

Abstract
Adenovirus (Ad) vectors are the most commonly used system for gene therapy applications, due in part to their ability to infect a wide array of cell types and tissues. However, many therapies would benefit from the ability to target the Ad vector only to specific cells, such as tumor cells for cancer gene therapy. In this study, we investigated the utility of capsid protein IX (pIX) as a platform for the presentation of single-chain variable-fragment antibodies (scFv) and single-domain antibodies (sdAb) for virus retargeting. We show that scFv can be displayed on the capsid through genetic fusion to native pIX but that these molecules fail to retarget the virus, due to improper folding of the scFv. Redirecting expression of the fusion protein to the endoplasmic reticulum (ER) results in correct folding of the scFv and allows it to recognize its epitope; however, ER-targeted pIX-scFv was incorporated into the Ad capsid at a very low level which was not sufficient to retarget virus infection. In contrast, a pIX-sdAb construct was efficiently incorporated into the Ad capsid and enhanced virus infection of cells expressing the targeted receptor. Taken together, our data indicate that pIX is an effective platform for presentation of large targeting polypeptides on the surface of the virus capsid, but the nature of the ligand can significantly affect its association with virions.
Adenovirus (Ad) vectors have many desirable characteristics which have allowed them to become popular gene transfer vehicles (2). Many of the gene therapy “successes” when using Ads in animal models involve transduction of the liver either to restore a functional deficiency to hepatocytes or to use this organ as a protein production factory to produce large amounts of secreted protein. There is a practical reason for this: when Ad is injected systemically, greater than 80% of the virus is retained in the liver (27). While this may be an advantage in many studies, it is one of the limitations to effective Ad therapy in many other disease models for which tissue-specific gene expression is required. Nonspecific vector transduction is undesirable for several reasons. First, less vector is available to interact with the target tissue, necessitating higher doses of Ad to achieve a given level of therapeutic protein expression. Second, acute toxicity caused by Ad is, at least in part, due to activation of the innate immune response, possibly mediated by Kupffer cells of the liver (62). Third, promiscuous vector transduction can include infection of antigen-presenting cells which will enhance the formation of antivector and antitransgene immune responses (31). Thus, the development of novel strategies which lead to greater efficiency and specificity of infection of target tissue and reduced infection of nontarget tissues is required.
The majority of gene therapy studies utilize Ad vectors based on serotype 2 or 5 (Ad2 or Ad5, respectively). Ad5 infection initiates with the capsid protein fiber binding to the cell surface coxsackie-adenovirus receptor (CAR) (4, 76), followed by a secondary interaction between penton protein and avb3 and avb5 integrins, which triggers internalization of the virus by endocytosis (81). Other studies have suggested that Ad can enter cells by using heparan sulfate proteoglycans as an alternative receptor through a bridging interaction between Ad and blood factors such as factor IX and complement component C4-binding protein (66, 72). Recently, the mechanism of high-efficiency uptake of Ad by the liver has been elucidated: the virus hexon capsid protein binds to blood factor X, which then interacts with heparan sulfate on the surface of hepatocytes (33, 80). Importantly, swapping the hypervariable regions of the Ad5 hexon with those of Ad48, which does not interact with factor X, reduces hepatocyte uptake 600-fold. This later work represents a paradigm shift in our understanding of Ad infection in vivo and clearly shows that detargeting Ad can circumvent the problem of liver sequestration.
There are two main strategies for retargeting Ad infection specificity: covalent or noncovalent attachment of targeting ligands to the capsid and genetic modification of capsid proteins. Covalent and noncovalent methods involve the addition of targeting ligands after the virus has been purified, through the use of bispecific antibodies (one binding the Ad virion and the other binding the desired cellular ligand) or antibody-receptor ligand complexes or by mixing of chemically modified Ad virions with a reactive ligand (37, 57, 71, 82). Alternatively, genetic modification involves cloning of the targeting ligand directly into one of the virion coat proteins (19, 36, 83). These two strategies have been combined to produce a metabolically biotinylated vector that can be combined with a variety of targeting ligands to achieve cell-type-specific targeting (9, 10). Since the natural protein for virus attachment to the cell is the capsid fiber protein, many groups have focused upon genetic modification of this protein in order to redirect virus attachment (24). Targeting moieties placed on fiber can be combined with other mutations that abolish binding to the native cellular receptor (30, 35, 56), thereby reducing Ad promiscuity as well as redirecting binding.
Other capsid proteins besides fiber can also be modified and used for virus retargeting. Incorporation of an arginine-glycine-aspartic acid (RGD) motif into the hexon of Ad resulted in enhanced transduction of cells expressing low levels of CAR (79). The Ad penton protein was modified to incorporate a FLAG epitope tag, and this virus was subsequently used in a bispecific-antibody strategy (82). Minor capsid protein IX (pIX) has also served as a platform for presentation of targeting ligands, including a polylysine motif (20), an RGD motif (78), or a biotin acceptor peptide for subsequent addition of targeting ligands (10). Taken together, these studies demonstrate that the Ad capsid can be modified through several different proteins to redirect Ad binding to the cell type of interest.
We and others have shown that pIX can be modified genetically to incorporate large polypeptides into the Ad capsid (38, 47, 53, 85). pIX is a minor capsid protein that stabilizes the hexon on the facets of the capsid (14, 23). Recent studies showed that the N-terminal domains of three pIX monomers form a triskelion structure that cements three hexon proteins together, whereas the C-terminal domains are located near the edge between two facets and form a tetramer (22, 45, 60, 68). Three of the four C-terminal domains associate together in a parallel structure, whereas the fourth domain, which stretches across from one facet to the adjacent facet, associates with the trimer in an antiparallel manner. Capsids which contained a pIX-green fluorescent protein (GFP) fusion showed normal growth characteristics and could be visualized by fluorescence microscopy both in vitro and in vivo (47). Curiel and coworkers (39, 46) used pIX for the attachment of luciferase and herpes simplex virus (HSV) thymidine kinase for visualization of virus in vivo and to generate a single virus containing three different pIX isoforms displaying a FLAG tag, 6×His tag, and monomeric red fluorescent protein (RFP) (74). Hoeben and colleagues (18, 77) showed that pIX fused to a hyperstable antibody directed to β-galactosidase or a single-chain T-cell receptor targeted to MAGE-A1 antigen was efficiently incorporated into the Ad capsid and bound its ligand. Other research groups have used pIX to display complement-inhibiting polypeptides (64, 65). Finally, C-terminal cysteines added to pIX have been used as a target for the chemical addition of targeting ligands bearing reactive thiol groups (15).
The ability to fuse large polypeptides to pIX suggests that it may serve as a platform for the addition of other large targeting ligands, such as single-chain variable-fragment antibodies (scFv) or single-domain antibodies (sdAb) (17), which are likely capable of providing greater specificity of infection than many previously utilized, smaller ligands. scFv are heterodimers of the variable light (VL) and heavy (VH) chains of an antibody joined by a peptide linker and retain the binding specificity of a monoclonal antibody. sdAb are derived from camels and llamas and are formed by heavy chains only; the antigen-binding site of these antibodies consists of one single domain (VHH). scFv and sdAb have exquisite specificity and high antigen-binding affinity, and many different antibodies that recognize surface receptors on different cell types have been described (3). Presentation of scFv or sdAb on the capsid of Ad should improve the specificity with which Ad infects a target cell. In this study, we describe our efforts designed to redirect Ad infection specificity through display of scFv and sdAb on the surface of the Ad capsid by genetic fusion to the pIX capsid protein.
MATERIALS AND METHODS
Cell culture.
All cell culture media and reagents were obtained from Invitrogen (Burlington, ON, Canada). 293 (26), 293N3S (25), and A549 cells were grown in monolayers in minimum essential medium supplemented with 100 U of penicillin per ml, 100 μg of streptomycin per ml, 2.5 μg amphotericin B (Fungizone) per ml, and 10% fetal bovine serum (FBS). Chinese hamster ovary (CHO) cells were grown in Ham's F-10 medium, supplemented with 10% FBS and antibiotics. To generate a CHO cell line which expressed the human epidermal growth factor receptor version III (EGFRvIII), CHO cells were transfected with plasmid pRL86, which contains an EGFRvIII cDNA upstream of an internal ribosome entry site and hygromycin resistance gene and under the regulation of the murine cytomegalovirus (CMV) immediate-early promoter and thymidine kinase polyadenylation sequence. After ∼2 weeks under selection in 200 μg/ml hygromycin, colonies were isolated and analyzed for expression of EGFRvIII by immunoblot analysis. One of these clones, CHO-EGFRvIII clone 11, demonstrated high-level EGFRvIII expression that was stable for at least 30 cell doublings.
Plasmid and virus construction.
The viruses used in this study are described in Table 1, and all viruses were constructed using a combination of conventional cloning and bacterial RecA-mediated recombination (13). They were propagated in 293 and 293N3S cells and their titers in these cells were determined as previously described (59). MR1 is a single-chain antibody that recognizes a variant of the epidermal growth factor receptor (EGFRvIII) and has been described previously (40, 41). The MR1 sequence was amplified from a plasmid by PCR, using Platinum Hi-Fidelity Taq (Invitrogen), with synthetic oligonucleotides 5′GCGGCTAGCGGGGCCCAGCCGGCCATGGCCCAG and 5′GCGACGCGTGGTTCGAACTATGCGGCCGCTTTGATTTCCAGC that flanked the coding sequence and introduced unique NheI/MluI restriction sites. The resulting fragment was cloned into NheI/MluI-digested pRP2288 (47), replacing enhanced GFP (EGFP) and creating a pIX-MR1 fusion construct, designated pRL4. An NheI/BstBI fragment from pRL4 was cloned into NheI/BstBI-digested pRP2306 (47), replacing EGFP in this plasmid with MR1; the resulting construct was designated pRL5. pRL5 was subjected to bacterial RecA-mediated recombination (13, 28) with an Ad genomic plasmid, pRP2014 (61), creating an infectious plasmid, pRL6. Thus, pRL6 and the resulting virus, AdRL6, encode the pIX-MR1 fusion protein replacing the native pIX coding sequence within the Ad genome and under regulation by the native pIX promoter. A BamHI/MfeI fragment from pRP2202, containing the murine secreted alkaline phosphatase (mSEAP) reporter gene under regulation by the mouse phosphoglycerate kinase promoter and simian virus 40 (SV40) polyadenylation sequence (43), was used to replace a BamHI/MfeI fragment in pRL5, generating pRL7, which was subsequently recovered as an infectious plasmid designated pRL34. AdRP2218, an E1/E3-deleted Ad that contains an mSEAP expression cassette identical to that of pRL34 and an unmodified pIX gene, has been described previously (43, 49).
