we examined whether caspase(s) activation was involved in doxycycline-induced melanoma cell apoptosis. A variety of caspase inhibitors, including Z-WHED-FMK (caspase-1 inhibitor), Z-DEVD-FMK (caspase-3/-7 inhibitor), Z-IETD-FMK (caspase-8 inhibitor) and Z-VAD-FMK (pan-caspase inhibitor), were used to examine whether they could block doxycycline-induced melanoma cell death. Among these inhibitors, only Z-DEVD-FMK and Z-VAD-FMK partially reversed doxycycline-induced melanoma cell death (Figure 3A). Z-VAD-FMK exhibited a relatively stronger effect than Z-DEVD-FMK, suggesting that caspase-3/-7 and other caspases but not caspase-1 and -8 were partially involved in the melanoma cell apoptosis, induced by doxycycline. The cleavage of PARP and caspase activation during apoptosis were then investigated. In Figure 3B, doxycycline treatment caused PARP cleavage in melanoma cells, a full 116-kDa PARP was cleaved into an 89-kD fragment. Moreover, caspase-3, -7 and -9 were found to be cleaved (activated) during melanomacell apoptosis, demonstrating that the apoptosis involved caspase activation.
Figure 3.
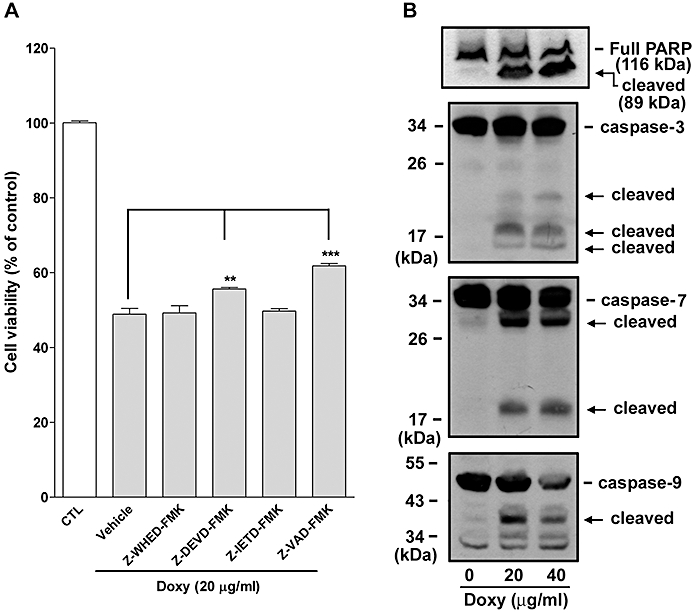
Caspases are involved in doxycycline-induced melanoma cell death. (A) Effect of caspase inhibitors on doxycycline (doxy) -induced melanoma cell death. Human melanoma cells were pre-treated with vehicle (DMSO) or caspase inhibitors (50 µM) for 30 min. Pre-treated cells were then incubated with or without doxycycline (20 µg·mL−1) for 24 h in the presence or absence of selective caspase inhibitors. Cell viability was measured by MTT assay and data were presented as percentage of control (n = 5–7). **P < 0.01, ***P < 0.001 versus doxycycline treatment alone (+ vehicle). (B) Effect of doxycycline on caspase activation and PARP cleavage. Melanoma cells were treated with the indicated concentration of doxycycline for 24 h. Cells were collected and cell lysates were analyzed by Western blotting (n = 2). The cleaved (activated) caspases and PARP are indicated by arrows.
Cell death contributes to doxycycline inhibition of MMP activity and migration of melanoma cells
Seftor et al. (1998) suggested that the effects of doxycycline and CMTs on melanoma cell invasion correlated with their inhibitory activities on MMPs, but not on cell viability. In the following experiments, first, we examined doxycycline effect on melanoma cell MMP-2 activity. It was found that MMP-2 secreted by melanoma cells was inhibited by doxycycline but was partially reversed by Z-DEVD-FMK and Z-VAD-FMK (Figure 4A). Second, we examined whether doxycycline-induced inhibition on growth/viability of melanoma cells affected their migration. As previously reported, melanoma cell migration was markedly inhibited by doxycycline. However, the inhibition was also partially reversed by the pan-caspase inhibitor Z-VAD-FMK (Figure 4B). Taken together, our results demonstrate that doxycycline inhibited melanoma cell MMP activity and migration, partially through induction of cell death.
Figure 4.
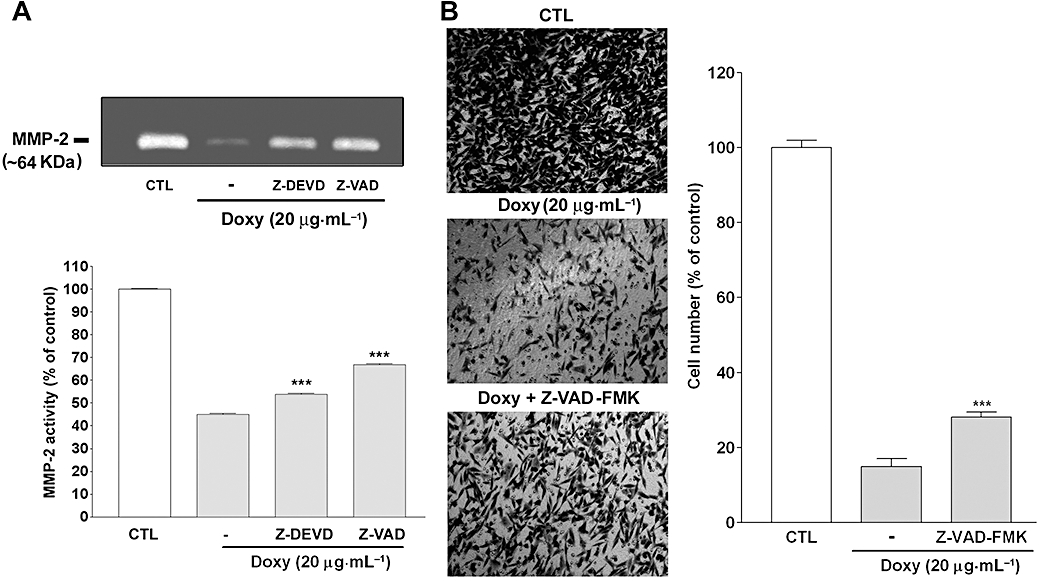
Cell death contributes to inhibition of melanoma cell MMP activity and migration, by doxycycline (doxy) (A) Gelatin zymographic analysis of conditioned media from doxycycline-treated melanoma cells. Human melanoma cells were incubated with or without doxycycline (20 µg·mL−1) in the presence or absence of the indicated caspase inhibitors (50 µM) for 24 h and conditioned media were analyzed by gelatin zymography (n = 10). CTL: control. ***P < 0.001 versus doxycycline treatment alone. (B) Cell migration assay. Melanoma cells were seeded in the upper chamber in the presence or absence of doxycycline (20 µg·mL−1) and the pancaspase inhibitor. Migration assay was performed as described in Methods and migrated cells were photographed (left panel) and quantified under the microscope (right panel). Data were presented as percentage of control (n = 5–10). ***P < 0.001 versus doxycycline treatment alone.
