

Abstract
Herpesviral virions contain a tegument layer that consists primarily of viral proteins. The delivery of fully functional proteins to infected cells upon virion envelope fusion to the plasma membrane allows herpesviruses to modulate cellular activities prior to viral gene expression. Certain tegument proteins can also regulate viral processes. For example, the pp71 tegument protein encoded by the UL82 gene of human cytomegalovirus (HCMV) stimulates viral immediate early (IE) gene expression and thus acts to initiate the productive lytic infectious cycle. In terminally differentiated fibroblasts infected with HCMV, tegument-delivered pp71 traffics to the nucleus and degrades the cellular transcriptional corepressor Daxx to initiate viral IE gene expression and lytic replication. However, when HCMV infects incompletely differentiated cells, tegument-delivered pp71 remains in the cytoplasm, allowing the nucleus-localized Daxx protein to silence viral IE gene expression and promote the establishment of a latent infection in certain cell types. We sought to determine whether undifferentiated cells block the trafficking of tegument-delivered pp71 to the nucleus or whether differentiated cells facilitate the nuclear transport of tegument-delivered pp71. Heterogenous cell fusion experiments demonstrated that tegument-delivered pp71 found in the cytoplasm of undifferentiated NT2 cells could be driven into the nucleus by one or more factors provided by fully differentiated fibroblasts. Our data raise the intriguing possibility that latency is the default program launched by HCMV upon viral entry into cells and that lytic infection is initiated only in certain (differentiated) cells that can facilitate the delivery of incoming pp71 to the nucleus.
Human cytomegalovirus (HCMV) is a ubiquitous betaherpesvirus that infects 60 to 90% of the world's population (38). Though infections are typically asymptomatic for healthy individuals, HCMV is the leading cause of virus-induced birth defects, it causes severe disease in immunocompromised and immunosuppressed individuals, and it has been associated with several proliferative diseases, including atherosclerosis, restenosis, and certain types of cancer (37, 53, 54). Upon entry into a cell, HCMV can either initiate a productive lytic infection or establish a latent infection in which the viral genome is maintained without progeny virion production (24, 38, 52, 55). Latently infected cells help the virus avoid immune detection and clearance. Reactivation events produce new virions for dissemination among and between hosts (51). In general, lytic infections are initiated when the virus infects terminally differentiated cells, such as fibroblasts, and latent infections are established when the virus infects certain incompletely differentiated cells of the myeloid lineage, such as CD34+ hematopoietic progenitor cells.
The double-stranded DNA genome of HCMV is packaged in an icosahedral capsid that in turn is surrounded by a lipid envelope. Located between the capsid and envelope of infectious virions is a proteinaceous layer known as the tegument (27). Fusion of the virion envelope to the cell membrane during viral entry introduces the fully formed and active tegument proteins into the infected cell, where they perform multiple functions that include immune evasion and assisting viral-genome delivery to the nucleus (26). A critical activity of the tegument is to initiate the lytic replication cycle by activating the expression of the first set of viral lytic-phase genes that encode the viral immediate early (IE) proteins. The most prominent IE proteins (IE1 and IE2) are encoded by a single locus whose transcription is controlled by the major immediate early promoter (MIEP) and activated by a tegument-delivered viral protein named pp71 (3, 5-7, 18, 32, 33, 35, 43, 46, 49, 57, 63).
The general mechanism through which pp71 activates IE gene expression is well established and involves counteracting the effects of a cellular intrinsic immune defense designed to silence the incoming viral genome (44, 58). Upon entry into the nucleus, the viral genome becomes associated with histones (10, 41, 63), as well as cellular proteins that normally localize to promyelocytic leukemia nuclear body (PML-NB) structures (23). PML-NBs regulate multiple activities, such as transcription, DNA repair, and apoptosis (4, 31). HCMV genomes associated with PML-NBs at very early times after infection display a chromatin structure reminiscent of transcriptionally silent heterochromatin, and viral gene expression is not observed (63). In cells destined to initiate a lytic infection (such as terminally differentiated fibroblasts), tegument-delivered pp71 traffics to the nucleus and counteracts the intrinsic PML-NB defense (17, 46).
A major target of pp71 is the cellular Daxx protein, a transcriptional corepressor found in PML-NBs that silences gene expression through the recruitment of histone deacetylases (HDACs) to targeted promoters (6, 18, 43, 46). pp71 neutralizes the ability of Daxx to silence HCMV IE gene expression by displacing the corepressor ATRX (33) and induces Daxx sumoylation (21) and eventually its proteasome-dependent, ubiquitin-independent degradation (22, 46). Activation of IE1 expression by pp71 allows this protein to disrupt PML-NB structures, further weakening this intrinsic defense, amplifying IE gene expression, and fully activating the lytic replication cycle (1, 2, 29, 62).
When experimental latent infections are established in vitro in CD34+ cells, the PML-NB intrinsic defense is not neutralized and Daxx is not degraded (47). In these latent infections, Daxx silences viral IE gene expression in cooperation with an additional, uncharacterized mechanism apparently encoded only by clinical-strain viruses. Daxx remains stable in HCMV-infected CD34+ cells because tegument-delivered pp71 remains in the cytoplasm, failing to accumulate in the nucleus. If CD34+ cells are terminally differentiated into dendritic cells prior to HCMV infection, tegument-delivered pp71 traffics to the nucleus, degrades Daxx, and initiates viral IE gene expression and the lytic replication cycle (47).
Thus, pp71 appears to be an important viral determinant for outcome of infection, as there is a strong correlation between the subcellular localization of tegument-delivered pp71 and initiation of a lytic or latent infection (46, 47). This correlation extends to quiescent infections (45) that resemble latency, except that efficient reactivation triggers, if they exist, have not yet been identified. Examples of cell types supporting quiescent HCMV infections include THP-1 monocytes and NTERA-2 (NT2) teratoma cells (12, 13, 30, 36, 45, 59, 61). In these poorly differentiated cells, tegument-delivered pp71 is found in the cytoplasm and viral genes are repressed by the intrinsic PML-NB defense (45) as well as additional, unknown mechanisms (14, 47). Terminal differentiation of these cells prior to infection allows tegument-delivered pp71 to accumulate in the nucleus, degrade Daxx, and initiate viral IE gene expression and productive lytic infection (12, 13, 36, 45, 59).