TABLE 1.
Viruses used in this study
| Virus | Transgene type | Promotera | ER signal peptideb | pIX fusion proteinc | NLSd |
|---|---|---|---|---|---|
| AdRP2330 | pIX | − | − | ||
| AdAVH6 | GFP | pIX | − | − | |
| AdRP2218 | mSEAP | pIX | − | − | |
| AdRP2310 | pIX | − | GFP | − | |
| AdRL6 | pIX | − | MR1 | − | |
| AdRL34 | mSEAP | pIX | − | MR1 | − |
| AdRL28 | mSEAP | pIX | + | GFP | − |
| AdRL29 | mSEAP | pIX | + | GFP | + |
| AdRL31 | mSEAP | pIX | + | MR1 | − |
| AdRL32 | mSEAP | pIX | + | MR1 | + |
| AdRL39 | mSEAP | pIX | + | − | |
| AdRL71 | CMV | + | GFP | + | |
| AdRL74 | CMV | + | MR1 | + | |
| AdRP2687 | RFP | MLP | + | MR1 | + |
| AdRP2698 | RFP | MLP | + | GFP | + |
| AdRP2595 | mSEAP | pIX | − | AFAI | + |
| AdRP2823 | GFP | pIX | − | AFAI | + |
Promoter used to regulate expression of the pIX expression cassette.
Presence (+) or absence (−) of a endoplasmic reticulum signal peptide on the pIX expression cassette.
With the exception of AdRP2218 and AdAVH6, all viruses contain a FLAG epitope tag on the C terminus of pIX (located between pIX and GFP, MR1, or AFAI).
Presence (+) or absence (−) of a C-terminal nuclear localization sequence (NLS) on the pIX construct.
To facilitate cloning of the various pIX-MR1 derivatives into Ad vectors, we modified pΔE1sp1A as follows (5). pΔE1sp1A was digested with BglII, the ends were filled in by Klenow polymerase, and it was recircularized, generating pΔE1sp1AΔBglII. The pIX coding sequence of this plasmid was removed by PCR using oligonucleotides 5′GCGAGATCTACGTACTAGTAAAACATAAATAAAAAACCAGACTCTG and 5′GCGAGATCTGCAAAACAGATACAAAACTACATAAG, corresponding to the sequences immediately 3′ and 5′, respectively, of the pIX gene. PCR around pΔE1sp1AΔBglII produces a linear fragment with BglII ends, from which the pIX coding sequence is deleted. Recircularization of this plasmid produced pΔE1sp1AΔpIX, which was used as a base plasmid for all of our subsequent viral constructs.
pIX, pIX-GFP, or pIX-MR1 was targeted to the endoplasmic reticulum (ER) through incorporation of an amino-terminal ER signal peptide (SP), with or without a carboxy-terminal nuclear localization sequence. The ER SP was derived from the beta interferon gene and was added to pIX by PCR using the synthetic oligonucleotide 5′CGCGGATCCGCCGCCATGACCAACAAGTGTCTCCTCCAAATTGCTCTCCTGTTGTGCTTCTCCACGACAGCTCTTTCCATGAGCACCAACTCGTTTGATG, which also introduces a BamHI site. This common oligonucleotide was used in combination with the following oligonucleotides to generate the derivative pIX genes shown below, and the resulting PCR products were cloned into BamHI/XbaI-digested pcDNA3 (to generate the plasmids indicated in parentheses): for SP-pIX (pRL36), 5′CCTGGATCCTTCATTCTCGTCGTCATCCTTGTAATCAACCGCATTGGGAGGGGAGGAAGC; for SP-pIX-GFP (pRL10), 5′CGCTCTAGACTAATCGATGTTGTACAGTTCATC; for SP-pIX-GFP-nuclear localization signal (NLS) (pRL11), 5′CGCTCTAGACTATGGGTCCTCTACCTTGCGCTTCTTTTTAGGCGGGGATGCATCGATGTTGTACAGTTCATC; for SP-pIX-MR1 (pRL13), 5′CGCTCTAGACTATGCGGCCGCTTTGATTTCCAG; and for SP-pIX-MR1-NLS (pRL14), 5′CGCTCTAGACTATGGGTCCTCTACCTTGCGCTTCTTTTTAGGCGGGGATGCTGCGGCCGCTTTGATTTCCAG. The nuclear localization sequence was derived from simian virus 40 large tumor antigen (32). All PCR products were confirmed by sequencing. BamHI/XbaI fragments from pRL10, -11, -13, and -14 were cloned into BglII/SpeI-digested pΔE1Sp1AΔpIX, generating pRL16, -17, -19, and -20, respectively. These plasmids were digested with BamHI/EcoRV, and a BamHI/NotI (filled in with Klenow polymerase) mSEAP reporter gene under regulation by the mouse phosphoglycerate kinase promoter and SV40 polyadenylation sequence (43) was inserted, generating pRL22, -23, -24, and -25, respectively. These plasmids were recombined into an Ad infectious plasmid and recovered as viruses designated AdRL28 (SP-pIX-GFP), AdRL29 (SP-pIX-GFP-NLS), AdRL31 (SP-pIX-MR1), and AdRL32 (SP-pIX-MR1-NLS). A BamHI fragment from pRL36 was cloned into pΔE1Sp1AΔpIX, generating pRL37, which was subsequently recombined into an Ad infectious plasmid and recovered as a virus designated AdRL39 (SP-pIX).
To enhance expression of the SP-pIX-GFP-NLS and SP-pIX-MR1-NLS proteins, we constructed viruses that contained the corresponding fusion genes under regulation of the human CMV promoter. BglII/NheI fragments from pRL11 and pRL14 were cloned into BamHI/NheI-digested pRL17 and pRL20, respectively, to generate the shuttle plasmids pRL70 and pRL72, respectively. These shuttle plasmids were recombined with an Ad infectious plasmid and used to generate viruses designated AdRL71 (CMV-SP-pIX-GFP-NLS) and AdRL74 (CMV-SP-pIX-MR1-NLS). These two viruses have the contiguous E1-to-pIX coding sequence deleted but utilize the native pIX polyadenylation sequence for termination of the pIX fusion transcript. These viruses do not coexpress native pIX.
To place SP-pIX-MR1-NLS under late gene expression, the fusion gene was cloned into the E3 region of an Ad. pBluescript KS was digested with SpeI, and synthetic oligonucleotides (5′CTAGCGATCGGCAGGCGCAATCTTCGCATTTCTTTTTTCCAGGAAG and its complement), which contain a splice acceptor site derived from the Ad40 long fiber gene as previously described (11), were ligated into the plasmid, generating pDC3. pDC3 was digested with XhoI, the ends were repaired with Klenow polymerase, and it was recircularized, generating pDC4. pDC4 was digested with ClaI, the ends were repaired with Klenow polymerase, and it was recircularized, generating pDC5. A BamHI/XbaI fragment (filled in with Klenow polymerase) from pRL14 was cloned into BamHI/EcoRV-digested pDC5, generating pRP2654. A PvuI fragment from pRP2654 containing the splice acceptor upstream of the SP-pIX-MR1-NLS fusion gene was cloned into the PacI site of pBHG10 (5), thus replacing the E3 that was deleted, generating pRP2656. The E3-replacing expression cassette was recombined into an Ad infectious plasmid (generating pRP2658), and a CMV-monomeric RFP (mRFP; kindly provided by Roger Tsien, University of California at San Diego [8]) expression cassette was added in place of the E1 region. The resulting infectious plasmid was recovered as a virus and designated AdRP2687. A similar strategy was used to generate a virus containing SP-pIX-GFP-NLS in the E3 region, designated AdRP2698.
pAFAI encodes a single-domain antibody (sdAb) identified through panning of a llama sdAb phage library on the non-small-cell lung carcinoma cell line A549 (86), and it was a kind gift of Jianbing Zhang (National Research Council, Ottawa, ON, Canada). This sdAb recognizes a variant of CD66c (carcinoembryonic antigen-related cell adhesion molecule 6 [CEACAM6 or CEA6]) (44). The sdAb from pAFAI was PCR amplified with synthetic oligonucleotides 5′GAGGCTAGCGATGTGCAGCTGCAGGCGTCTG and 5′GCATAGCGGCCGCCTATGGGTCCTCTACCTTGCGCTTCTTTTTAGGCGGGGATGCTGAGGAGACGGTGACCTGGGTC containing NheI and NotI restriction sites, in addition to encoding a C-terminal nuclear localization sequence derived from the simian virus 40 large tumor antigen (ASPPKKKRKVEDP) (32), and cloned in frame with pIX in a pcDNA3-derived plasmid, generating pRP2583. The pIX-AFAI fusion gene from pRP2583 was used to replace native pIX in an Ad shuttle vector carrying an mSEAP reporter gene (43, 49) replacing the E1 region, generating pRP2587. A 45-Å alpha-helical linker was cloned between the pIX gene and the AFAI gene, essentially as described by Vellinga et al. (78), generating pRP2587-45A. pRP2587-45A was cloned into an Ad E1/E3-deleted infectious plasmid by using homologous recombination in bacteria, generating pRP2595, which was subsequently rescued into a virus, AdRP2595, in 293 cells. The mSEAP reporter gene in pRP2587-45A was replaced with a GFP reporter gene under the regulation of the human CMV promoter and bovine growth hormone polyadenylation sequence, and the construct was recovered as a virus designated AdRP2823. AdAVH6 contains a GFP reporter cassette identical to that of AdRP2823 and native pIX, and it has been previously described (29).
Pull-down assay and immunoblot analysis.