Effect of doxycycline on MAPKs and ASK1 activation and intra-cellular ROS production in melanoma cells
To elucidate the possible mechanism of doxycycline in causing melanoma cell apoptosis, we examined its effects on MAPK signalling. Since melanoma cell viability was not obviously affected by doxycycline within 1.5-h treatment (Figure 1C), the effect of doxycycline on MAPK activation was examined in this period. As shown in Figure 5A, JNK (JNK1, 46 kDa) and p38 MAPK but not ERK1/2 were activated, by doxycycline treatment, whereas their corresponding total proteins and α-tubulin were unaffected. It was noted that JNK was activated at 15 and 30 min but decreased at 45 min and thereafter, whereas p38 MAPK was only activated at 15 min in human melanoma cells. A similar JNK activation profile was also observed in mouse B16F10 melanoma cells treated with doxycycline (Figure 5B).
Figure 5.
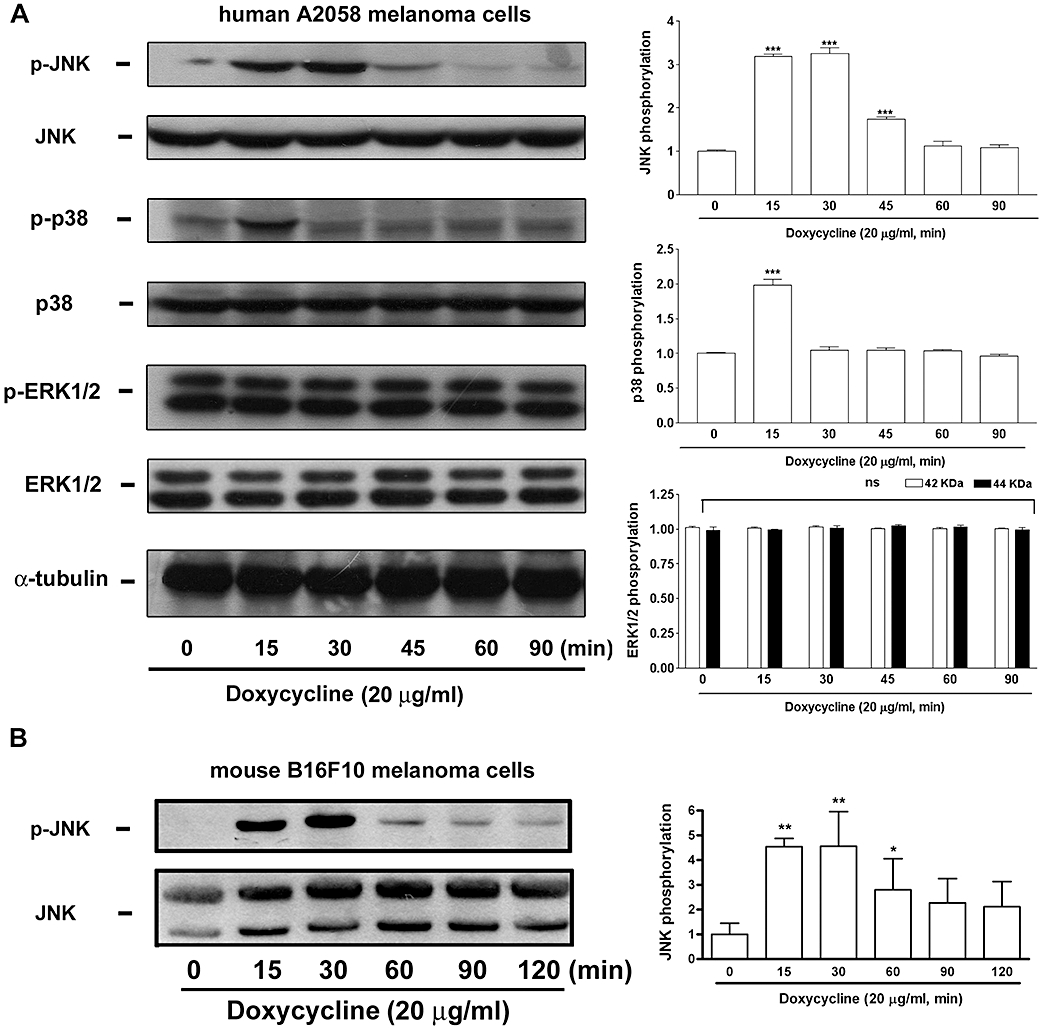
Doxycycline induces JNK and p38 MAPK activation in melanoma cells. (A) Human A2058 and (B) mouse B16F10 melanoma cells were incubated with doxycycline (20 µg·mL−1) for the indicated times. Cells were collected and cell lysates were analyzed by Western blotting. Each blot (on the left) is representative of four independent experiments. Quantitative analysis of MAPK activation was performed by densitometry and is shown in the right hand histograms. Activation (phosphorylation) is expressed as fold-control. *P < 0.05, **P < 0.01, and ***P < 0.001 versus basal level (0 min).