Interestingly, de novo-expressed pp71 in CD34+, THP-1, or NT2 cells (by plasmid transfection or recombinant adenoviral transduction) enters and accumulates in the nucleus (45, 47). In THP-1 and NT2 cells, newly expressed pp71 can degrade Daxx and activate viral IE gene expression upon subsequent infection by HCMV (45). Thus, tegument-delivered pp71 is cytoplasmic, but newly expressed pp71 is nuclear in the incompletely differentiated cells where HCMV establishes quiescent or latent infections.
The mechanisms controlling pp71's subcellular localization are poorly understood. Mutations at specific sites of pp71 phosphorylation affect the subcellular localization of the mutant proteins in transfection assays (49). It is presently unclear if phosphorylation affects pp71 subcellular localization during HCMV infection. Furthermore, whether tegument-delivered pp71 fails to accumulate in the nuclei of undifferentiated cells because of a block of nuclear import or because of highly efficient nuclear export is not known. We sought to determine if tegument-delivered pp71 is found in the cytoplasm of poorly differentiated cells because of a dominant gain-of-function phenotype (i.e., because they express one or more factors that actively trap pp71 in the cytoplasm) or because of a recessive, loss-of-function phenotype (i.e., because they are missing one or more factors expressed in terminally differentiated cells that facilitate the trafficking of tegument-delivered pp71 to the nucleus). Here we describe a series of classic heterogenous cell-cell fusion experiments that led us to conclude that terminally differentiated cells express one or more factors that facilitate the transport of tegument-delivered pp71 to the nucleus. Artificial modulation of pp71 subcellular localization has the potential for therapeutic use in HCMV-infected individuals. This work represents a critical initial step toward determining how pp71 subcellular localization is controlled during HCMV infection.
MATERIALS AND METHODS
Cells, transfections, virus, and infections.
Normal human dermal fibroblasts (NHDFs) and NTERA-2 (NT2) embryonal carcinoma cells purchased from the ATCC were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (Gemini), 100 U/ml penicillin, and 100 μg/ml streptomycin plus 0.292 mg/ml glutamine (Gibco) at 37°C in a 5% CO2 atmosphere. One million NHDFs were transfected with a total of 3 to 4 μg of DNA using the Amaxa Biosystems Nucleofector and the NHDF transfection kit (Lonza; VPD-1001) by following the manufacturer's protocol. Where indicated, transfection cocktails included 1 μg of pEGFPN-1 and 3 μg of pCDNA3-p14, with the empty pCDNA3 vector as both a filler and a substitute for the other plasmids when appropriate. NT2 cells cultured on 10-cm plates were transfected with 20 μg of DNA using the calcium phosphate protocol. The individual amounts of DNA transfected were 2 μg green fluorescent protein (GFP), 5 μg pCDNA3-p14, and 13 μg the empty pCDNA3 vector. The HCMV strain utilized was AD169. UV inactivation was performed as previously described (46). NT2 cell infections were at a multiplicity of infection (MOI) of 5 PFU/cell (titrated on fibroblasts) for fusion experiments and 1 PFU/cell for nonfusion experiments. Where indicated, NT2 cells were treated with 2 ng/ml leptomycin B (LMB) (Calbiochem; 431050) for 1 h prior to HCMV infection.
Cell-cell fusion experiments.
Transfected cells plated on coverslips were incubated for either 6 h (NHDFs) or 24 h (NT2 cells) prior to being overseeded with infected NT2 cells, which were washed with TDE (25 mM Tris, 134 mM NaCl, 5 mM KCl, 0.75 mM Na2HPO4, and 0.5 mM EDTA, pH 7.5) and collected by trypsinization. Cells were then cocultured for 15 to 18 h. Figure 2 contains a visual depiction of the setup of these experiments.
Immunofluorescence, Western blots, and antibodies.
Coverslips were washed with phosphate-buffered saline (PBS; Gibco) and then fixed with 1% paraformaldehyde in PBS. After a 30-min blocking period in PBST (PBS plus 0.1% Triton X-100 and 0.005% Tween 20) plus 0.5% bovine serum albumin (BSA) and 5% goat serum, coverslips were incubated with the appropriate primary antibody for 1 h at room temperature in the same blocking solution. After being washed three times for 5 min each with PBST, coverslips were stained with a secondary antibody and then subsequently washed as described above. Next, the coverslips were washed twice with distilled water, and then nuclei were counterstained with Hoechst 33342 diluted in water for 10 min at room temperature, followed by two washes with distilled water. Finally, coverslips were mounted on glass slides using Fluoromount-G (Southern Biotech). Images were taken using a Zeiss Axiovert 200 M microscope and camera system. Note that multiple Z-stack sections are compressed into a single image to allow for visualization of punctate pp71. Lysate preparation and Western blotting were performed as previously described (46). Antibodies against pp71 (IE-233) and IE1 (1B12) have been previously described (28, 42, 65). Oct4 (sc-9081) and NF-κB (sc-372) antibodies were purchased from Santa Cruz. Tubulin (DM 1A) and Daxx (D7810) antibodies were purchased from Sigma. The pp65 antibody (8F5) was a gift from Tom Shenk. Secondary antibodies were anti-mouse IgG conjugated to Alexa Fluor 488, Alexa Fluor 594 (Molecular Probes), or horseradish peroxidase (Chemicon).
RESULTS
The subcellular localization of tegument-delivered pp71 depends upon the differentiation state of the infected cell.
Previous work has demonstrated that tegument-delivered pp71 traffics to the nuclei of fully differentiated cells, such as fibroblasts (17, 35, 46), but remains in the cytoplasm of incompletely differentiated cells, such as THP-1, NT2, and CD34+ cells (45, 47). These easily distinguishable subcellular localizations are illustrated here in fibroblasts and NT2 cells (Fig. 1). In normal human dermal fibroblasts (NHDFs), tegument-delivered pp71 is found in the nucleus (Fig. 1A) and initiates the lytic replication cycle by activating the expression of the viral IE genes, shown here by detection of the IE1 protein (Fig. 1B). Another tegument protein, pp65, also traffics to the nucleus in HCMV-infected fibroblasts (Fig. 1C). In NT2 cells, tegument-delivered pp71 remains in the cytoplasm as punctate spots (Fig. 1D and E), and IE genes are not expressed (Fig. 1F). Fibroblasts and NT2 cells are readily distinguished because only NT2 cells express the Oct4 protein (Fig. 1), a marker for incompletely differentiated cells. Upon differentiation of NT2 cells, Oct4 expression is downregulated and the protein rapidly disappears from the cell (11). Interestingly, pp65 is also sequestered in the cytoplasm upon HCMV infection of NT2 cells (Fig. 1G), indicating that pp71 is not the only tegument-delivered protein to achieve different subcellular localization upon infection of fully or incompletely differentiated cells.