To verify whether the various pIX-MR1 proteins were functional and capable of binding the appropriate epitope of EGFRvIII, we employed a pull-down assay similar to that described by Lorimer and coworkers (41). 293 cells in 60-mm dishes were transfected with 5 μg of an expression construct or infected with a virus at a multiplicity of infection (MOI) of 10 virus particles per cell. Twenty-four hours later, the cells were lysed on ice with a modified radioimmunoprecipitation assay (RIPA) extraction buffer (50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, 1% glycerol, 1% NP-40, protease inhibitors), and the cell debris was removed by centrifugation. A 20-μl aliquot of the resulting cell supernatant was removed for later analysis. To preclear the lysate, 20 μl of streptavidin-coated magnetic beads (Dynabeads M280; Dynal, Oslo, Norway) was added to each sample, the mixture was incubated for 1 h at 4°C on a rotating rack, the beads were pelleted in a magnetic rack, and the cleared lysate was transferred to a fresh tube. The supernatant was then incubated with biotinylated peptide LEEKKGNYVVTDHSGGK-biotin (41) at a final concentration of 100 mM for 1 h at 4°C in a rotating rack. Following the incubation, 20 μl of streptavidin-coated magnetic beads was added and incubated for 15 min at 4°C in a rotating rack. The beads were pelleted and washed four times with modified RIPA buffer. The beads were finally resuspended in 50 μl SDS-PAGE loading buffer (62.5 mM Tris, pH 6.8, 25% glycerol, 2% SDS, 0.1% bromophenol blue, 5% 2-mercaptoethanol). All protein samples were boiled for 10 min prior to SDS-PAGE. For all experiments, 5% of the total input lysate and 30% of the final bound protein were applied to the gel. After separation, the proteins were transferred to a polyvinylidene difluoride membrane (Immuno-Blot PVDF; Bio-Rad, Hercules, CA), and the membrane was probed with an anti-FLAG antibody (Sigma), anti-fiber protein antibody (MS-1027-P0, 1/1,000; NeoMarkers), ATF4 (sc-200, 1/5,000; Santa Cruz Biotechnology)- or tubulin (CP06, 1/5,000; Oncogene Research Products)-specific antibody, or goat polyclonal antisera to Rauscher leukemia virus GP69/71 (anti-SU, serum 80S000024, 1/1,000; Quality Biotech, Camden, NJ). Binding of the primary antibody was detected using a goat anti-mouse (Bio-Rad, Hercules, CA) or rabbit anti-goat (Sigma) secondary antibody conjugated to horseradish peroxidase and visualized by a chemiluminescent reaction (Amersham ECL Plus) and autoradiography. Preparation and analysis of protein constituents of purified Ad virions were as previously described (47, 70).
Immunofluorescence analysis.
Twenty-four hours prior to infection, 293 cells were seeded on 1-cm rounded glass coverslips (Fisher, Ottawa, ON, Canada) at a density of 0.1 × 106 cells per 35-mm dish. The next day, the cells were infected with the test viruses or transfected using Superfect reagent (Qiagen), and 24 h later, the medium was removed and the cells were washed once with cold phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde in PBS for 15 min, and then washed immediately with PBS containing 1% horse serum (HS) (Invitrogen). After two additional washes with PBS plus 1% HS, the cells were permeabilized with PBS plus 1% HS containing 0.5% Triton X-100 for 10 min. The cells were washed three times with PBS plus 1% HS. The coverslips were then placed in a humidified chamber and incubated with 100 μl of mouse monoclonal anti-FLAG antibody M2 (1:1,500; Sigma Genosys, Oakville, ON, Canada) in PBS plus 1% HS for 1 h at room temperature. The coverslips were washed three times for 5 min with PBS plus 1% HS, placed in a humidified chamber, and incubated with 100 μl of tetramethylrhodamine isothiocyanate (TRITC)- or fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibody (1:200; Jackson Immuno Research Laboratories, West Grove, PA) in PBS plus 1% HS and containing 0.2 μg/ml Hoechst stain (Sigma Genosys, Oakville, ON, Canada) for 1 h at room temperature. The coverslips were washed three times for 5 min with PBS plus 1% HS and then applied to glass slides by using a drop of Dako fluorescent mounting medium (Dako, Glostrup, Denmark). Immunofluorescent detection was performed using a Zeiss Axioplan 2 fluorescence microscope, and the pictures were taken using an Axiocam HR charge-coupled-device (CCD) camera. The images were processed using Adobe Photoshop imaging software. A plasmid expressing enhanced yellow fluorescent protein (EYFP)-tagged calreticulin was a kind gift of John Copeland (University of Ottawa).
Infection of A549 cells in the presence of soluble knob or AFAI.
The knob domains of Ad2 and AFAI sdAb were prepared in bacteria and purified as previously described (7, 86). A549 cells were seeded into 35-mm dishes and, the next day, were preincubated with various quantities of knob or AFAI protein for 15 min at 37°C, at which time AdRP2218, AdRP2595, or AdRP2823 was added at an MOI of 10 PFU/cell. After a 1-h attachment period at 37°C, the virus inoculum was removed, the plates were washed once with PBS, and the monolayers were overlaid with 2 ml of fresh media. Twenty-four hours later, the media were removed and the cells were lysed in reporter lysis buffer (Promega), with the quantity of alkaline phosphatase (AP) activity determined using a SEAP chemiluminescence assay kit (Roche), or lysed in protein loading buffer for immunoblot analysis as described above. For the reporter gene studies, the background level of activity was subtracted and the value expressed relative to the AP activity observed for cells not treated with knob.
RESULTS
MR1 fused to native pIX does not recognize the EGFRvIII epitope.
Previously, we showed that large polypeptides can be attached to the carboxy terminus of pIX, and these proteins are efficiently incorporated into the Ad capsid without affecting virus growth (47). This suggested that pIX may act as a platform for presentation of large targeting ligands, such as scFv. The MR1 scFv binds specifically to a mutant form of the epidermal growth factor receptor (EGFRvIII) that is found only on cancer cells (40, 41). Presentation of MR1 on the surface of Moloney murine leukemia virus (MMLV) and measles virus allowed both viruses to specifically target tumor cells (42, 50, 73), suggesting that MR1 is an ideal scFv for proof-of-principle studies of Ad retargeting to cancer cells. Our first attempt at presenting MR1 on the surface of the Ad capsid involved genetically fusing it to the C terminus of pIX, similar to our previous strategy for pIX-GFP. The resulting virus, which encoded either pIX-GFP or pIX-MR1, was designated AdRP2310 or AdRL6, respectively. Upon infection of 293 cells, these viruses efficiently produced the fusion proteins (Fig. 1 A), and both pIX-GFP and pIX-MR1 were incorporated into the Ad capsid (Fig. 1B). These two viruses contain an unaltered Ad fiber protein and bind to many cells through the native Ad receptor CAR. To test our pIX-MR1-modified virus for enhanced infection of cells that express EGFRvIII, we generated a CHO cell line which stably expresses EGFRvIII. CHO cells do not express CAR and are relatively refractory to Ad infection (4). We infected A549 (CAR-positive [CAR+]), CHO (CAR-negative [CAR−]), or CHO-EGFRvIII cells with virus carrying the mSEAP reporter gene and displaying either native pIX (AdRP2218) or pIX-MR1 (AdRL34) and examined AP activity 24 h later. Presentation of MR1 on the surface of the Ad capsid did not enhance infection of CHO cells expressing EGFRvIII on the cell surface (Fig. 1C).
FIG. 1.

MR1 fused to native pIX does not enhance infection of EGFRvIII-expressing cells. (A) 293 cells were infected with AdRP2310 or AdRL6 at an MOI of 1, and 24 h later, crude cell extracts were prepared, separated by SDS-PAGE, and subjected to immunoblotting for expression of FLAG epitope on the pIX fusion proteins or for fiber. Both fusion proteins are efficiently expressed in infected cells. α, anti. (B) Purified virions of AdRP2310 or AdRL6 (2 × 108 particles) were separated by SDS-PAGE and subjected to immunoblotting for expression of FLAG epitope on the pIX fusion proteins or for fiber. Both fusion proteins are incorporated into the Ad capsid. (C) A549, CHO, and CHO-EGFRvIII cells were infected with equal quantities of AdRP2218 (native pIX) or AdRL34 (pIX-MR1), and crude protein lysates were assayed for the level of mSEAP reporter gene expression 24 h later. The addition of MR1 to the Ad capsid did not enhance infection of EGFRvIII-expressing cells. RLU, relative light units. (D) 293 cells were infected with AdRP2310 or AdRL6, and 24 h later, the cells were lysed in a modified RIPA buffer and subjected to a pull-down assay with biotinylated EGFRvIII peptide as described in Materials and Methods. For each virus, lysates from input (I) and from the bound fraction (B) were separated by SDS-PAGE and immunoblotted for the FLAG epitope present on the pIX fusion protein. In parallel, 293 cells were transfected with a plasmid expressing MR1 fused to the MMLV envelope glycoprotein (ENV-MR1) and 24 h later, the cells were lysed in a modified RIPA buffer and subjected to a pull-down assay with biotinylated EGFRvIII peptide; aliquots were analyzed by immunoblotting with an antibody specific to the envelope protein (αSU). Five percent of the total input fraction and 30% of the total bound fraction were applied to the gel. Pull-down of ENV-MR1 by the biotinylated EGFRvIII peptide results in enrichment of protein in the bound fraction. For pIX-MR1, no enrichment is observed in the bound fraction, indicating that pIX-MR1 cannot bind the EGFRvIII epitope.
Previous studies showed that fusion of MR1 to the envelope protein of Moloney murine leukemia virus (ENV-MR1) resulted in a protein that was efficiently incorporated into the virion, that bound its target ligand, and that was capable of redirecting virus binding (42). In MMLV, MR1 was expressed in the disulfide-bonded surface loop near the native receptor binding site of the envelope glycoprotein (ENV-MR1) (42) and therefore trafficked through the endoplasmic reticulum (ER). For Ad, pIX-MR1 would be synthesized in the cytoplasm, a reducing environment, and whether or not an scFv is functional in the cytoplasm of mammalian cells cannot be predicted. To test whether MR1 fused to pIX was capable of binding ligand, we infected cells with virus and performed a pull-down assay with biotinylated EGFRvIII peptide. A small quantity of pIX-GFP was nonspecifically pelleted in the pull-down assay, and a similar fraction of pIX-MR1 was also pelleted, suggesting that pIX-MR1 did not specifically bind to the EGFRvIII peptide (Fig. 1D). In contrast, ENV-MR1 effectively bound the peptide, leading to enrichment of the protein in the bound fraction. These results indicate that fusion of MR1 to native pIX did not produce a protein capable of binding to the EGFRvIII epitope.