As ASK1 could activate MKK3 and MKK4, which in turn activate JNK and p38 MAPK (Matsuzawa and Ichijo, 2001), we next investigated whether doxycycline induced ASK1 activation in melanoma cells and found that doxycycline at 20 and 40 µg·mL−1 induced ASK1 phosphorylation in a time-dependent manner. The phosphorylation increased rapidly at 7.5 min, was maintained at 15 and 30 min but decreased at 60 min. In contrast, α-tubulin expression was unaffected (Figure 6A). In parallel, as oxidative stress may activate ASK1 and MAPKs, we then tested whether doxycycline induced production of intra-cellular reactive oxygen species (ROS). Figure 6B shows that only a few cells were stained with fluorescence in unstimulated condition (control) but the majority of cells were stained with fluorescence when treated with H2O2 (as a positive control) or doxycycline, as determined by fluorescence microscopy (upper panel). The result was confirmed by the quantitation of fluorescence intensity from the fluorescence reader (lower panel).
Figure 6.
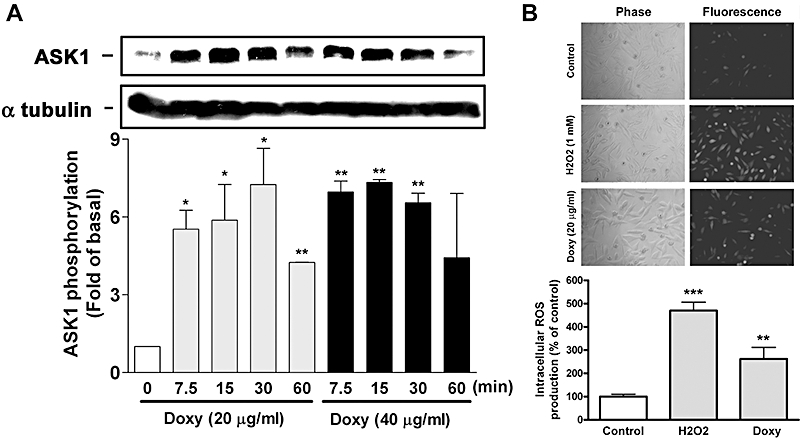
Doxycycline (doxy) induces ASK1 activation and intra-cellular ROS production in melanoma cells. (A) Human melanoma cells were incubated with doxycycline for the indicated times. Cells were collected and cell lysates were analyzed by Western blotting. Quantitative analysis of ASK1 phosphorylation was performed by densitometry (n = 4) (B) Intra-cellular ROS production in melanoma cells at an early stage of doxycycline treatment. Fluorescent dye-loaded human melanoma cells were treated with PBS, doxycycline, or H2O2 (as a positive control) for 10 min. Cells were analyzed by fluorescence microscopy (upper panel) or fluorescence plate reader (lower panel) (n = 3). **P < 0.01 and ***P < 0.001 versus control.
Doxycycline induces cytochrome c release and mitochondrial membrane potential (ΔΨm) change in melanoma cells
We have shown that caspases were activated during doxycycline-induced melanoma cell apoptosis and the decrease of cell viability was partially reversed by the caspase inhibitors (Figure 3). As caspases can be triggered by two distinct mechanisms including mitochondrial and death receptor pathway (Hengartner, 2000), we investigated whether cytochrome c was released from mitochondria into cytosol during apoptosis. Figure 7A shows that cytosolic cytochrome c was hardly detectable in cells treated with doxycycline for 4 h, but it was significantly increased in cells treated with doxycycline for 8 and 16 h.
Figure 7.
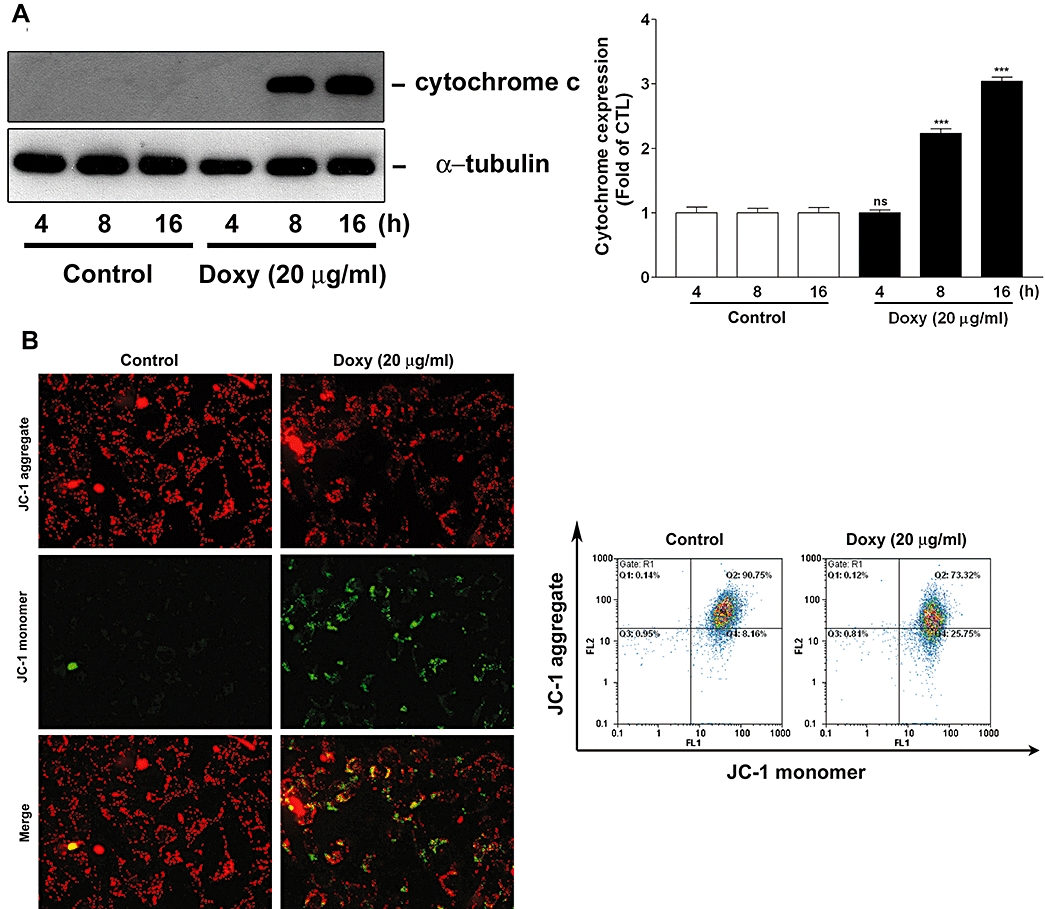