FIG. 1.

Tegument-delivered pp71 is nuclear in differentiated but not undifferentiated cells. NHDFs (A, B, and C) and NT2 cells (D, E, F, and G) were plated on coverslips, infected at an MOI of 1 for 6 h, and then analyzed by indirect immunofluorescence for pp71, IE1, or pp65 and Oct4, as indicated. The boxed area of the merged panel of row D is shown as an enlarged image in row E for easier visualization of pp71 localization. In coculture experiments, NHDFs were transfected with either GFP and p14 (H) or GFP and an empty vector (I). Six hours later, they were overseeded with NT2 cells. These heterogenous cell cultures were fixed 15 h later and then analyzed by indirect immunofluorescence for Oct4. Nuclei were counterstained with Hoechst dye. NHDF nuclei in the second and fourth panels of row I are marked with asterisks.
Heterogenous cell fusion experiments are a classic method of differentiating dominant and recessive phenotypes.
Tegument-delivered pp71 may be trapped in the cytoplasm of incompletely differentiated cells because they possess a dominant restriction of pp71 nuclear trafficking, such as a cellular protein that traps pp71 in the cytoplasm. Alternatively, incompletely differentiated cells may display a recessive phenotype in which tegument-delivered pp71 is unable to localize to the nucleus due to the absence of one or more factors required for pp71 nuclear transport. Cell fusion experiments are often used to differentiate between dominant and recessive phenotypes under such circumstances (9, 15, 16, 34, 39, 50, 60). They provide the critical information (whether permissive or nonpermissive cells control the observed phenotype) that allows the eventual identification of the cellular factor that restricts or facilitates viral infection (20, 40, 48, 56). For example, much earlier cell fusion experiments paved the way for the recent identification of Trim5α (9, 39, 56), APOBEC3G (34, 48, 50), and Tetherin (40, 60), cellular proteins that restrict human immunodeficiency virus type 1 (HIV-1) replication, as well as TMX2 (15, 16, 20), an apparent inhibitor of herpes simplex virus type 1 (HSV-1).
Therefore, we undertook a series of fibroblast and NT2 cell fusion experiments to determine if the NT2 cells (and by extension incompletely differentiated cells in general) display a dominant or recessive phenotype in terms of the cytoplasmic localization of tegument-delivered pp71. To efficiently induce fusion between these two cell types, we utilized the reovirus p14 FAST protein (8).
The reovirus p14 FAST protein induces cell-to-cell fusion.
p14 FAST is a small transmembrane protein that induces membrane fusion in adjacent cells (8). For example, when GFP- and p14-transfected NHDFs are cocultured with NT2 cells, syncytia form between these two heterogenous cell types (Fig. 1H). Syncytia are easily distinguished from single cells because they have multiple nuclei, and the nuclei tend to aggregate. Both cell types must contribute to the syncytia because the fused cells express both GFP (contributed by the fibroblasts) and Oct4 (contributed by the NT2 cells). When p14 is left out of the transfection, the cocultured cells do not fuse, and thus individual cells are either GFP positive (fibroblasts) or Oct4 positive (NT2s) but never both (Fig. 1I). Thus, transfection of p14 facilitates rapid and substantial heterogenous cell-cell fusion and syncytium formation.
Fusion of HCMV-infected NT2 cells with uninfected fibroblasts results in the nuclear accumulation of pp71, formerly localized in the NT2 cell cytoplasm.
To determine whether incompletely differentiated cells display a dominant or recessive phenotype in terms of the cytoplasmic localization of tegument-delivered pp71, we coplated HCMV-infected NT2 cells with GFP- and p14-transfected NHDFs, allowed syncytia to form, and then determined the subcellular localization of pp71 by indirect immunofluorescence. A flowchart of the experimental design is presented in Fig. 2 A. Heterogenous syncytia that formed between HCMV-infected NT2 cells and cocultured GFP- and p14-transfected NHDFs clearly displayed pp71 predominantly within nuclei (Fig. 3 A). The syncytia formed during this experiment were defined as heterogenous fusions of NT2 and NHDFs because they expressed both GFP and Oct4 (Fig. 3B). Because pp71 originally located in the NT2 cell cytoplasm was found in nuclei after fusion with uninfected fibroblasts, we conclude that the fibroblasts provide one or more factors that facilitate tegument-delivered pp71 accumulation within the nucleus. Thus, with regard to the subcellular localization of tegument-delivered pp71, undifferentiated cells show a recessive phenotype, whereas differentiated cells show a dominant phenotype.
FIG. 2.