Targeting pIX-MR1 to the endoplasmic reticulum produces a protein capable of recognizing the EGFRvIII epitope.
scFv typically contain cysteine bonds that are crucial for proper folding of the protein (84). These bonds are likely formed correctly in the ER (as in the ENV-MR1 protein) but not in the cytoplasm, which is a strongly reducing environment. Most Ad proteins, including pIX, are translated in the cytoplasm (i.e., not in association with the ER), suggesting that the reason MR1 fused to native pIX does not bind ligand is improper folding of MR1 in the cytoplasm. To solve this problem, we investigated whether inclusion of an ER signal peptide (SP) on the N terminus of pIX-MR1, which would target the fusion protein for expression in the ER, could result in properly folded protein that is incorporated into capsid. Initially, plasmids pRP2288 (pIX-GFP), pRL4 (pIX-MR1), pRL13 (SP-pIX-MR1), and pRL14 (SP-pIX-MR1-NLS) were transiently transfected into 293 cells and the ability of pIX-MR1 to bind its epitope was assessed using our pull-down assay. Plasmids encoding the ER-targeted versions of pIX-MR1 produced proteins slightly larger than those produced in the cytoplasm (Fig. 2 A), possibly due to posttranslational modification(s) (55). As expected, pIX-GFP and pIX-MR1 did not bind the EGFRvIII epitope. In contrast, when pIX-MR1 was expressed with an amino-terminal signal peptide, it efficiently bound the EGFRvIII peptide, indicating that the scFv had indeed folded correctly. The presence of an NLS on the carboxy terminus of ER-pIX-MR1 had no significant effect on its ability to bind the EGFRvIII peptide.
FIG. 2.

Targeting pIX-MR1 to the endoplasmic reticulum produces a protein capable of recognizing the EGFRvIII epitope. (A) 293 cells were transfected with the indicated plasmids, and 24 h later, the cells were lysed and subjected to a pull-down assay with biotinylated EGFRvIII peptide as described in Materials and Methods. For each plasmid, lysates from input and the bound fraction were separated by SDS-PAGE and immunoblotted for the FLAG epitope present on the pIX fusion protein. Inclusion of an ER SP leads to proper folding of MR1 and recognition of its epitope, resulting in enrichment of the fusion protein in the pull-down assay. (B) Purified virions of AdRP2330 or AdRL39 (2 × 108 particles) were separated by SDS-PAGE and immunoblotted for expression of FLAG epitope on the pIX fusion proteins or for fiber. The SP on pIX does not affect incorporation of pIX into the capsid, suggesting that the SP is efficiently cleaved from the protein. (C) Purified virions from the indicated viruses (2 × 108 particles) were separated by SDS-PAGE and immunoblotted for expression of FLAG epitope on the pIX fusion proteins or for fiber. ER-targeted pIX-MR1 is not incorporated into the Ad capsid. (D) 293 cells were infected with the indicated viruses, and 24 h later, crude protein extracts were prepared, separated by SDS-PAGE, and immunoblotted for the FLAG epitope on the pIX fusion proteins or for fiber. 293 cells were infected with the indicated viruses (panel E) or cotransfected with pRL14 (SP-pIX-MR1-NLS) or pRP2266 (pIX-FLAG) along with a plasmid expressing EYFP-tagged calreticulin (Cal) or EGFP (panel F). Twenty-four hours later, the cells were fixed and the localization of pIX was examined by indirect immunofluorescence (red). The cells were also counterstained with Hoescht stain to identify the nuclei (blue).
We next determined if the addition of an SP on native pIX resulted in a protein that was incorporated into capsid. If the N-terminal SP is not efficiently cleaved from SP-pIX, the SP may sterically interfere with the ability of pIX to interact with the capsid, effectively producing pIX-deficient virus. Virus containing the ER-targeted pIX, AdRL39, produced pIX of the correct size, and this protein was efficiently incorporated into the Ad capsid (Fig. 2B), suggesting that the SP was cleaved from the protein. We next constructed viruses encoding SP-pIX-GFP without or with an NLS (AdRL28 or AdRL29, respectively) or SP-pIX-MR1 without or with an NLS (AdRL31 or AdRL32, respectively) and examined expression and incorporation of the pIX fusion. An examination of capsid protein content showed that pIX-GFP was efficiently incorporated into the Ad capsid regardless of whether it contained an ER SP or NLS; however, pIX-MR1 was not found associated with capsids (Fig. 2C). An examination of protein expression from the viruses showed that all of the viruses efficiently expressed the fusion proteins, with the exception of AdRL32 (SP-pIX-MR1-NLS), suggesting that the presence of the NLS adversely affected expression or accumulation of the protein when expressed from the virus (Fig. 2D). Immunofluorescence analysis of cells infected with the various viruses showed that pIX-MR1 produced in AdRL32-infected cells was located predominately outside of the nucleus, possibly retained in the endoplasmic reticulum, likely contributing to the inability of the protein to be incorporated into the Ad capsid (Fig. 2E). To determine in which cellular compartment the SP-pIX-MR1 resided, we cotransfected cells with plasmids expressing FLAG-tagged pIX or SP-pIX-MR1 in combination with enhanced yellow fluorescent protein (EYFP)-tagged calreticulin or EGFP. Calreticulin resides in the lumen of the ER and is commonly used as a marker of ER localization (48). While SP-pIX-MR1 showed complete colocalization with calreticulin around the periphery of the nucleus, it showed only partial colocalization with EGFP, which was found throughout the cytoplasm and nucleus (Fig. 2F). When expressed from a plasmid, pIX was found in both the cytoplasm and the nucleus, as we have previously observed (47), and showed only partial localization with calreticulin. Taken together, these results indicate that routing pIX-MR1 through the ER results in significant retention of the protein in this organelle.
All of the viruses used thus far carry the modified pIX genes under the regulation of the endogenous pIX promoter, which is active at intermediate times after infection (after early genes but before late genes) but is increasingly active after DNA replication occurs (6). To enhance expression of the pIX fusion proteins within the infected cells, we replaced the pIX promoter with the CMV promoter, which is known for very-high-level expression in Ad-infected cells (1). As expected, we observed very good expression of SP-pIX-GFP-NLS (AdRL71) and SP-pIX-MR1-NLS (AdRL74) (Fig. 3 A) and a proportionate increase in incorporation of the SP-pIX-GFP-NLS protein into the Ad capsid (Fig. 3B), suggesting that we did not have complete occupancy of pIX-GFP in our previous constructs. We did not observe incorporation of pIX-MR1 into the Ad capsid for AdRL74.
FIG. 3.

Increasing the expression level of ER-targeted pIX-MR1 does not result in its incorporation into the Ad capsid. (A) 293 cells were infected with the indicated viruses, and 24 h later, crude protein extracts were prepared, separated by SDS-PAGE, and immunoblotted for the FLAG epitope on the pIX fusion proteins or for fiber. Use of the CMV promoter improves expression of the ER-targeted pIX-MR1-NLS fusion protein. (B) Purified virions from the indicated viruses (2 × 108 particles) were separated by SDS-PAGE and immunoblotted for expression of FLAG epitope on the pIX fusion proteins or for fiber. Increased expression of SP-pIX-MR1 does not lead to incorporation of the protein into the Ad capsid. (C) 293 cells were infected with AdRL71, AdRL74, or wild-type Ad (AdWT) and crude protein extracts were prepared at various times postinfection and assayed for expression of ATF4 or tubulin by immunoblot analysis. As a control for induction of the UPR, cells were treated with 1 μM thapsigargin (Tg) for 16 h. Forced high-level expression of ER-targeted pIX-MR1 does not result in enhanced activation of UPR.
The folding structure of pIX-MR1 being more complex than that of pIX-GFP, combined with high-level expression from the CMV promoter, may have saturated the ability of the ER to process the protein, leading to activation of the unfolded-protein response (UPR) within the cell (34). To determine if AdRL71 and AdRL74 exhibited a difference in their abilities to activate the UPR, we examined induction of activating transcription factor 4 (ATF4) a marker of UPR within the cell. 293 cells were infected with AdRL71 or AdRL74, and protein samples were isolated at 6, 12, and 24 h postinfection or treated with thapsigargin (Tg), a common chemical inducer of the UPR (52). We did not observe a difference in the timings or levels of ATF4 expression in AdRL71- and AdRL74-infected cells, and the two viruses produced similar responses, similar to what was observed in wild-type Ad-infected cells (Fig. 3C). Thus, expression of SP-pIX-MR1 within the infected cell does not lead to enhanced activation of the UPR.
Coexpression of native pIX and pIX-MR1 enhances incorporation.
Recent imaging studies have indicated that, within the Ad capsid, the amino termini of pIX are arranged as a trimer within the group of nine, while the carboxy termini of each of these pIX proteins extend away from the trimer and form tetramers with four different pIX molecules (22, 60). This complex structure helps to stabilize the hexon structure of the capsid. It is possible that the complex folding structure of MR1 may sterically inhibit the ability of pIX to trimerize or tetramerize, thus affecting its incorporation. We therefore investigated whether coexpression of native pIX with pIX-MR1 would allow formation of heteromeric pIX molecules that could be effectively incorporated into the Ad capsid. We constructed a virus, AdRP2687, which contained a CMV-RFP expression cassette replacing the deleted E1, a wild-type pIX in its native position, and an SP-pIX-MR1 expression cassette replacing the deleted E3 (Fig. 4 A). To allow high-level expression of the pIX-MR1 fusion protein at late times during infection, we placed a splice acceptor site, derived from the Ad40 long fiber gene as previously described (11), upstream of the SP-pIX-MR1 coding sequence. Thus, alternative splicing of the transcript derived from the Ad major late promoter (MLP) should give rise to expression of pIX-MR1. We also designed a similar virus that encoded GFP in place of MR1, designated AdRP2698.
FIG. 4.