Doxycycline (doxy) induces cytochrome c release and ΔΨm change in melanoma cells. (A) Human melanoma cells were treated with doxycycline (20 µg·mL−1) for the indicated times. Cytochrome c release from mitochondria to cytosol was determined by Western blotting. Quantitative analysis of cytochrome c expression was performed by densitometry (n = 3). (B) Human melanoma cells were treated with or without doxycycline for 16 h. ΔΨm change in melanoma cells was monitored by loading with JC-1 and was analyzed by fluorescence microscopy (left panels) or flow cytometry (right panels). Each panel and blot is representative of three similar experiments. ***P < 0.001versus control.
Next, we used JC-1 to examine whether changes in ΔΨm occurred in melanoma cells treated with doxycycline. JC-1 is a cationic dye that exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (∼525 nm) to red (∼590 nm). Therefore, JC-1 has been widely used to detect mitochondrial depolarization during apoptosis, which is indicated by a decrease in the red/green fluorescence intensity ratio (Smiley et al., 1991). Fluorescence microscopy and flow cytometry were performed to examine ΔΨm change in melanoma cells. Fluorescence microscopy shows that most untreated melanoma cells (control) had strong J-aggregation (red) and weak JC-1 monomer. However, in doxycycline-treated cells, many cells were increased with strong JC-1 monomer (green staining) and concomitantly decreased of J-aggregation (red staining) due to low ΔΨm (Figure 7B, left panel). A similar result was observed by flow cytometry, showing that JC-1 aggregates (FL2) was apparently decreased in doxycycline-treated cells (Figure 7B, right panel). In brief, our results indicate that doxycycline induced mitochondrial cytochrome c release and ΔΨm change in melanoma cells during apoptosis.
JNK activation is necessarily required for doxycycline-induced melanoma cell apoptosis and ΔΨm change
We next examined whether doxycycline-induced of MAPKs was involved in melanoma cell apoptosis. The following MAPK inhibitors were used, SP600125, SP202190, and PD98059, which are known to target and abolish JNK, p38 MAPK and ERK1/2 signalling respectively. Among these inhibitors, only SP600125 was found to significantly attenuate doxycycline-induced melanoma cell death, as determined by MTT assay (Figure 8A) and flowcytometric analysis (Figure 8B). In parallel, we also tested whether SP600125 affected doxycycline-induced ASK1 activation. In Figure 8C, inhibition of JNK activation by SP600125 was demonstrated but this inhibitor did not affect doxycycline-induced ASK1 activation, suggesting that ASK1 was activated prior to JNK in melanoma cells treated with doxycycline. In Figure 8D, siRNA targeting JNK was used to validate the JNK function in doxycycline-induced melanoma cell death. It was found that the JNK siRNA reduced JNK (46 kDa) expression and doxycycline-induced JNK activation, while the negative control siRNA did not (upper panel). MTT assay revealed that JNK knockdown reduced doxycycline-induced melanoma cell death (lower panel).
Figure 8.