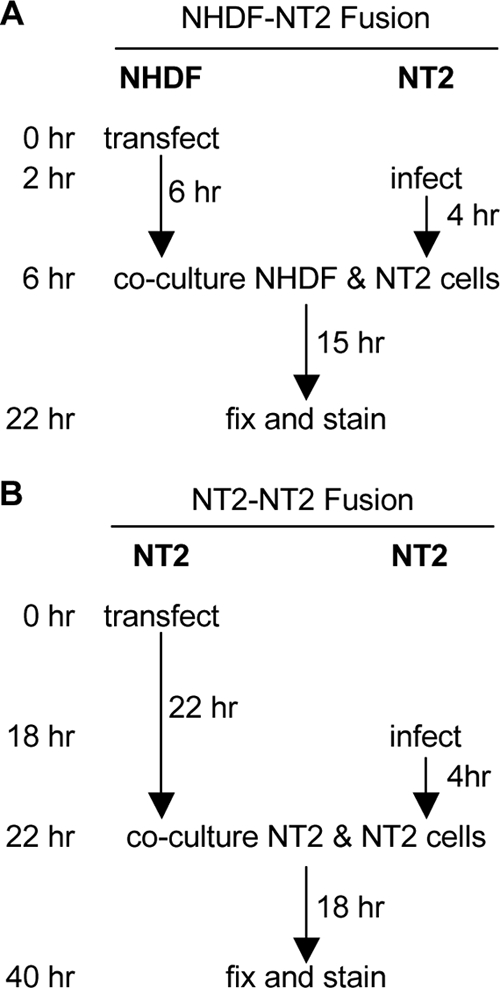
Experimental design for fusion experiments. (A) NHDFs were transfected with either GFP and p14, GFP and an empty vector, or p14 alone, plated on coverslips, and then cultured for 6 h. Separately, NT2 cells were infected at an MOI of 5 for 4 h, collected with trypsin, and subsequently coplated with the transfected NHDFs. Cells were incubated for 15 h to allow syncytium formation and then fixed and analyzed by indirect immunofluorescence. (B) NT2 cells were transfected with either GFP and p14 or GFP and an empty vector, plated on coverslips, and then cultured for 22 h. Separately, NT2 cells were infected at an MOI of 5 for 4 h, collected with trypsin, and subsequently coplated with the transfected NT2 cells. Cells were incubated for 18 h to allow syncytium formation and then fixed and analyzed by indirect immunofluorescence.
FIG. 3.

pp71 localizes to the nuclei of NHDF-NT2 syncytia but not NT2-NT2 syncytia. NHDFs transfected with GFP and p14 (A and B) or GFP and an empty vector (C and D) were overseeded with infected NT2 cells (MOI = 5) and then fixed and analyzed for pp71 (A and C) or Oct4 (B and D) by indirect immunofluorescence. NT2 cells transfected with p14 and GFP (E and F) were overseeded with infected NT2 cells (MOI = 5) and then fixed and analyzed for pp71 (E) or Oct4 (F) expression by indirect immunofluorescence. Nuclei were counterstained with Hoechst dye. Asterisks in panel D indicate GFP-positive, Oct4-negative NHDF nuclei. Arrows in panel E and H point to punctate pp71. To allow for easier visualization of pp71, enlargements of the merged images from panels C (#) and D (##) are shown as panels G and H, respectively.
When HCMV-infected NT2 cells were coplated with NHDFs transfected with GFP but not p14, pp71 remained punctate in the cytoplasm of NT2 cells (Fig. 3C; an enlargement of the merged image [enlargement #] is shown in Fig. 3G) and was not observed within the fibroblasts because syncytia did not form. The lack of cell fusion is evident because the coplated cells display either Oct4 (NT2 cells) or GFP (NHDFs) but never both (Fig. 3D). This control experiment indicates that cell-cell fusion is required to permit cytoplasmic pp71 to accumulate in the nucleus in this assay and argues against the possibility that extracellular virions attached to NT2 cells can directly enter cocultured NHDFs and thereby deliver pp71 to the nucleus (that mechanism would not require the presence of p14).
To confirm that fusion of HCMV-infected NT2 cells to a differentiated cell type was required to shift the steady-state localization of tegument-delivered pp71 from the cytoplasm of NT2 cells into the nuclei of heterogenous syncytia, we performed a homologous cell-cell fusion experiment as outlined by the flowchart in Fig. 2B. The coculture of HCMV-infected NT2 cells with GFP- and p14-transfected NT2 cells resulted in syncytium formation (evidenced by multiple, aggregated nuclei in a single, large cell) but failed to allow cytoplasmic pp71 to accumulate in the nucleus (Fig. 3E; an enlargement of the merged image [enlargement ##] is shown in Fig. 3H). Oct4 staining confirmed that the cells remained undifferentiated during the time course of the experiment (Fig. 3F). These experiments show that tegument-delivered pp71 localized in the cytoplasm of NT2 cells is free to accumulate in the nucleus upon fusion with fibroblasts. Approximately 60% of heterogenous (NT2-NHDF) syncytia showed pp71 nuclear staining, whereas pp71 was found in the nuclei of less than 1% of homologous (NT2-NT2) syncytia (Fig. 4). From this we conclude that terminally differentiated cells express at least one factor that facilitates the steady-state nuclear accumulation of tegument-delivered pp71.
FIG. 4.

Quantification of pp71 localization and IE gene expression in syncytia. Syncytia formed by HCMV-infected NT2 cells and either NHDFs (left) or NT2 cells (right) were examined for pp71 localization and IE gene expression. The average percentages of syncytia with tegument-delivered pp71 in the cytoplasm (bars labeled C) or nucleus (bars labeled N) or that express IE1 (bars labeled IE) are shown for 3 independent experiments, in which a total of at least 250 syncytia were analyzed. Error bars represent 1 standard deviation.
Productive replication initiates when HCMV-infected NT2 cells are fused with fibroblasts.
We next wanted to determine if the pp71 formerly found in the cytoplasm could initiate the productive, lytic replication cycle after it accumulates in the nuclei of heterogenous NT2-NHDF syncytia. Therefore, we stained control and fused cells for the viral IE1 protein. IE1 is not expressed upon quiescent (Fig. 1E) or latent infection but is expressed at the start of lytic infection (Fig. 1B). Furthermore, IE1 expression strictly correlates with, and is activated by, nuclear pp71 (45-47). Along with IE2, IE1 activates viral and cellular gene expression to drive the productive, lytic replication cycle of the virus (38). The coculture of HCMV-infected NT2 and GFP-transfected NHDFs (in the absence of p14) did not permit IE1 expression (Fig. 5 A), as expected. However, the coculture of HCMV-infected NT2 with GFP- and p14-transfected NDHFs that induced heterogenous syncytium formation and permitted pp71 nuclear accumulation (Fig. 3A) also activated IE1 expression (Fig. 5B). The coculture of HCMV-infected NT2 cells with GFP- and p14-transfected NT2 cells that induced homogenous syncytia where pp71 remained in the cytoplasm (Fig. 3E) did not support IE1 expression (Fig. 5C). We determined that approximately 25% of heterogenous (NT2-NHDF) syncytia expressed IE1 but that less than 2% of homogenous (NT2-NT2) syncytia were IE1 positive (Fig. 4). From these experiments we conclude that pp71 found in the cytoplasm of HCMV-infected incompletely differentiated cells, when allowed to accumulate in the nucleus, can initiate the lytic replication cycle.