Coexpression of native pIX leads to low-level incorporation of pIX-MR1. (A) Structure of AdRP2687, which encodes red fluorescent protein (RFP) under the regulation of the human cytomegalovirus promoter (CMV) and bovine growth hormone polyadenylation sequence (pA), native pIX in its endogenous locus, and an SP-pIX-MR1-NLS expression cassette in the E3 region. Immediately 5′ of the pIX expression cassette is a splice acceptor (SA) derived from the Ad40 long fiber gene. (B) Kinetics of pIX-MR1 expression from viruses with different promoters. 293 cells were infected with the indicated viruses at an MOI of 1, and at 6, 12, or 24 h postinfection, crude cell extracts were prepared. At the end of the time course, samples were separated by SDS-PAGE and immunoblotted for the FLAG epitope on the pIX fusion proteins or for fiber. (C) 293 cells were infected with the indicated viruses, and 24 h later, crude protein extracts were prepared, separated by SDS-PAGE, and immunoblotted for the FLAG epitope on the pIX fusion proteins or for fiber. pIX-MR1 is efficiently expressed from AdRP2687. (D) 293 cells were infected with the indicated viruses, and 24 h later, the cells were lysed in a modified RIPA buffer and subjected to a pull-down assay with biotinylated EGFRvIII peptide as described in Materials and Methods. ER-targeted pIX-MR1 expressed from the E3 region of the virus is capable of binding the EGFRvIII epitope. (E) 293 cells were infected with AdRP2698 or AdRP2687, and 24 h later, the cells were fixed and analyzed by immunofluorescence for expression of the FLAG epitope tag present on the pIX fusion proteins. The resulting images were pseudocolored red to be consistent with images in Fig. 2. (F) Purified virions from the indicated viruses (2 × 108 particles) were separated by SDS-PAGE and immunoblotted for expression of the FLAG epitope on the pIX fusion proteins or for fiber. Coexpression of native pIX and SP-pIX-MR1 resulted in limited incorporation of the protein into the Ad capsid.
To examine the kinetics of expression from the various promoters in our viruses, we infected cells with AdRL6 (pIX promoter), AdRL74 (CMV promoter), or AdRP2687 (major late promoter), harvested total cell protein at various times postinfection (6, 12, and 24 h), and performed immunoblotting for expression of pIX-MR1 and fiber (Fig. 4B). The fiber gene is a late gene and should demarcate the onset of replication and expression of late genes. As expected, expression from the pIX promoter in AdRL6 at early time points was very low and was undetectable at 6 h but increased at 12 and 24 h. In contrast, expression from the CMV promoter in AdRL74 initiated early and at very high levels and increased throughout the infection. However, there were two notable observations for AdRL74. First, there was a significant amount of degraded protein in the pIX-MR1 immunoblot that was not observed for the other two viruses. Second, fiber gene expression from AdRL74 was not detectable at 12 h but was at roughly normal levels at 24 h. These data suggest that use of the high-level-activity CMV promoter leads to initiation of degradation of the transgene protein within the cell and that use of this promoter can adversely affect the timing and regulation of virus replication. For AdRP2687, expression initiated within 6 h of infection, prior to the onset of late gene expression, and continued to increase thereafter. Early expression of pIX-MR1 from AdRP2687 may be due to expression from the early region 3 promoter, which is upstream of the SP-pIX-GFP-NLS expression cassette in this virus. AdRP2687 produced roughly the same amount of pIX fusion protein as AdRL6 but significantly less than AdRL74 at 24 h postinfection (Fig. 4B and C). The pIX-MR1 produced from AdRP2687 was able to bind the EGFRvIII epitope (Fig. 4D), indicating that the MR1 was correctly folded. We also examined localization of the protein within infected cells. Native pIX forms district speckles in the nucleus (Fig. 2E), as does ER-targeted pIX-GFP expressed from AdRP2698 (Fig. 4E). For AdRP2687, pIX-MR1 was observed in both the cytoplasm and the nucleus (Fig. 4E). This increased localization of pIX-MR1 in the nucleus was sufficient to allow incorporation of the protein into the capsid of AdRP2687, albeit at significantly reduced levels compared to those for AdRP2310 (pIX-GFP in the native position of the genome) and AdRP2698 (SP-pIX-GFP-NLS within the E3 region and under late regulation) (Fig. 4F). Taken together, these data suggest that rerouting pIX-MR1 to the ER results in correct folding of the scFv, but only a small proportion of this protein can reach the nucleus and is available for incorporation into the Ad virion.
Presentation of pIX-MR1 on the Ad virion does not enhance infection of cells expressing EGFRvIII.
We have shown that a functional scFv can be expressed from an Ad vector and incorporated into the capsid through fusion to pIX, provided the protein is routed through the ER to allow proper folding. However, the efficiency of incorporation of the fusion protein is low. We next wished to determine if virus which displayed scFv on the capsid surface could enhance cell-type-specific infection by Ad: if pIX-MR1 can mediate attachment to EGFRvIII and internalization, we should see improved infection of the CHO-EGFRvIII cell line relative to infection by normal Ad. A549, CHO, or CHO-EGFRvIII cells were infected with AdRP2698 or AdRP2687 (at equal quantities), and protein extracts from the infected cells were prepared 24 h later and assayed for expression of the RFP transgene by immunoblot analysis. The two viruses infected A549 cells (CAR+) equally well and CHO cells (CAR−) poorly (Fig. 5 A). Although we observed a small improvement in transgene expression in the CHO-EGFRvIII-expressing cells when they were infected with AdRP2687, we did not observe enhanced transduction of this cell line relative to our control virus (Fig. 5B). We conclude that pIX fusion molecules with complex folding patterns which require processing in the ER are not efficiently incorporated in the Ad capsid and the small amount of protein that is incorporated is not capable of significantly redirecting infection.
FIG. 5.
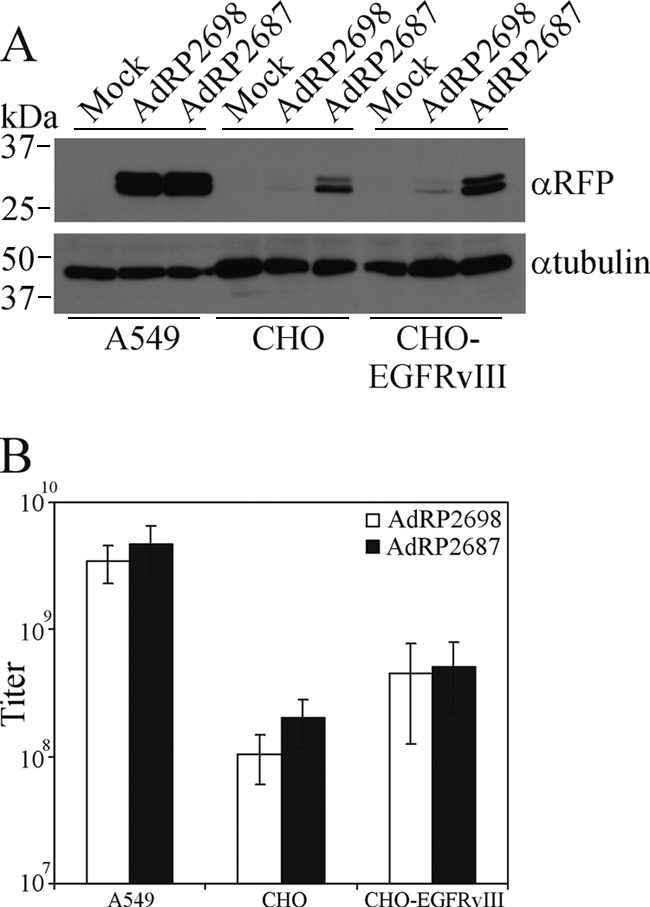
Presentation of MR1 through fusion to pIX does not enhance infection of cells expressing EGFRvIII. (A) A549 cells, CHO cells, or CHO cells engineered to express EGFRvIII were infected with AdRP2698 or AdRP2687 at an MOI of 10. Twenty-four hours later, crude protein extracts were prepared and assayed for reporter gene expression (RFP gene) by immunoblotting or for tubulin expression. (B) A549 cells, CHO cells, or CHO-EGFRvIII cells were infected with serial dilutions of AdRP2698 or AdRP2687, and 48 h later, the titers were determined by immunofluorescence and visual inspection (n = 3). The limited incorporation of pIX-MR1 in the Ad capsid is insufficient to enhance infection of EGFRvIII-expressing cells.
sdAb displayed on pIX can retarget vector transduction.
Our studies suggest that scFv fused to pIX are not effectively incorporated into the Ad capsid, likely due to the more complex folding requirements of the scFv. Another class of antibody that may be more amenable to incorporation into the Ad capsid is sdAb. To determine if pIX can be used to display sdAd on the capsid surface and lead to altered transduction characteristics, we generated a virus that contained a fusion protein composed of pIX and AFAI, an sdAb identified through phage panning of the A549 non-small-cell lung carcinoma cell line (86), and examined its ability to infect A549 cells. AFAI binds to CD66c (carcinoembryonic antigen-related cell adhesion molecule family member 6 [CEACAM6]), which is expressed on a subset of A549 cells in the culture (69, 86). To determine if pIX-AFAI was efficiently incorporated into the Ad capsid, we examined the protein content of purified capsid by immunoblot analysis for the FLAG-tagged pIX-AFAI and fiber (loading control). As shown in Fig. 6 A, pIX-AFAI was present in the Ad capsid and was of the expected size (∼34 kDa).
FIG. 6.

Incorporation of a pIX-sdAb into the Ad capsid enhances infection of target cells. (A) Purified virions from the indicated viruses (2 × 108 particles) were separated by SDS-PAGE and immunoblotted for expression of the FLAG epitope on the pIX fusion proteins or for fiber. pIX-AFAI is efficiently incorporated into the Ad capsid. (B) A549 cells were infected with AdAVH6 (native pIX) or AdRP2823 (pIX-AFAI) at an MOI of 500, and 24 h later, total cell protein extracts were isolated in modified RIPA buffer and analyzed by coimmunoprecipitation for interaction of pIX-AFAI with CD66c. IP, immunoprecipitation; IB, immunoblotting. (C) A549 cells were pretreated with either soluble AFAI or knob protein for 15 min, followed by infection with AdRP2823, and the relative level of infection was determined by measuring reporter gene expression 24 h later by immunoblot analysis. (D) A549 cells were infected with AdRP2218 (native pIX) or AdRP2595 (pIX-AFAI) at an MOI of 10 either in the absence or in the presence of an increasing concentration of soluble knob protein. Twenty-four hours later, the cells were harvested and assayed for alkaline phosphatase activity. Data are presented relative to the AP activity observed for cells not treated with knob protein. Incorporation of pIX-AFAI into the Ad capsid leads to enhanced, CAR-independent infection of target cells.