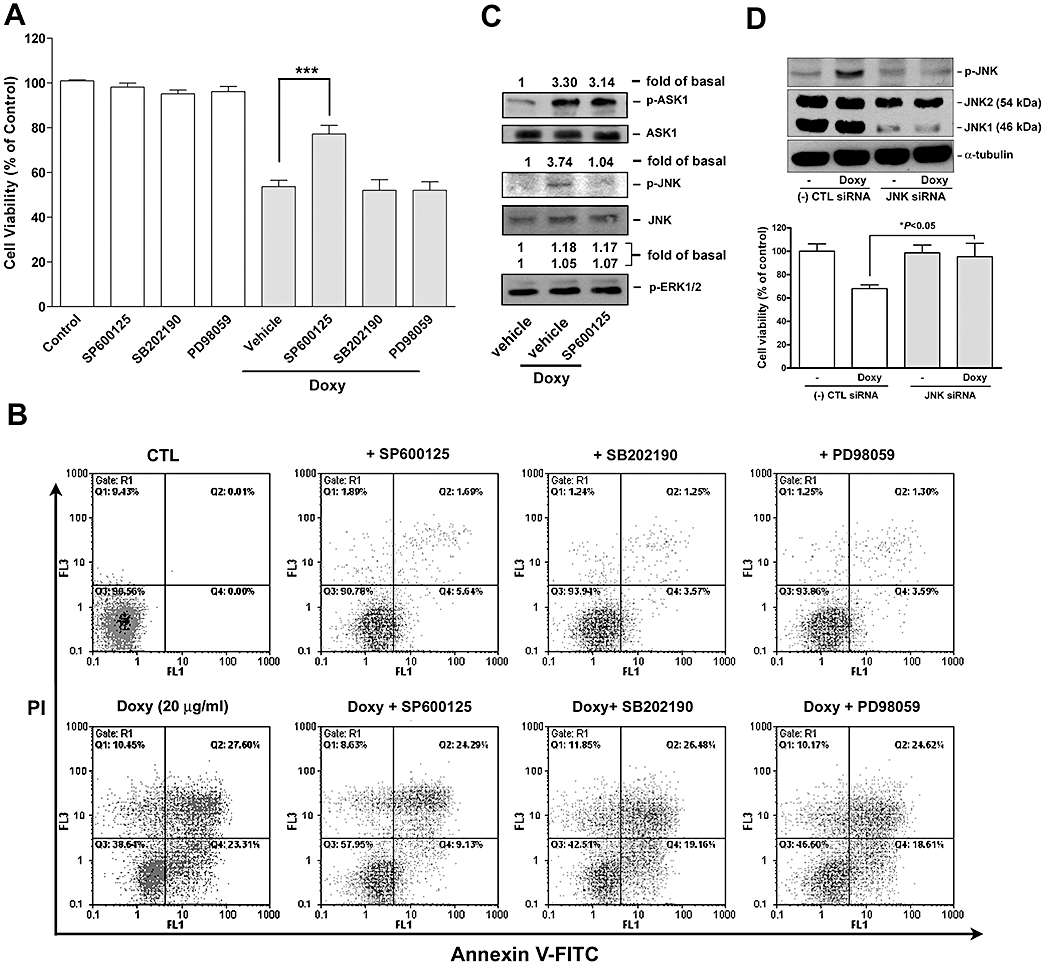
Effect of MAPK inhibitors and JNK siRNA on doxycycline-induced melanoma cell death and signalling. (A and B) Human melanoma cells were pre-treated with the indicated MAPK inhibitors (20 µM for each) for 30 min and followed by incubation with or without doxycycline (doxy; 20 µg·mL−1) for 24 h. The extent of cell death was determined by (A) MTT assay (n = 5–7) or (B) flow cytometry (n = 2–3). ***P < 0.001versus doxycycline treatment alone. (C) Cells were pre-treated with vehicle (DMSO) or SP600125 (10 µM) for 30 min and followed by the addition of doxycycline (20 µg·mL−1) for 30 min. Activation and expression of the kinases were determined by Western blotting and quantified by densitometry (n = 2). (D) Melanoma cells were transfected with negative control [(-) CTL] or JNK siRNA for 72 h. Upper panel: JNK expression and activation was determined by Western blotting. Lower panel: cell growth/viability after treated with or without doxycycline (20 µg·mL−1) for an additional 24 h was determined by MTT assay (n = 3).
Further, we examined whether doxycycline-induced JNK activation was required for the induction of ΔΨm change by flow cytometry and fluorescence microscopy. Again, only SP600125 was able to reverse doxycycline-induced ΔΨm change (Figure 9), indicating that JNK was activated upstream to ΔΨm change during doxycycline-induced melanoma cell apoptosis.
Figure 9.
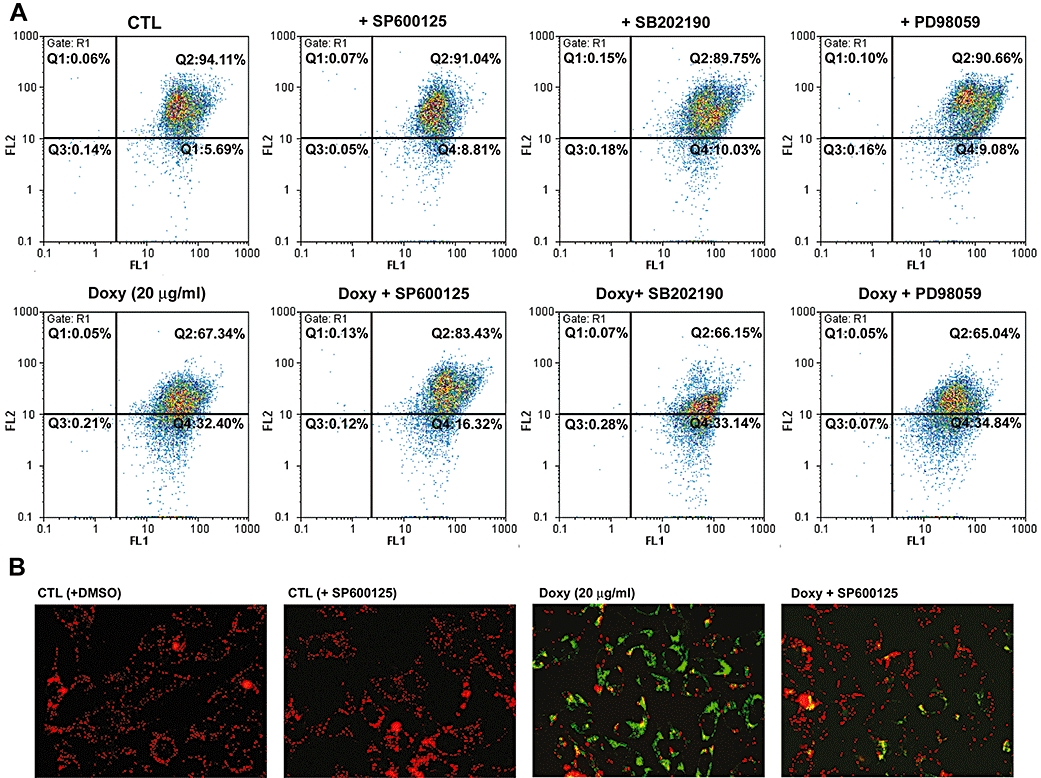
Effect of MAPK inhibitors on ΔΨm change induced by doxycycline (doxy). Human melanoma cells were pre-treated with the indicated MAPK inhibitors (20 µM for each) for 30 min and followed by incubation with or without doxycycline (20 µg·mL−1) for 16 h. Cells were then loaded with JC-1 and analyzed by (A) flow cytometry or (B) fluorescence microscopy. Each panel is representative of three similar experiments.