FIG. 5.

pp71 can activate IE gene expression when transported to the nuclei of NHDF-NT2 syncytia. NHDFs transfected with either GFP and an empty vector (A) or GFP and p14 (B) were overseeded with infected NT2 cells (MOI = 5) and examined for IE1 expression by indirect immunofluorescence. (C) NT2 cells were transfected with GFP and p14, overseeded with infected NT2 cells (MOI = 5), and examined for IE1 expression by indirect immunofluorescence. (D) NHDFs transfected with GFP and p14 were overseeded with NT2 cells infected with UV-inactivated virus (MOI = 5) and then examined for pp71 by indirect immunofluorescence. Nuclei were counterstained with Hoechst dye.
The initiation of the lytic replication program in these syncytia, evidenced by the IE1 protein, raised the possibility that early genes, such as pp71, could also be expressed. Thus, some portion of the nuclear pp71 that we observed in the syncytia may not be tegument delivered but in fact be newly synthesized. Because the expression of the viral IE genes (which is absolutely required for early gene expression) is enhanced by nuclear but not cytoplasmic pp71, we hypothesized that tegument-delivered pp71 must have entered the nuclei in these syncytia in order for early gene expression to become activated. To directly test this prediction, we infected NT2 cells with UV-inactivated HCMV prior to performing fusion experiments as outlined in Fig. 2A. Treatment of HCMV virions with UV light damages the viral DNA genome, rendering it unable to produce any new protein products. Thus, the pp71 observed in cells infected with UV-inactivated virus must be tegument delivered. In heterogenous syncytia, tegument-delivered pp71 introduced into NT2 cells by UV-inactivated HCMV clearly localized to the nucleus after fusion with fibroblasts (Fig. 5D). Thus, both indirect reasoning and direct experimental evidence support the conclusion that fusion of HCMV-infected NT2 cells to uninfected fibroblasts drives the transport of tegument-delivered pp71 from the cytoplasm into the nucleus.
Nuclear pp71 or IE1 can be observed simultaneously with Oct4 or Daxx in heterogenous syncytia not expressing GFP.
Initial experiments used transfected GFP as an easy way to identify syncytia. One drawback of this approach was our ability to image only one other protein (pp71, IE1, or Oct4) along with Hoechst nuclear stain in these experiments. Therefore, we repeated this series of experiments with GFP omitted from the transfection. The coculture of HCMV-infected NT2 cells with p14-transfected NHDFs permitted syncytium formation, accumulation of the previously cytoplasmic tegument-delivered pp71 to the nucleus (Fig. 6 A), and the initiation of lytic infection, shown by the expression of IE1 (Fig. 6B). Without GFP, we were able to image pp71 (Fig. 6A) or IE1 (Fig. 6B) in the nuclei of Oct4-positive syncytia (both the pp71- and IE1-specific sera represent mouse monoclonal antibodies; thus, these two proteins were not simultaneously visualized). This control experiment helps confirm that the syncytia in which pp71 accumulates in the nucleus, and in which IE1 is expressed, represent the fusion of NHDFs with undifferentiated (Oct4-expressing) NT2 cells.
FIG. 6.

Tegument-delivered pp71 localizes to the nuclei of Oct4-positive syncytia and colocalizes with Daxx. NHDFs transfected with p14 were overseeded with NT2 cells infected with wild-type (A and B) or UV-inactivated (C and D) virus (MOI = 5) and then fixed and analyzed for pp71 and Oct4 (A), pp71 and IE1 (B), or pp71 and Daxx (C and D) by indirect immunofluorescence. Nuclei were counterstained with Hoechst dye. (D) Enlargement of the boxed area in panel C.
Similarly, in the absence of cotransfected GFP, we were able to visualize the colocalization of pp71 and Daxx in heterogenous syncytia (Fig. 6C and D). As was predicted from its colocalization with pp71 and the subsequent expression of IE1, Daxx levels were substantially reduced in the heterogenous syncytia formed from fibroblasts and HCMV-infected NT2 cells compared to levels in heterogenous syncytia formed with uninfected NT2 cells (Fig. 7). These results show that after translocating to the nuclei of the heterogenous syncytia, pp71 colocalizes with and induces the degradation of Daxx to stimulate IE gene expression, as it does during direct infection of differentiated cell types (45-47).
FIG. 7.

Daxx is degraded in NHDF-NT2 syncytia. NHDFs transfected with p14 were overseeded with mock-infected (lane M) or HCMV-infected (lane V) NT2 cells. Fifteen hours after being overseeded, cells were trypsinized and lysed and Western blotting was performed. Syncytial lysates were blotted for Daxx and tubulin (Tub).
pp71 subcellular localization appears to be controlled at the stage of nuclear import and not nuclear export.
It is unknown whether tegument-delivered pp71 fails to accumulate in the nuclei of incompletely differentiated cells because it is unable to enter the nucleus or because it enters but is rapidly and efficiently exported from the nucleus, and therefore its steady-state localization is in the cytoplasm. The data presented above indicate that NHDFs express one or more factors that facilitate the nuclear accumulation of tegument-delivered pp71. Mechanistically, this could occur by facilitating the import of cytoplasmic tegument-delivered pp71 into the nucleus or by preventing its nuclear export. To distinguish between these two possibilities, we inhibited nuclear export with leptomycin B (LMB) and examined the subcellular localization of tegument-delivered pp71 in NT2 cells. We used the NF-κB protein to monitor nuclear export, as this protein is known to accumulate within the nuclei of cells treated with LMB (19, 25). Our LMB-treated NT2 cells were clearly deficient in nuclear export, as the normally cytoplasmic NF-κB transcription factor (Fig. 8 A) accumulated in the nucleus in the presence of the drug (Fig. 8B). Although HCMV is known to induce NF-κB nuclear translocation in fibroblasts (64), infection of NT2 cells did not result in the accumulation of NF-κB in the nucleus (Fig. 8C) but also did not prevent the nuclear accumulation of NF-κB in the presence of LMB (Fig. 8D). Importantly, tegument-delivered pp71 remained in the cytoplasm of infected NT2 cells treated with LMB (Fig. 8D). Control experiments that detected Oct4 indicated that LMB treatment did not induce cellular differentiation either in the absence (Fig. 8E) or in the presence (Fig. 8F) of HCMV. In total, we conclude that differentiated cells express one or more positively acting factors that act in a dominant fashion to facilitate the import of tegument-delivered pp71 into the nucleus (Fig. 9) and thus also to activate the productive lytic replication program of HCMV.