We next examined whether AFAI fused to pIX still retained the ability to bind CD66c: A549 cells were infected at an MOI of 500 with AdRP2823 (sufficient to allow replication of the E1-deleted vector and de novo expression of pIX-AFAI [51]) or control virus and, 24 h later, subjected to coimmunoprecipitation analysis for interaction of pIX-AFAI with CD66c. In A549 cells, CD66c is variably glycosylated and appears as a diffuse region of immunoreactivity by immunoblot analysis (Fig. 6B), as previously reported (69, 86). AFAI is known to interact with a specific glycosylation variant of CD66c (86), and we were readily able to detect the interaction between the two proteins, suggesting that fusion of AFAI to pIX did not alter its ability to recognize CD66c. To determine if virus presenting pIX-AFAI on the capsid bound A549 cells through CD66c, we pretreated cells with either bacterially expressed and purified soluble AFAI protein or the knob domain of fiber, which should inhibit binding and infection of cells through CD66c and CAR, respectively, and examined the level of AdRP2823 infection by reporter gene expression 24 h later. While we observed robust reporter gene expression in mock-treated cells, we observed significant reductions in infection and gene expression in cells treated with AFAI or knob (Fig. 6C). Thus, presentation of AFAI on the surface of the Ad capsid through fusion to pIX allows binding and infection through the CD66c receptor. This conclusion was further supported by an examination of infection efficiency of pIX-AFAI-containing virus in A549 cells in the presence of various concentrations of soluble knob (Fig. 6D). Since the ability of AdRP2595 to bind CAR is not ablated and A549 cells express CAR (63), the presence of soluble knob will block virus binding to cells through CAR. Infection of A549 cells in 35-mm dishes with AdRP2218 in the presence of 30 μg soluble knob resulted in an 85% reduction in infection, as monitored by reporter gene expression, with a >90% reduction at higher concentrations of soluble knob (120 μg) (Fig. 6D). In contrast, infection of A549 cells with AdRP2595 was decreased by only 60% by inclusion of soluble knob, and there was no further decrease in infection with increasing concentrations of fiber knob. That we observed an initial reduction in infection of A549 cells with AdRP2595 in the presence of soluble knob was not unexpected, since only approximately 40 to 50% of the cells in the culture express CD66c (69, 86). Taken together, these data indicate that sdAb can be displayed on the Ad capsid through fusion to pIX and can be used to redirect virus infection.
DISCUSSION
In this study, we investigated the utility of pIX as a platform for presentation of large targeting ligands on the surface of the Ad capsid for redirecting virus infection. We showed that scFv can be fused to pIX but must be routed through the ER in order to achieve proper folding of the scFv (Fig. 2A). ER-targeted native pIX (i.e., without a C-terminal addition) was able to reach the nucleus and was incorporated into the Ad capsid as efficiently as native pIX (Fig. 2B). However, the addition of the scFv to ER-targeted pIX inhibited translocation of the protein into the nucleus (Fig. 2E and 4E), resulting in a poor level of incorporation (Fig. 2C and 4F). The resulting virus was unable to effectively retarget infection to cells expressing the appropriate cell surface ligand (Fig. 5). In contrast, a fusion protein of pIX and an sdAb was efficiently incorporated into the Ad capsid and mediated enhanced, CAR-independent infection of target cells (Fig. 6). Taken together, our results indicate that pIX is an effective platform for presentation of large targeting polypeptides on the surface of the virus capsid, but the nature of the ligand can significantly affect its association with virions.
Ad capsid proteins are synthesized in the cytoplasm and transported to the nucleus for assembly (67). Routing pIX to the ER did not affect the ability of pIX to traffic to the nucleus, nor did it inhibit the protein's incorporation into the Ad capsid (Fig. 2B). Similarly, we were able to produce an ER-targeted pIX-GFP that was efficiently incorporated into the capsid (Fig. 2C). Targeting of this protein, or pIX-MR1, to the ER did not activate the unfolded protein response within the cell, at least not to any degree greater than observed for the wild-type Ad (Fig. 3C). Thus, we did not exhaust the ability of the ER to process these proteins, as has been observed in other viral systems (16). Importantly, our study has shown that, under the correct conditions, pIX that is routed through the ER is still capable of reaching the nucleus and can be incorporated into the capsid, and this approach can be used to aid in the correct folding of complex molecules fused to the C terminus of pIX.
pIX normally forms speckles in the nucleus, described as clear amorphous structures by electron microscopy (58). ER-targeted pIX-GFP did not form distinct speckles in the nucleus when expressed alone but did form speckles when coexpressed with native pIX (compare Fig. 2E and 4E). This phenomenon has been observed previously: Rosa-Calatrava et al. (58) showed that mutation of the C-terminal leucine repeat (which prevents self-association) or the addition of a large C-terminal tag to pIX (which presumably sterically inhibited self-association) causes pIX to accumulate primarily in the cytoplasm, with microspeckles in the nucleus, and coexpression of native pIX was able to rescue this phenotype. The functional significance of these speckles is unclear. These data underscore the importance of the leucine repeat and self-association in proper localization of pIX within the cell. Coexpression of native pIX with ER-targeted pIX-MR1 did not rescue the speckling pattern (Fig. 4E). MR1 may obscure the leucine repeat region, preventing its association with itself or native pIX. Inclusion of a 75-Å spacer region between pIX and MR1 (78) did not improve the efficiency of pIX-MR1 incorporation (data not shown). Thus, the nature of the C-terminal addition to pIX is crucially important for its function and is difficult to predict. This method may be more amenable to other scFv or polypeptides that do not adversely affect pIX self-association and localization.
AFAI is an sdAb, identified by panning a llama sdAb phage library, on the non-small-cell lung carcinoma A549 cell line and recognizes a variant of CD66c (86). Although not exclusive to cancer cells, CD66 protein expression is upregulated on many cancers, including colon, stomach, pancreas, lung, and breast cancer and leukemia (12). In a screen of 139 neoplastic lesions of lungs and other organs, AFAI showed strong immunoreactivity to nonsquamous large-cell lung carcinomas, poorly differentiated lung adenocarcinomas, and undifferentiated large-cell lung carcinomas and weaker reactivity to many other types (44). Importantly, AFAI did not react with normal tissue. We showed that a virus that encoded AFAI fused to the C terminus of pIX demonstrated enhanced, CAR-independent infection of A549 cells (Fig. 6). Virus containing pIX-AFAI should be valuable for development of novel therapeutics directed toward diverse lung tumors. Perhaps more importantly, our study serves as a proof of principle that sdAb can be attached to pIX and used to retarget Ad infection. Libraries of camelid-derived sdAb are available (75), and such libraries can be panned against any target cell or tissue to identify specific sdAb. Alternatively, scFv libraries that have enhanced stability in the reducing environment of the cytoplasm, which would circumvent the need for ER-associated expression of this class of molecule, have been developed (54). Thus, Ads that express a wide array of polypeptides for specific targeting of a variety of cells and tissues can be developed.
Although gene therapy holds great promise for the treatment of many genetic and acquired diseases, most current vector systems suffer from an inability to specifically infect a desired target tissue. Ad vectors have proven to be one of the most efficient vehicles to mediate gene delivery in gene therapy studies, and they have repeatedly demonstrated high-level transduction of a variety of tissues; however, this promiscuity reduces their effectiveness in applications in which cell-type-specific transduction is required. We have developed an approach to present large targeting ligands, such as scFv and sdAb, on the surface of the Ad capsid, and our data indicate that such modifications improve virus infection of cells expressing the targeted cell surface epitope. Ad promiscuity can be reduced through mutation of the Ad fiber protein to prevent CAR binding (56), penton protein to prevent integrin binding (21), or hexon protein to prevent liver cell transduction (33, 80), and for maximum detargeting, mutation of all three capsid proteins is likely required. Undoubtedly, the ability to target Ad to specific cell types will enhance many research efforts and provide a technology which will be invaluable to many researchers investigating the use of Ads for gene delivery in various preclinical models and, ultimately, clinical trials involving genetic and acquired diseases.
Acknowledgments
We thank Jianbing Zhang, Jim Dimitroulakos, Roger Tsien, John Copeland, and Scott Gray-Owen for providing reagents and Michael A. Kennedy, Scott D. Ryan, and David J. Picketts for helpful discussions and critical evaluation of the manuscript.
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) and the Jesse Davidson Foundation for Gene and Cell Therapy, a CIHR/Muscular Dystrophy Canada/Amyotrophic Lateral Sclerosis Society of Canada Partnership grant, and the Cancer Research Society of Canada.
Footnotes
Published ahead of print on 14 July 2010.