Role of ROS in doxycycline-induced melanoma cell death
We used two ROS scavengers, Trolox and vitamin C, to examine the role of intra-cellular ROS production in doxycycline-induced melanoma cell death. MTT assay indicated that the ROS scavengers significantly reduced doxycycline-induced death in human and mouse melanoma cells (Figure 10A). The effect of vitamin C on doxycycline-induced caspase cleavage showed that this ROS scavenger also attenuated doxycycline-induced caspase-7 and -9 cleavage (Figure 10B). Finally, we showed that vitamin C reduced doxycycline-induced JNK activation (Figure 10C), suggesting that intra-cellular ROS production was responsible for JNK activation during melanoma cell apoptosis.
Figure 10.
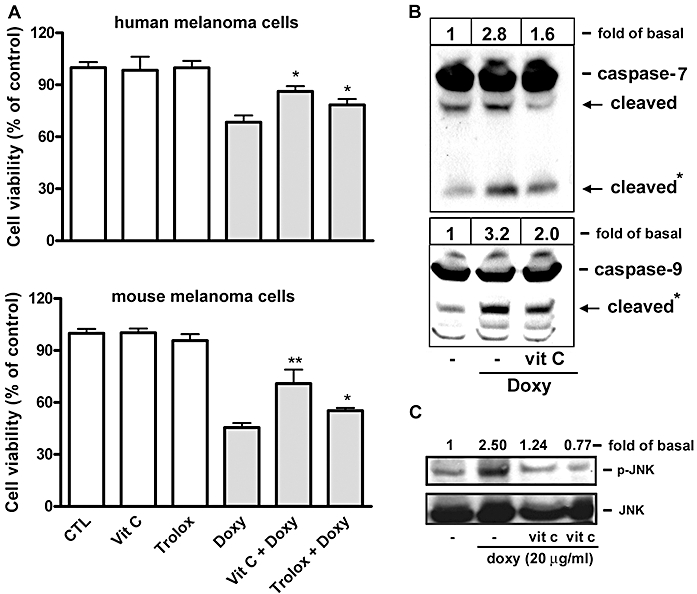
Effect of ROS scavengers on doxycycline-induced melanoma cell death, caspase activation and signalling. Human melanoma cells were treated with doxycycline (20 µg·mL−1) in the presence or absence of the ROS scavengers (trolox or vitamin C (vit C); 100 µM for each) for 30 min. (A) Cell growth was determined by MTT assay (n = 3–4). *P < 0.05 and **P < 0.01 versus doxycycline treatment alone. (B) Caspase and (C) JNK activation were determined by Western blotting (n = 2–3). Band intensities of the cleaved caspases (*) and activated JNK were determined by densitometry and expressed as fold-basal.
Discussion
The tetracyclines were discovered in the 1940s and introduced into clinical practice in the 1950s. They were first widely prescribed by dermatologists in the early 1950s when it was discovered that they were effective as a treatment for acne and have been proven to be useful in the management of a diverse range of infections (Roberts, 2003). Recently, the therapeutic effects of tetracycline and its analogues in various diseases have been investigated, including several skin diseases, neutrophilic diseases, aortic aneurysms, cancer metastasis, periodontitis, and autoimmune disorders (Sapadin and Fleischmajer, 2006). In this study, we have shown that doxycycline was cytotoxic for melanoma and lung epithelial carcinoma cells. Interestingly, doxycycline appeared more cytotoxic on melanoma than on lung epithelial carcinoma cells (Figure 1), as 2 µg·mL−1 doxycycline was sufficient to show cytotoxicity in melanoma cells. A marked inhibitory effect was observed when cells were treated with doxycycline (20 µg·mL−1) for 24 h. Moreover, cell death was observed when incubation time with doxycycline was prolonged. In addition to being cytotoxic on human A2058 and A375 melanoma cells, a similar effect of doxycycline was also observed on mouse B16F10 melanoma cells but not on vascular SMCs. In humans, peak serum concentration of doxycycline of about 1.7–2.8 µg·mL−1 can be achieved after a single 100 mg dose given to adults by oral or intravenous administration (Saivin and Houin, 1988) and this could be increased if a higher dose was given. Therefore, doxycycline, but not minocycline, possibly could be considered for development as an anti-melanoma agent, if an appropriate in vivo serum concentration could be achieved.
Regarding the mechanism of action of doxycycline in causing melanoma cell death, we demonstrated that JNK activation was necessary for ΔΨm change and melanoma cell apoptosis (Figures 8 and 9). The hypothesis was also supported by the evidence that doxycycline rapidly caused JNK activation at a very early stage of treatment (within 1 h) (Figure 5). There are two possibilities to explain how doxycycline affects melanoma cells. One is that doxycycline acts through activation of the cell membrane death receptor and the other is that it acts through direct activation of intra-cellular signalling molecules. A previous study has shown that doxycycline-induced Jurkat T lymphocyte apoptosis is associated with increased Fas/Fas ligand (FasL) expression but this expression was observed after 24 h treatment with doxycycline (20 µg·mL−1) (Liu et al., 1999). This mechanism seems to be less likely in this study because doxycycline rapidly (within 60 min) activated ASK1 and JNK (Figures 5 and 6). Moreover, the inhibitor Z-IETD-FMK targeting caspase-8, which is known to be induced following death receptor activation, failed to reverse doxycycline-induced melanoma cell death (Figure 3). The Fcγ receptors are decreased in leukocytes incubated with doxycycline or from patients receiving doxycycline and this effect can be reversed by addition of divalent cations in vitro. The authors suggested that the divalent cation chelating effect of tetracyclines may be important for its action (Naess et al., 1985). In the present study, doxycycline does not appear to act in this way because the tetracyclines (minocycline and doxycycline) we used exerted differential cytotoxic effects on melanoma cells (Figure 1). Our data revealed that ASK1 was activated at a very early stage of doxycycline treatment (Figure 6A), indicating that ASK1 might be responsible for JNK activation. According to our observations, the selective JNK inhibitor (SP600125) was effective in reversing doxycycline-induced ΔΨm change and melanoma cell death (Figures 8 and 9) but did not affect ASK1 activation. So we would suggest that ASK1 was activated before JNK and JNK was activated prior to ΔΨm change during melanoma cell apoptosis. Moreover, since an increase in intra-cellular ROS production was observed at this very early stage of doxycycline treatment (Figure 6B), we suggest the role of a ROS-ASK1-JNK pathway in doxycycline-induced melanoma cell apoptosis. The further analyses support this hypothesis. The ROS scavengers (trolox and vitamin C) reduced doxycycline-induced JNK activation, caspase cleavage, and melanoma cell death (Figure 10), demonstrating that intra-cellular ROS generation is responsible for doxycycline-induced JNK activation and melanoma cell apoptosis.