FIG. 8.

Tegument-delivered pp71 remains cytoplasmic in NT2 cells treated with leptomycin B (LMB). NT2 cells were either untreated (A and C) or pretreated (B, D, E, and F) for 1 h with LMB and then mock infected (A, B, and E) or infected with HCMV (C, D, and F) at an MOI of 2. pp71 and either NF-κB (A, B, C, and D) or Oct4 (E and F) were visualized by indirect immunofluorescence. Nuclei were counterstained with Hoechst dye.
FIG. 9.

Model for nuclear localization of tegument-delivered pp71. Tegument-delivered pp71 could be actively blocked from entering the nuclei of undifferentiated cells (A), efficiently exported from the nuclei of undifferentiated cells (B), or driven into the nuclei of differentiated cells (C) by a positively acting, dominant factor (X). Data presented here support the model depicted in panel C.
DISCUSSION
Nuclear pp71 degrades Daxx and inactivates the cellular intrinsic immune defense that silences viral IE gene expression through the actions of histone deacetylases (44, 58). Absolute correlations exist between the differentiation status of cells, the subcellular localization of tegument-delivered pp71, and the outcome of the infectious event (45-47). Incompletely differentiated cells infected with HCMV display tegument-delivered pp71 in the cytoplasm, fail to express viral IE genes, and establish either a quiescent or a latent infection. Fully differentiated cells infected with HCMV display tegument-delivered pp71 in the nucleus, express viral IE genes, and complete a productive, lytic replication program. These relationships extend beyond correlative to causative. Forced nuclear expression of pp71, HDAC inhibition, or Daxx knockdown permits IE gene expression in undifferentiated cells infected with the AD169 laboratory-adapted strain of HCMV (45, 47). Interestingly, clinical viral strains appear to encode an additional HDAC-independent, trans-acting dominant impediment to IE gene expression not related to pp71 or Daxx (47). Thus, while the functionality of the pp71-induced inactivation of the Daxx-mediated intrinsic immune defense controls, in part, the outcome of an infection (lytic versus latent), other mechanisms also appear to regulate this important decision point (14, 47).
As pp71 subcellular localization clearly plays a role in determining whether lytic infection is initiated or latency is established, it is important to identify viral and/or cellular determinants that regulate it. Unfortunately, little is known about how pp71 subcellular localization is controlled. Mutation of the threonine residue at amino acid position 223 of pp71 to an alanine prevents the accumulation of de novo-expressed (through transient transfection) pp71 within the nucleus (49). These data indicate that the phosphorylation state of pp71 may be one mechanism through which pp71 subcellular localization is controlled. However, it is not known if phosphorylation affects the subcellular localization of pp71 during a viral infection. Furthermore, our unpublished preliminary experiments with multiple small-molecule kinase or phosphatase inhibitors have failed to reveal any differences in tegument-delivered pp71 subcellular localization in undifferentiated and differentiated cells treated with these drugs. As pp71 is not the only tegument-delivered protein whose subcellular localization differs between fully and incompletely differentiated cells (Fig. 1), it is possible that pp71 itself may not be the direct target of the mechanism that restricts the trafficking of tegument-delivered proteins to the nuclei of undifferentiated cells. Therefore, we plan on undertaking an unbiased approach to identify cellular factors that control pp71 subcellular localization.
Here we show that the inhibition of Crm-1-dependent nuclear export with LMB does not allow tegument-delivered pp71 to accumulate in the nuclei of HCMV-infected NT2 cells (Fig. 8). While it is possible that tegument-delivered pp71 enters the nucleus but is rapidly exported in a Crm-1-independent manner in undifferentiated cells, we favor a model in which the import of tegument-delivered pp71 into the nuclei of undifferentiated cells is defective (Fig. 9). We reasoned that undifferentiated cells might express a factor that blocks the import of tegument-delivered pp71 into the nucleus. If this were true, then the fusion of undifferentiated, HCMV-infected NT2 cells with fully differentiated NHDFs would result in syncytia where tegument-delivered pp71 was still actively trapped in the cytoplasm. However, we clearly observed tegument-delivered pp71 within the nucleus under these experimental conditions (Fig. 3A and 5D). Thus, we conclude that differentiated cells express a dominantly acting factor that facilitates the entry of tegument-delivered pp71 into the nucleus, although we cannot exclude the possibility that syncytium formation substantially dilutes the concentration of an undifferentiated cellular factor that traps tegument-delivered pp71 in the cytoplasm and thus allows nuclear entry.
The type of fusion experiments that we employed are typically used as the very first step in identifying cellular proteins differentially expressed in various cell types that modulate some viral process or phenotype (9, 15, 16, 34, 39, 50, 60). The subsequent approaches that others have used to eventually identify the desired cellular factor include screens for differential expression, with subsequent candidate validation or direct selection of cDNAs that convert ectopically expressing cells from one phenotype to the other (20, 40, 48, 56). We plan to use both techniques to identify a fibroblast-expressed protein that, when ectopically expressed in an undifferentiated cell, allows tegument-delivered pp71 to enter the nucleus. Additional future work might then develop small molecules directed against this protein with which we could modulate the subcellular localization of pp71 to impact the outcome of an HCMV infection at the cellular level. For example, treatments that prevent tegument-delivered pp71 from entering the nucleus might be valuable inhibitors of HCMV lytic replication, which could potentially be used in combination with the current lytic inhibitor of choice for HCMV, ganciclovir. Alternatively, treatments that drive pp71 to the nuclei of undifferentiated cells may prevent the virus from establishing a latent infection. Over time, such a drug could reduce or eliminate the latent reservoir of virus and perhaps help the immune system to completely clear the infection.
Acknowledgments
R.R.P. was supported by NIH training grant T32 GM07215-33. R.F.K. is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease. This work was supported by a grant from the NIH (AI074984) to R.F.K.