REFERENCES
- 1.Addison, C. L., M. Hitt, D. Kunsken, and F. L. Graham. 1997. Comparison of the human versus murine cytomegalovirus immediate early gene promoters for transgene expression by adenoviral vectors. J. Gen. Virol. 78:1653-1661. [DOI] [PubMed] [Google Scholar]
- 2.Amalfitano, A., and R. J. Parks. 2002. Separating fact from fiction: assessing the potential of modified adenovirus vectors for use in human gene therapy. Curr. Gene Ther. 2:111-133. [DOI] [PubMed] [Google Scholar]
- 3.Beckman, R. A., L. M. Weiner, and H. M. Davis. 2007. Antibody constructs in cancer therapy: protein engineering strategies to improve exposure in solid tumors. Cancer 109:170-179. [DOI] [PubMed] [Google Scholar]
- 4.Bergelson, J. M., J. A. Cunningham, G. Droguett, E. A. Kurt-Jones, A. Krithivas, J. S. Hong, M. S. Horwitz, R. L. Crowell, and R. W. Finberg. 1997. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320-1323. [DOI] [PubMed] [Google Scholar]
- 5.Bett, A. J., W. Haddara, L. Prevec, and F. L. Graham. 1994. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. U. S. A. 91:8802-8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boulanger, P., P. Lemay, G. E. Blair, and W. C. Russell. 1979. Characterization of adenovirus protein IX. J. Gen. Virol. 44:783-800. [DOI] [PubMed] [Google Scholar]
- 7.Bramson, J. L., N. Grinshtein, R. A. Meulenbroek, J. Lunde, D. Kottachchi, I. A. Lorimer, B. J. Jasmin, and R. J. Parks. 2004. Helper-dependent adenoviral vectors containing modified fibre for improved transduction of developing and mature muscle cells. Hum. Gene Therapy 15:179-188. [DOI] [PubMed] [Google Scholar]
- 8.Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 99:7877-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campos, S. K., and M. A. Barry. 2006. Comparison of adenovirus fiber, protein IX, and hexon capsomeres as scaffolds for vector purification and cell targeting. Virology 349:453-462. [DOI] [PubMed] [Google Scholar]
- 10.Campos, S. K., M. B. Parrott, and M. A. Barry. 2004. Avidin-based targeting and purification of a protein IX-modified, metabolically biotinylated adenoviral vector. Mol. Ther. 9:942-954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carette, J. E., H. C. Graat, F. H. Schagen, M. A. Abou El Hassan, W. R. Gerritsen, and V. W. van Beusechem. 2005. Replication-dependent transgene expression from a conditionally replicating adenovirus via alternative splicing to a heterologous splice-acceptor site. J. Gene Med. 7:1053-1062. [DOI] [PubMed] [Google Scholar]
- 12.Chan, C. H., and C. P. Stanners. 2007. Recent advances in the tumour biology of the GPI-anchored carcinoembryonic antigen family members CEACAM5 and CEACAM6. Curr. Oncol. 14:70-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chartier, C., E. Degryse, M. Gantzer, A. Dieterle, A. Pavirani, and M. Mehtali. 1996. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 70:4805-4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colby, W. W., and T. Shenk. 1981. Adenovirus type 5 virions can be assembled in vivo in the absence of detectable polypeptide IX. J. Virol. 39:977-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corjon, S., A. Wortmann, T. Engler, N. van Rooijen, S. Kochanek, and F. Kreppel. 2008. Targeting of adenovirus vectors to the LRP receptor family with the high-affinity ligand RAP via combined genetic and chemical modification of the pIX capsomere. Mol. Ther. 16:1813-1824. [DOI] [PubMed] [Google Scholar]
- 16.Cottet, S., and B. Corthesy. 1997. Cellular processing limits the heterologous expression of secretory component in mammalian cells. Eur. J. Biochem. 246:23-31. [DOI] [PubMed] [Google Scholar]
- 17.Demarest, S. J., and S. M. Glaser. 2008. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr. Opin. Drug Discov. Devel. 11:675-687. [PubMed] [Google Scholar]
- 18.de Vrij, J., T. G. Uil, S. K. van den Hengel, S. J. Cramer, D. Koppers-Lalic, M. C. Verweij, E. J. Wiertz, J. Vellinga, R. A. Willemsen, and R. C. Hoeben. 2008. Adenovirus targeting to HLA-A1/MAGE-A1-positive tumor cells by fusing a single-chain T-cell receptor with minor capsid protein IX. Gene Ther. 15:978-989. [DOI] [PubMed] [Google Scholar]
- 19.Dmitriev, I., V. Krasnykh, C. R. Miller, M. Wang, E. Kashentseva, G. Mikheeva, N. Belousova, and D. T. Curiel. 1998. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J. Virol. 72:9706-9713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dmitriev, I. P., E. A. Kashentseva, and D. T. Curiel. 2002. Engineering of adenovirus vectors containing heterologous peptide sequences in the C terminus of capsid protein IX. J. Virol. 76:6893-6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Einfeld, D. A., R. Schroeder, P. W. Roelvink, A. Lizonova, C. R. King, I. Kovesdi, and T. J. Wickham. 2001. Reducing the native tropism of adenovirus vectors requires removal of both CAR and integrin interactions. J. Virol. 75:11284-11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fabry, C. M., M. Rosa-Calatrava, C. Moriscot, R. W. Ruigrok, P. Boulanger, and G. Schoehn. 2009. The C-terminal domains of adenovirus serotype 5 protein IX assemble into an antiparallel structure on the facets of the capsid. J. Virol. 83:1135-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furcinitti, P. S., J. van Oostrum, and R. M. Burnett. 1989. Adenovirus polypeptide IX revealed as capsid cement by difference images from electron microscopy and crystallography. EMBO J. 8:3563-3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glasgow, J. N., M. Everts, and D. T. Curiel. 2006. Transductional targeting of adenovirus vectors for gene therapy. Cancer Gene Ther. 13:830-844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham, F. L. 1987. Growth of 293 cells in suspension culture. J. Gen. Virol. 68:937-940. [DOI] [PubMed] [Google Scholar]
- 26.Graham, F. L., J. Smiley, W. C. Russell, and R. Nairn. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59-74. [DOI] [PubMed] [Google Scholar]
- 27.Guo, Z. S., L. H. Wang, R. C. Eisensmith, and S. L. Woo. 1996. Evaluation of promoter strength for hepatic gene expression in vivo following adenovirus-mediated gene transfer. Gene Ther. 3:802-810. [PubMed] [Google Scholar]
- 28.He, T. C., S. Zhou, L. T. da Costa, J. Yu, K. W. Kinzler, and B. Vogelstein. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. U. S. A. 95:2509-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubberstey, A. V., M. Pavliv, and R. J. Parks. 2002. Cancer therapy utilizing an adenoviral vector expressing only E1A. Cancer Gene Ther. 9:321-329. [DOI] [PubMed] [Google Scholar]
- 30.Jakubczak, J. L., M. L. Rollence, D. A. Stewart, J. D. Jafari, D. J. Von Seggern, G. R. Nemerow, S. C. Stevenson, and P. L. Hallenbeck. 2001. Adenovirus type 5 viral particles pseudotyped with mutagenized fiber proteins show diminished infectivity of coxsackie B-adenovirus receptor-bearing cells. J. Virol. 75:2972-2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jooss, K., Y. Yang, K. J. Fisher, and J. M. Wilson. 1998. Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers. J. Virol. 72:4212-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalderon, D., W. D. Richardson, A. F. Markham, and A. E. Smith. 1984. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature 311:33-38. [DOI] [PubMed] [Google Scholar]
- 33.Kalyuzhniy, O., N. C. Di Paolo, M. Silvestry, S. E. Hofherr, M. A. Barry, P. L. Stewart, and D. M. Shayakhmetov. 2008. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc. Natl. Acad. Sci. U. S. A. 105:5483-5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim, I., W. Xu, and J. C. Reed. 2008. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 7:1013-1030. [DOI] [PubMed] [Google Scholar]
- 35.Kirby, I., E. Davison, A. J. Beavil, C. P. Soh, T. J. Wickham, P. W. Roelvink, I. Kovesdi, B. J. Sutton, and G. Santis. 2000. Identification of contact residues and definition of the CAR-binding site of adenovirus type 5 fiber protein. J. Virol. 74:2804-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krasnykh, V., I. Dmitriev, G. Mikheeva, C. R. Miller, N. Belousova, and D. T. Curiel. 1998. Characterization of an adenovirus vector containing a heterologous peptide epitope in the HI loop of the fiber knob. J. Virol. 72:1844-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kreppel, F., J. Gackowski, E. Schmidt, and S. Kochanek. 2005. Combined genetic and chemical capsid modifications enable flexible and efficient de- and retargeting of adenovirus vectors. Mol. Ther. 12:107-117. [DOI] [PubMed] [Google Scholar]
- 38.Le, L. P., M. Everts, I. P. Dmitriev, J. G. Davydova, M. Yamamoto, and D. T. Curiel. 2004. Fluorescently labeled adenovirus with pIX-EGFP for vector detection. Mol. Imaging 3:105-116. [DOI] [PubMed] [Google Scholar]
- 39.Li, J., L. Le, D. A. Sibley, J. M. Mathis, and D. T. Curiel. 2005. Genetic incorporation of HSV-1 thymidine kinase into the adenovirus protein IX for functional display on the virion. Virology 338:247-258. [DOI] [PubMed] [Google Scholar]
- 40.Lorimer, I. A. 2002. Mutant epidermal growth factor receptors as targets for cancer therapy. Curr. Cancer Drug Targets 2:91-102. [DOI] [PubMed] [Google Scholar]
- 41.Lorimer, I. A., A. Keppler-Hafkemeyer, R. A. Beers, C. N. Pegram, D. D. Bigner, and I. Pastan. 1996. Recombinant immunotoxins specific for a mutant epidermal growth factor receptor: targeting with a single chain antibody variable domain isolated by phage display. Proc. Natl. Acad. Sci. U. S. A. 93:14815-14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lorimer, I. A., and S. J. Lavictoire. 2000. Targeting retrovirus to cancer cells expressing a mutant EGF receptor by insertion of a single chain antibody variable domain in the envelope glycoprotein receptor binding lobe. J. Immunol. Methods 237:147-157. [DOI] [PubMed] [Google Scholar]
- 43.Maelandsmo, G. M., P. J. Ross, M. Pavliv, R. A. Meulenbroek, C. Evelegh, D. A. Muruve, F. L. Graham, and R. J. Parks. 2005. Use of a murine secreted alkaline phosphatase as a non-immunogenic reporter gene in mice. J. Gene Med. 7:307-315. [DOI] [PubMed] [Google Scholar]
- 44.Mai, K. T., D. G. Perkins, J. Zhang, and C. R. Mackenzie. 2006. ES1, a new lung carcinoma antibody—an immunohistochemical study. Histopathology 49:515-522. [DOI] [PubMed] [Google Scholar]
- 45.Marsh, M. P., S. K. Campos, M. L. Baker, C. Y. Chen, W. Chiu, and M. A. Barry. 2006. Cryoelectron microscopy of protein IX-modified adenoviruses suggests a new position for the C terminus of protein IX. J. Virol. 80:11881-11886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matthews, Q. L., D. A. Sibley, H. Wu, J. Li, M. A. Stoff-Khalili, R. Waehler, J. M. Mathis, and D. T. Curiel. 2006. Genetic incorporation of a herpes simplex virus type 1 thymidine kinase and firefly luciferase fusion into the adenovirus protein IX for functional display on the virion. Mol. Imaging 5:510-519. [PMC free article] [PubMed] [Google Scholar]
- 47.Meulenbroek, R. A., K. L. Sargent, J. Lunde, B. J. Jasmin, and R. J. Parks. 2004. Use of adenovirus protein IX to display large polypeptides on the virion—generation of fluorescent virus through incorporation of pIX-GFP. Mol. Ther. 9:617-624. [DOI] [PubMed] [Google Scholar]
- 48.Michalak, M., J. Groenendyk, E. Szabo, L. I. Gold, and M. Opas. 2009. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 417:651-666. [DOI] [PubMed] [Google Scholar]
- 49.Muruve, D. A., M. J. Cotter, A. K. Zaiss, L. R. White, Q. Liu, T. Chan, S. A. Clark, P. J. Ross, R. A. Meulenbroek, G. M. Maelandsmo, and R. J. Parks. 2004. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J. Virol. 78:5966-5972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakamura, T., K. W. Peng, M. Harvey, S. Greiner, I. A. Lorimer, C. D. James, and S. J. Russell. 2005. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 23:209-214. [DOI] [PubMed] [Google Scholar]
- 51.Nelson, J. E., and M. A. Kay. 1997. Persistence of recombinant adenovirus in vivo is not dependent on vector DNA replication. J. Virol. 71:8902-8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niknejad, N., M. Morley, and J. Dimitroulakos. 2007. Activation of the integrated stress response regulates lovastatin-induced apoptosis. J. Biol. Chem. 282:29748-29756. [DOI] [PubMed] [Google Scholar]
- 53.Parks, R. J. 2005. Adenovirus protein IX: a new look at an old protein. Mol. Ther. 11:19-25. [DOI] [PubMed] [Google Scholar]
- 54.Philibert, P., A. Stoessel, W. Wang, A. P. Sibler, N. Bec, C. Larroque, J. G. Saven, J. Courtete, E. Weiss, and P. Martineau. 2007. A focused antibody library for selecting scFvs expressed at high levels in the cytoplasm. BMC Biotechnol. 7:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pihkala, P., M. Kawahara, H. Ueda, and T. Nagamune. 2004. An antigen-mediated selection system for mammalian cells that produce glycosylated single-chain Fv. Biochem. Biophys. Res. Commun. 324:1165-1172. [DOI] [PubMed] [Google Scholar]
- 56.Roelvink, P. W., G. Mi Lee, D. A. Einfeld, I. Kovesdi, and T. J. Wickham. 1999. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adenoviridae. Science 286:1568-1571. [DOI] [PubMed] [Google Scholar]
- 57.Rogers, B. E., J. T. Douglas, C. Ahlem, D. J. Buchsbaum, J. Frincke, and D. T. Curiel. 1997. Use of a novel cross-linking method to modify adenovirus tropism. Gene Ther. 4:1387-1392. [DOI] [PubMed] [Google Scholar]
- 58.Rosa-Calatrava, M., L. Grave, F. Puvion-Dutilleul, B. Chatton, and C. Kedinger. 2001. Functional analysis of adenovirus protein IX identifies domains involved in capsid stability, transcriptional activity, and nuclear reorganization. J. Virol. 75:7131-7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ross, P. J., and R. J. Parks. 2006. Construction of first-generation adenoviral vectors. In T. Freidman and J. Rossi (ed.), Gene therapy vectors: a techniques manual. Cold Spring Harbor, Cold Spring Harbor, NY.