Onoda et al. (2006) reported that doxycycline and tetracycline analogues (TCNAs) induced caspase-dependent and -independent apoptosis in human colon cancer cells. Since the pan-caspase inhibitor Z-VAD-FMK partially reversed doxycycline-induced melanoma cell death (Figure 3A), we suggest that doxycycline may induce caspase-dependent and -independent apoptosis (and possibly necrosis) in melanoma cells. This is supported by the evidence that cytochrome c was released into cytosol and ΔΨm was changed during apoptosis (Figure 7). It has been shown that the release of cytochrome c into cytosol activates Apaf-1 and caspase-9 and subsequently leads to activation of caspase-3, -6 and -7 (Liu et al., 1996). Since caspase-3/-7 and pan-caspase inhibitors could reduce doxycycline-induced melanoma death (Figure 3A), caspase-3/-7 and possible other caspases were involved in this process. Indeed, the caspase-3, -7 and -9 were activated and one of their downstream substrates, PARP, was cleaved during melanoma cell apoptosis (Figure 3B). Although we did not determine the activity of AIF (apoptosis-inducing factor) and endoG, which are known to induce caspase-independent apoptosis (Lipton and Bossy-Wetzel, 2002), they might be released into the cytosol when mitochondrial homeostasis, such as. ΔΨm, was disrupted by doxycycline.
Seftor et al. (1998) have shown that doxycycline and CMT inhibit melanoma cell invasion in vitro mainly through affecting gelatinases (MMP-2 and -9) activity. As previously reported by them, our results also show that melanoma cell MMP-2 activity was markedly reduced by treatment with doxycycline. However, it could be partially reversed (about 10–20%) by the caspase inhibitors, suggesting that in addition to direct MMP-2 inhibition, cell apoptosis (death) contributed to the inhibition of melanoma cell MMP activity, induced by doxycycline. A similar effect was also observed on melanoma cell migration (Figure 4). Taken together, we conclude here that the death contributes to the inhibitory effects of doxycycline on melanoma cell function, including MMP activity and migration.
In conclusion, in this study we have shown that doxycycline and minocycline differentially induce melanoma cell death (apoptosis). We also have demonstrated that induction of cell death contributes to the inhibitory effects of doxycycline on melanoma cell functions, such as secreted MMP activity and cell migration. The most important finding was supported by the evidence that ROS and ASK1 are possible initiators responsible for JNK activation, which is necessary for doxycycline-induced ΔΨm change and death in melanoma cells. As doxycycline is widely used in dermatology and useful in the management of a diverse range of infections, the concept proposed here not only elucidates the possible mechanism of action for doxycycline in causing melanoma cell death but may also provide us with information for consideration of doxycycline as an anti-melanoma agent.
Glossary
Abbreviations:
- ASK1
apoptosis signal-regulating kinase 1
- CMT
chemically modified tetracyclines
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
- PARP
poly(ADP-ribose) polymerase
- TCNAs
tetracycline analogues
Conflict of interest
The authors declare no conflict of interest.