We thank our lab manager, Phil Balandyk, for expert technical assistance, Bill Sugden for the use of his microscope, the members of our laboratory for their useful comments, Tom Shenk and Trish Robinson (Princeton University) for the pp65 antibody, Roy Duncan (Dalhousie University) for the p14 expression plasmid, and Jim Smiley (University of Alberta), Karen Mossman (McMaster University), and the members of their laboratories for helpful discussions.
Footnotes
Published ahead of print on 4 August 2010.
REFERENCES
- 1.Ahn, J. H., and G. S. Hayward. 2000. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 274:39-55. [DOI] [PubMed] [Google Scholar]
- 2.Ahn, J. H., and G. S. Hayward. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 71:4599-4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldick, C. J., Jr., A. Marchini, C. E. Patterson, and T. Shenk. 1997. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J. Virol. 71:4400-4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernardi, R., and P. P. Pandolfi. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8:1006-1016. [DOI] [PubMed] [Google Scholar]
- 5.Bresnahan, W. A., and T. E. Shenk. 2000. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. U. S. A. 97:14506-14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cantrell, S. R., and W. A. Bresnahan. 2006. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 80:6188-6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantrell, S. R., and W. A. Bresnahan. 2005. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J. Virol. 79:7792-7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corcoran, J. A., J. Salsman, R. de Antueno, A. Touhami, M. H. Jericho, E. K. Clancy, and R. Duncan. 2006. The p14 fusion-associated small transmembrane (FAST) protein effects membrane fusion from a subset of membrane microdomains. J. Biol. Chem. 281:31778-31789. [DOI] [PubMed] [Google Scholar]
- 9.Cowan, S., T. Hatziioannou, T. Cunningham, M. A. Muesing, H. G. Gottlinger, and P. D. Bieniasz. 2002. Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc. Natl. Acad. Sci. U. S. A. 99:11914-11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuevas-Bennett, C., and T. Shenk. 2008. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J. Virol. 82:9525-9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deb-Rinker, P., D. Ly, A. Jezierski, M. Sikorska, and P. R. Walker. 2005. Sequential DNA methylation of the Nanog and Oct-4 upstream regions in human NT2 cells during neuronal differentiation. J. Biol. Chem. 280:6257-6260. [DOI] [PubMed] [Google Scholar]
- 12.Dosa, R., K. Burian, and E. Gonczol. 2005. Human cytomegalovirus latency is associated with the state of differentiation of the host cells: an in vitro model in teratocarcinoma cells. Acta Microbiol. Immunol. Hung. 52:397-406. [DOI] [PubMed] [Google Scholar]
- 13.Gonczol, E., P. W. Andrews, and S. A. Plotkin. 1985. Cytomegalovirus infection of human teratocarcinoma cells in culture. J. Gen. Virol. 66:509-515. [DOI] [PubMed] [Google Scholar]
- 14.Groves, I. J., and J. H. Sinclair. 2007. Knockdown of hDaxx in normally non-permissive undifferentiated cells does not permit human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 88:2935-2940. [DOI] [PubMed] [Google Scholar]
- 15.Hancock, M. H., J. A. Corcoran, and J. R. Smiley. 2006. Herpes simplex virus regulatory proteins VP16 and ICP0 counteract an innate intranuclear barrier to viral gene expression. Virology 352:237-252. [DOI] [PubMed] [Google Scholar]
- 16.Hancock, M. H., K. L. Mossman, and J. R. Smiley. 2009. Cell fusion-induced activation of interferon-stimulated genes is not required for restriction of a herpes simplex virus VP16/ICP0 mutant in heterokarya formed between permissive and restrictive cells. J. Virol. 83:8976-8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hensel, G. M., H. H. Meyer, I. Buchmann, D. Pommerehne, S. Schmolke, B. Plachter, K. Radsak, and H. F. Kern. 1996. Intracellular localization and expression of the human cytomegalovirus matrix phosphoprotein pp71 (ppUL82): evidence for its translocation into the nucleus. J. Gen. Virol. 77:3087-3097. [DOI] [PubMed] [Google Scholar]
- 18.Hofmann, H., H. Sindre, and T. Stamminger. 2002. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 76:5769-5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang, T. T., N. Kudo, M. Yoshida, and S. Miyamoto. 2000. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc. Natl. Acad. Sci. U. S. A. 97:1014-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hummel, J., J. Shoots, D. Cummings, D. Ilieva, and K. Mossman. 2009. TMX2 blocks the replication of HSV-1 lacing ICP0, abstr. 1.06. 34th International Herpesvirus Workshop, Ithaca, NY.