- 60.Saban, S. D., M. Silvestry, G. R. Nemerow, and P. L. Stewart. 2006. Visualization of alpha-helices in a 6-angstrom resolution cryoelectron microscopy structure of adenovirus allows refinement of capsid protein assignments. J. Virol. 80:12049-12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sargent, K., R. A. Meulenbroek, and R. J. Parks. 2004. Activation of adenoviral gene expression by protein IX is not required for efficient virus replication. J. Virol. 78:5032-5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schnell, M. A., Y. Zhang, J. Tazelaar, G. P. Gao, Q. C. Yu, R. Qian, S. J. Chen, A. N. Varnavski, C. LeClair, S. E. Raper, and J. M. Wilson. 2001. Activation of innate immunity in nonhuman primates following intraportal administration of adenoviral vectors. Mol. Ther. 3:708-722. [DOI] [PubMed] [Google Scholar]
- 63.Seidman, M. A., S. M. Hogan, R. L. Wendland, S. Worgall, R. G. Crystal, and P. L. Leopold. 2001. Variation in adenovirus receptor expression and adenovirus vector-mediated transgene expression at defined stages of the cell cycle. Mol. Ther. 4:13-21. [DOI] [PubMed] [Google Scholar]
- 64.Seregin, S. S., Y. A. Aldhamen, D. M. Appledorn, Z. C. Hartman, N. J. Schuldt, J. Scott, S. Godbehere, H. Jiang, M. M. Frank, and A. Amalfitano. 28 May 2010. Adenovirus capsid-display of the retro-oriented human complement inhibitor DAF reduces Ad-vector triggered immune responses in vitro and in vivo. Blood doi: 10.1182/blood-2010-03-276949. [DOI] [PMC free article] [PubMed]
- 65.Seregin, S. S., Z. C. Hartman, D. M. Appledorn, S. Godbehere, H. Jiang, M. M. Frank, and A. Amalfitano. 2010. Novel adenovirus vectors ‘capsid-displaying’ a human complement inhibitor. J. Innate Immun. 2:353-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shayakhmetov, D. M., A. Gaggar, S. Ni, Z. Y. Li, and A. Lieber. 2005. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 79:7478-7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shenk, T. 2001. Adenoviridae: the viruses and their replication, p. 2265-2300. In B. N. Fields, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, S. E. Strauss, and D. M. Knipe (ed.), Fields virology, vol. 67. Lippincott-Raven Publishers, Philadelphia, PA. [Google Scholar]
- 68.Silvestry, M., S. Lindert, J. G. Smith, O. Maier, C. M. Wiethoff, G. R. Nemerow, and P. L. Stewart. 2009. Cryo-electron microscopy structure of adenovirus type 2 temperature-sensitive mutant 1 reveals insight into the cell entry defect. J. Virol. 83:7375-7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singer, B. B., I. Scheffrahn, R. Kammerer, N. Suttorp, S. Ergun, and H. Slevogt. 2010. Deregulation of the CEACAM expression pattern causes undifferentiated cell growth in human lung adenocarcinoma cells. PLoS One 5:e8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith, A. C., K. L. Poulin, and R. J. Parks. 2009. DNA genome size affects the stability of the adenovirus virion. J. Virol. 83:2025-2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith, J. S., J. R. Keller, N. C. Lohrey, C. S. McCauslin, M. Ortiz, K. Cowan, and S. E. Spence. 1999. Redirected infection of directly biotinylated recombinant adenovirus vectors through cell surface receptors and antigens. Proc. Natl. Acad. Sci. U. S. A. 96:8855-8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith, T. A., N. Idamakanti, M. L. Rollence, J. Marshall-Neff, J. Kim, K. Mulgrew, G. R. Nemerow, M. Kaleko, and S. C. Stevenson. 2003. Adenovirus serotype 5 fiber shaft influences in vivo gene transfer in mice. Hum. Gene Ther. 14:777-787. [DOI] [PubMed] [Google Scholar]
- 73.Snitkovsky, S., T. M. Niederman, B. S. Carter, R. C. Mulligan, and J. A. Young. 2000. A TVA-single-chain antibody fusion protein mediates specific targeting of a subgroup A avian leukosis virus vector to cells expressing a tumor-specific form of epidermal growth factor receptor. J. Virol. 74:9540-9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang, Y., L. P. Le, Q. L. Matthews, T. Han, H. Wu, and D. T. Curiel. 2008. Derivation of a triple mosaic adenovirus based on modification of the minor capsid protein IX. Virology 377:391-400. [DOI] [PubMed] [Google Scholar]
- 75.Tanha, J., G. Dubuc, T. Hirama, S. A. Narang, and C. R. MacKenzie. 2002. Selection by phage display of llama conventional V(H) fragments with heavy chain antibody V(H)H properties. J. Immunol. Methods 263:97-109. [DOI] [PubMed] [Google Scholar]
- 76.Tomko, R. P., R. Xu, and L. Philipson. 1997. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. U. S. A. 94:3352-3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vellinga, J., J. de Vrij, S. Myhre, T. Uil, P. Martineau, L. Lindholm, and R. C. Hoeben. 2007. Efficient incorporation of a functional hyper-stable single-chain antibody fragment protein-IX fusion in the adenovirus capsid. Gene Ther. 14:664-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vellinga, J., M. J. Rabelink, S. J. Cramer, D. J. van den Wollenberg, H. Van der Meulen, K. N. Leppard, F. J. Fallaux, and R. C. Hoeben. 2004. Spacers increase the accessibility of peptide ligands linked to the carboxyl terminus of adenovirus minor capsid protein IX. J. Virol. 78:3470-3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vigne, E., I. Mahfouz, J. F. Dedieu, A. Brie, M. Perricaudet, and P. Yeh. 1999. RGD inclusion in the hexon monomer provides adenovirus type 5-based vectors with a fiber knob-independent pathway for infection. J. Virol. 73:5156-5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Waddington, S. N., J. H. McVey, D. Bhella, A. L. Parker, K. Barker, H. Atoda, R. Pink, S. M. K. Buckley, J. A. Greig, L. Denby, J. Custers, T. Morita, I. M. B. Francischetti, R. Q. Monteiro, D. H. Barouch, N. van Rooijen, C. Napoli, M. J. E. Havenga, S. A. Nicklin, and A. H. Baker. 2008. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 132:397-409. [DOI] [PubMed] [Google Scholar]
- 81.Wickham, T. J., P. Mathias, D. A. Cheresh, and G. R. Nemerow. 1993. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 73:309-319. [DOI] [PubMed] [Google Scholar]
- 82.Wickham, T. J., D. M. Segal, P. W. Roelvink, M. E. Carrion, A. Lizonova, G. M. Lee, and I. Kovesdi. 1996. Targeted adenovirus gene transfer to endothelial and smooth muscle cells by using bispecific antibodies. J. Virol. 70:6831-6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wickham, T. J., E. Tzeng, L. L. Shears, P. W. Roelvink, Y. Li, G. M. Lee, D. E. Brough, A. Lizonova, and I. Kovesdi. 1997. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins. J. Virol. 71:8221-8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wörn, A., and A. Pluckthun. 2001. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 305:989-1010. [DOI] [PubMed] [Google Scholar]
- 85.Zakhartchouk, A., W. Connors, A. van Kessel, and S. K. Tikoo. 2004. Bovine adenovirus type 3 containing heterologous protein in the C-terminus of minor capsid protein IX. Virology 320:291-300. [DOI] [PubMed] [Google Scholar]
- 86.Zhang, J., Q. Li, T. D. Nguyen, T. L. Tremblay, E. Stone, R. To, J. Kelly, and C. Roger MacKenzie. 2004. A pentavalent single-domain antibody approach to tumor antigen discovery and the development of novel proteomics reagents. J. Mol. Biol. 341:161-169. [DOI] [PubMed] [Google Scholar]