References
- Assefa Z, Vantieghem A, Garmyn M, Declercq W, Vandenabeele P, Vandenheede JR, et al. p38 mitogen-activated protein kinase regulates a novel, caspase-independent pathway for the mitochondrial cytochrome c release in ultraviolet B radiation-induced apoptosis. J Biol Chem. 2000;275:21416–21421. doi: 10.1074/jbc.M002634200. [DOI] [PubMed] [Google Scholar]
- Bettany JT, Wolowacz RG. Tetracycline derivatives induce apoptosis selectively in cultured monocytes and macrophages but not in mesenchymal cells. Adv Dent Res. 1998;12:136–143. doi: 10.1177/08959374980120010901. [DOI] [PubMed] [Google Scholar]
- Bettany JT, Peet NM, Wolowacz RG, Skerry TM, Grabowski PS. Tetracyclines induce apoptosis in osteoclasts. Bone. 2000;27:75–80. doi: 10.1016/s8756-3282(00)00297-0. [DOI] [PubMed] [Google Scholar]
- Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, Pouyssegur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Agostino P, Ferlazzo V, Milano S, La RM, Di BG, Caruso R, et al. Chemically modified tetracyclines induce cytotoxic effects against J774 tumour cell line by activating the apoptotic pathway. Int Immunopharmacol. 2003;3:63–73. doi: 10.1016/s1567-5769(02)00213-8. [DOI] [PubMed] [Google Scholar]
- Fife RS, Rougraff BT, Proctor C, Sledge GW., Jr Inhibition of proliferation and induction of apoptosis by doxycyclinecycline in cultured human osteosarcoma cells. J Lab Clin Med. 1997;130:530–534. doi: 10.1016/s0022-2143(97)90130-x. [DOI] [PubMed] [Google Scholar]
- Gilbertson-Beadling S, Powers EA, Stamp-Cole M, Scott PS, Wallace TL, Copeland J, et al. The tetracycline analogs minocycline and doxycycline inhibit angiogenesis in vitro by a non-metalloproteinase-dependent mechanism. Cancer Chemother Pharmacol. 1995;36:418–424. doi: 10.1007/BF00686191. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Hung CF, Huang TF, Chiang HS, Wu WB. (-)-Epigallocatechin-3-gallate, a polyphenolic compound from green tea, inhibits fibroblast adhesion and migration through multiple mechanisms. J Cell Biochem. 2005;96:183–197. doi: 10.1002/jcb.20509. [DOI] [PubMed] [Google Scholar]
- Ibrahim N, Haluska FG. Molecular pathogenesis of cutaneous melanocytic neoplasms. Annu Rev Pathol. 2009;4:551–579. doi: 10.1146/annurev.pathol.3.121806.151541. [DOI] [PubMed] [Google Scholar]
- Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, et al. Cancer statistics, 2004. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- Kroon AM, Dontje BH, Holtrop M, Van den BC. The mitochondrial genetic system as a target for chemotherapy: tetracyclines as cytostatics. Cancer Lett. 1984;25:33–40. doi: 10.1016/s0304-3835(84)80023-3. [DOI] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, van AL, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Bossy-Wetzel E. Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell. 2002;111:147–150. doi: 10.1016/s0092-8674(02)01046-2. [DOI] [PubMed] [Google Scholar]
- Liu J, Kuszynski CA, Baxter BT. doxycyclinecycline induces Fas/Fas ligand-mediated apoptosis in Jurkat T lymphocytes. Biochem Biophys Res Commun. 1999;260:562–567. doi: 10.1006/bbrc.1999.0929. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Lo HM, Hung CF, Tseng YL, Chen BH, Jian JS, Wu WB. Lycopene binds PDGF-BB and inhibits PDGF-BB-induced intracellular signaling transduction pathway in rat smooth muscle cells. Biochem Pharmacol. 2007;74:54–63. doi: 10.1016/j.bcp.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Lokeshwar BL, Selzer MG, Zhu BQ, Block NL, Golub LM. Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non-antimicrobial tetracycline analog (COL-3) in a metastatic prostate cancer model. Int J Cancer. 2002;98:297–309. doi: 10.1002/ijc.10168. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Ichijo H. Molecular mechanisms of the decision between life and death: regulation of apoptosis by apoptosis signal-regulating kinase 1. J Biochem. 2001;130:1–8. doi: 10.1093/oxfordjournals.jbchem.a002947. [DOI] [PubMed] [Google Scholar]
- Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- Mouratidis PX, Colston KW, Dalgleish AG. doxycyclinecycline induces caspase-dependent apoptosis in human pancreatic cancer cells. Int J Cancer. 2007;120:743–752. doi: 10.1002/ijc.22303. [DOI] [PubMed] [Google Scholar]
- Naess A, Glette J, Halstensen AI, Sandberg S, Solberg CO. In vivo and in vitro effects of doxycyclinecycline on leucocyte membrane receptors. Clin Exp Immunol. 1985;62:310–314. [PMC free article] [PubMed] [Google Scholar]
- Onoda T, Ono T, Dhar DK, Yamanoi A, Nagasue N. Tetracycline analogues (doxycycline and COL-3) induce caspase-dependent and -independent apoptosis in human colon cancer cells. Int J Cancer. 2006;118:1309–1315. doi: 10.1002/ijc.21447. [DOI] [PubMed] [Google Scholar]
- Peus D, Vasa RA, Beyerle A, Meves A, Krautmacher C, Pittelkow MR. UVB activates ERK1/2 and p38 signaling pathways via reactive oxygen species in cultured keratinocytes. J Invest Dermatol. 1999;112:751–756. doi: 10.1046/j.1523-1747.1999.00584.x. [DOI] [PubMed] [Google Scholar]
- Peus D, Vasa RA, Meves A, Beyerle A, Pittelkow MR. UVB-induced epidermal growth factor receptor phosphorylation is critical for downstream signaling and keratinocyte survival. Photochem Photobiol. 2000;72:135–140. doi: 10.1562/0031-8655(2000)072<0135:uiegfr>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Roberts MC. Tetracycline therapy: update. Clin Infect Dis. 2003;36:462–467. doi: 10.1086/367622. [DOI] [PubMed] [Google Scholar]
- Ryan ME, Ramamurthy S, Golub LM. Matrix metalloproteinases and their inhibition in periodontal treatment. Curr Opin Periodontol. 1996;3:85–96. [PubMed] [Google Scholar]
- Saivin S, Houin G. Clinical pharmacokinetics of doxycyclinecycline and minocyclinecycline. Clin Pharmacokinet. 1988;15:355–366. doi: 10.2165/00003088-198815060-00001. [DOI] [PubMed] [Google Scholar]
- Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54:258–265. doi: 10.1016/j.jaad.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Seftor RE, Seftor EA, De Larco JE, Kleiner DE, Leferson J, Stetler-Stevenson WG, et al. Chemically modified tetracyclines inhibit human melanoma cell invasion and metastasis. Clin Exp Metastasis. 1998;16:217–225. doi: 10.1023/a:1006588708131. [DOI] [PubMed] [Google Scholar]
- Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat Cell Biol. 2001;3:950–957. doi: 10.1038/ncb1101-950. [DOI] [PubMed] [Google Scholar]
- Wu WB, Chang SC, Liau MY, Huang TF. Purification, molecular cloning and mechanism of action of graminelysin I, a snake-venom-derived metalloproteinase that induces apoptosis of human endothelial cells. Biochem J. 2001;357:719–728. doi: 10.1042/0264-6021:3570719. [DOI] [PMC free article] [PubMed] [Google Scholar]