- 21.Hwang, J., and R. F. Kalejta. 2009. Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J. Virol. 83:6591-6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang, J., and R. F. Kalejta. 2007. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367:334-338. [DOI] [PubMed] [Google Scholar]
- 23.Ishov, A. M., R. M. Stenberg, and G. G. Maul. 1997. Human cytomegalovirus immediate early interaction with host nuclear structures: definition of an immediate transcript environment. J. Cell Biol. 138:5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarvis, M. A., and J. A. Nelson. 2002. Mechanisms of human cytomegalovirus persistence and latency. Front. Biosci. 7:d1575-d1582. [DOI] [PubMed] [Google Scholar]
- 25.Johnson, C., D. Van Antwerp, and T. J. Hope. 1999. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J. 18:6682-6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalejta, R. F. 2008. Functions of human cytomegalovirus tegument proteins prior to immediate early gene expression. Curr. Top. Microbiol. Immunol. 325:101-115. [DOI] [PubMed] [Google Scholar]
- 27.Kalejta, R. F. 2008. Tegument proteins of human cytomegalovirus. Microbiol. Mol. Biol. Rev. 72:249-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalejta, R. F., J. T. Bechtel, and T. Shenk. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23:1885-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korioth, F., G. G. Maul, B. Plachter, T. Stamminger, and J. Frey. 1996. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp. Cell Res. 229:155-158. [DOI] [PubMed] [Google Scholar]
- 30.Lee, C. H., G. C. Lee, Y. J. Chan, C. J. Chiou, J. H. Ahn, and G. S. Hayward. 1999. Factors affecting human cytomegalovirus gene expression in human monocyte cell lines. Mol. Cells 9:37-44. [PubMed] [Google Scholar]
- 31.Lindsay, C. R., V. M. Morozov, and A. M. Ishov. 2008. PML NBs (ND10) and Daxx: from nuclear structure to protein function. Front. Biosci. 13:7132-7142. [DOI] [PubMed] [Google Scholar]
- 32.Liu, B., and M. F. Stinski. 1992. Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis-acting elements. J. Virol. 66:4434-4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukashchuk, V., S. McFarlane, R. D. Everett, and C. M. Preston. 2008. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 82:12543-12554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madani, N., and D. Kabat. 1998. An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. J. Virol. 72:10251-10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marshall, K. R., K. V. Rowley, A. Rinaldi, I. P. Nicholson, A. M. Ishov, G. G. Maul, and C. M. Preston. 2002. Activity and intracellular localization of the human cytomegalovirus protein pp71. J. Gen. Virol. 83:1601-1612. [DOI] [PubMed] [Google Scholar]
- 36.Meier, J. L. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 75:1581-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michaelis, M., H. W. Doerr, and J. Cinatl, Jr. 2009. Oncomodulation by human cytomegalovirus: evidence becomes stronger. Med. Microbiol. Immunol. 198:79-81. [DOI] [PubMed] [Google Scholar]
- 38.Mocarski, E., T. Shenk, and R. Pass. 2007. Cytomegaloviruses, p. 2701-2772. In D. M. Knipe et al. (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 39.Munk, C., S. M. Brandt, G. Lucero, and N. R. Landau. 2002. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc. Natl. Acad. Sci. U. S. A. 99:13843-13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neil, S. J., T. Zang, and P. D. Bieniasz. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425-430. [DOI] [PubMed] [Google Scholar]
- 41.Nitzsche, A., C. Paulus, and M. Nevels. 2008. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 82:11167-11180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nowak, B., C. Sullivan, P. Sarnow, R. Thomas, F. Bricout, J. C. Nicolas, B. Fleckenstein, and A. J. Levine. 1984. Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 132:325-338. [DOI] [PubMed] [Google Scholar]
- 43.Preston, C. M., and M. J. Nicholl. 2006. Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 87:1113-1121. [DOI] [PubMed] [Google Scholar]
- 44.Saffert, R., and R. Kalejta. 2008. Promyelocytic leukemia-nuclear body proteins: herpesvirus enemies, accomplices, or both? Fut. Virol. 3:265-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saffert, R. T., and R. F. Kalejta. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 81:9109-9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saffert, R. T., and R. F. Kalejta. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863-3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saffert, R. T., R. R. Penkert, and R. F. Kalejta. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J. Virol. 84:5594-5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646-650. [DOI] [PubMed] [Google Scholar]
- 49.Shen, W., E. Westgard, L. Huang, M. D. Ward, J. L. Osborn, N. H. Chau, L. Collins, B. Marcum, M. A. Koach, J. Bibbs, O. J. Semmes, and J. A. Kerry. 2008. Nuclear trafficking of the human cytomegalovirus pp71 (ppUL82) tegument protein. Virology 376:42-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simon, J. H., N. C. Gaddis, R. A. Fouchier, and M. H. Malim. 1998. Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat. Med. 4:1397-1400. [DOI] [PubMed] [Google Scholar]
- 51.Sinclair, J. 2008. Human cytomegalovirus: latency and reactivation in the myeloid lineage. J. Clin. Virol. 41:180-185. [DOI] [PubMed] [Google Scholar]
- 52.Sinclair, J., and P. Sissons. 2006. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 87:1763-1779. [DOI] [PubMed] [Google Scholar]
- 53.Soderberg-Naucler, C. 2008. HCMV microinfections in inflammatory diseases and cancer. J. Clin. Virol. 41:218-223. [DOI] [PubMed] [Google Scholar]
- 54.Streblow, D. N., J. Dumortier, A. V. Moses, S. L. Orloff, and J. A. Nelson. 2008. Mechanisms of cytomegalovirus-accelerated vascular disease: induction of paracrine factors that promote angiogenesis and wound healing. Curr. Top. Microbiol. Immunol. 325:397-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Streblow, D. N., and J. A. Nelson. 2003. Models of HCMV latency and reactivation. Trends Microbiol. 11:293-295. [DOI] [PubMed] [Google Scholar]
- 56.Stremlau, M., C. M. Owens, M. J. Perron, M. Kiessling, P. Autissier, and J. Sodroski. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848-853. [DOI] [PubMed] [Google Scholar]
- 57.Tavalai, N., M. Kraiger, N. Kaiser, and T. Stamminger. 2008. Insertion of an EYFP-pp71 (UL82) coding sequence into the human cytomegalovirus genome results in a recombinant virus with enhanced viral growth. J. Virol. 82:10543-10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tavalai, N., and T. Stamminger. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta 1783:2207-2221. [DOI] [PubMed] [Google Scholar]
- 59.Turtinen, L. W., and B. J. Seufzer. 1994. Selective permissiveness of TPA differentiated THP-1 myelomonocytic cells for human cytomegalovirus strains AD169 and Towne. Microb. Pathog. 16:373-378. [DOI] [PubMed] [Google Scholar]
- 60.Varthakavi, V., R. M. Smith, S. P. Bour, K. Strebel, and P. Spearman. 2003. Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc. Natl. Acad. Sci. U. S. A. 100:15154-15159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weinshenker, B. G., S. Wilton, and G. P. Rice. 1988. Phorbol ester-induced differentiation permits productive human cytomegalovirus infection in a monocytic cell line. J. Immunol. 140:1625-1631. [PubMed] [Google Scholar]
- 62.Wilkinson, G. W., C. Kelly, J. H. Sinclair, and C. Rickards. 1998. Disruption of PML-associated nuclear bodies mediated by the human cytomegalovirus major immediate early gene product. J. Gen. Virol. 79:1233-1245. [DOI] [PubMed] [Google Scholar]
- 63.Woodhall, D. L., I. J. Groves, M. B. Reeves, G. Wilkinson, and J. H. Sinclair. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 281:37652-37660. [DOI] [PubMed] [Google Scholar]
- 64.Yurochko, A. D., and E. S. Huang. 1999. Human cytomegalovirus binding to human monocytes induces immunoregulatory gene expression. J. Immunol. 162:4806-4816. [PubMed] [Google Scholar]
- 65.Zhu, H., Y. Shen, and T. Shenk. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69:7960-7970. [DOI] [PMC free article] [PubMed] [Google Scholar]