

Abstract
All chemokines share a common structural scaffold that mediate a remarkable variety of functions from immune surveillance to organogenesis. Chemokines are classified as CXC or CC on the basis of conserved cysteines, and the two subclasses bind distinct sets of GPCR class of receptors and also have markedly different quaternary structures, suggesting that the CXC/CC motif plays a prominent role in both structure and function. For both classes, receptor activation involves interactions between chemokine N-loop and receptor N-domain residues (Site-I), and between chemokine N-terminal and receptor extracellular/transmembrane residues (Site-II). We engineered a CC variant (labeled as CC-CXCL8) of the chemokine CXCL8 by deleting residue X (CXC → CC), and found its structure is essentially similar to WT. In stark contrast, CC-CXCL8 bound poorly to its cognate receptors CXCR1 and CXCR2 (Ki > 1 μm). Further, CC-CXCL8 failed to mobilize Ca2+ in CXCR2-expressing HL-60 cells or recruit neutrophils in a mouse lung model. However, most interestingly, CC-CXCL8 mobilizes Ca2+ in neutrophils and in CXCR1-expressing HL-60 cells. Compared with the WT, CC-CXCL8 binds CXCR1 N-domain with only ∼5-fold lower affinity indicating that the weak binding to intact CXCR1 must be due to its weak binding at Site-II. Nevertheless, this level of binding is sufficient for receptor activation indicating that affinity and activity are separable functions. We propose that the CXC motif functions as a conformational switch that couples Site-I and Site-II interactions for both receptors, and that this coupling is critical for high affinity binding but differentially regulates activation.
Keywords: Chemokines, G Protein-coupled Receptors (GPCR), Neutrophil, NMR, Signal Transduction, Site-directed Mutagenesis, Structure-Function
Introduction
Chemokines mediate a wide variety of biological functions from recruiting leukocytes to the site of injury and infection to organ development, wound healing, and angiogenesis (1–4). Chemokines are characterized by four conserved cysteines that form disulfide bonds, and are classified into CXC and CC families based on cysteine pattern near the N terminus. CXC ligands bind and activate only CXC receptors and CC ligands bind and activate only CC receptors, indicating functional divergence could be as old as the molecules themselves. Structure-function studies consistently show that binding and receptor activation for both classes involves two interactions: between the ligand N-loop and receptor N-domain residues (Site-I) and between the ligand N-terminal and receptor extracellular/transmembrane residues (Site-II) (5–9).
Structures of several CXC and CC chemokines have been solved by NMR and x-ray crystallography, and reveal a common structural fold (known as chemokine fold) at the monomer level (10–15). The CXC/CC motif connects the functionally important N-loop and N-terminal residues, and also plays a structural role by forming disulfide bonds that tether the N-terminal and N-loop residues to the protein core (Fig. 1). In the CXC chemokine CXCL8 (also known as interleukin-8, IL-8), the residue corresponding to X (Gln) in the CXC motif has been mutated with no effect on receptor function, whereas mutating the cysteines result in complete loss of function (6, 16). Disulfides play a functional role in the folded CXCL8, as modifying disulfides by introducing non-natural cysteine homologs such as homocysteine retains native like structure but result in significant loss of function (17). These observations collectively indicate that disulfides are essential for function, but the molecular basis of the CXC motif and how disulfides influence the activity of CXC chemokines remains unknown.
FIGURE 1.
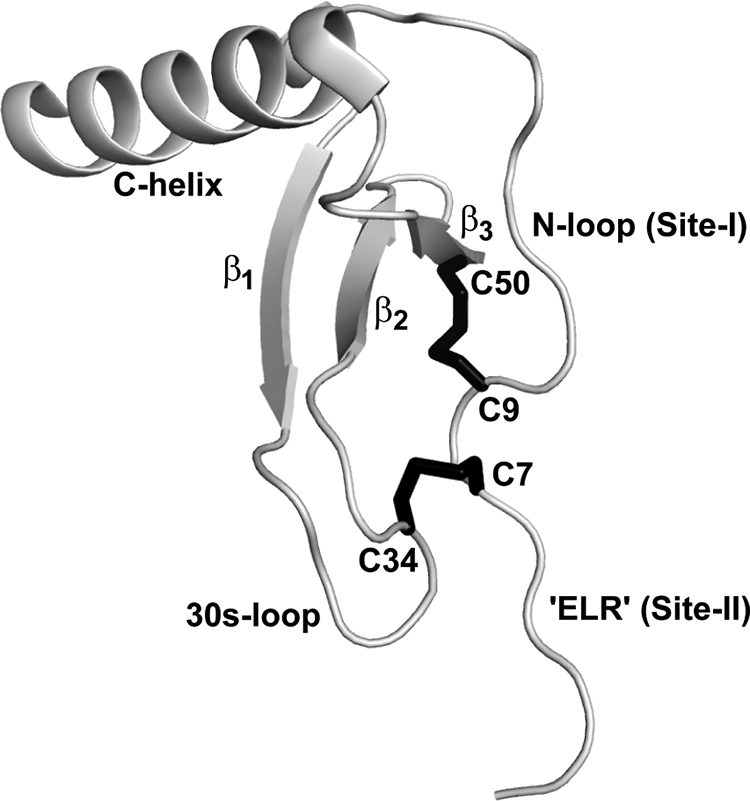
Molecular plot of CXCL8 monomer. The CXC motif and the secondary structural regions are highlighted. Disulfide bonds are shown in black. Molecular plot was generated using Pymol (34).
A characteristic property of chemokines is their propensity to form dimers. Structures reveal that the CXC and CC chemokines dimerize using different regions of the protein. CXC chemokines form globular dimers through residues from the 1st β-strand and the α-helix, whereas CC chemokines form extended dimers through residues from the N-loop. Trapped non-dissociating CC dimers have been shown incapable of binding their receptors as dimerization blocks the N-loop binding site, whereas trapped CXC chemokine dimers can bind their receptors and elicit function (18–21). Chemokine function also involves binding to soluble and cell surface glycosaminoglycans (GAG),2 and structure-function studies have shown that dimerization and GAG binding are coupled (22–24). These observations suggest that the CXC/CC motif influences the mode of dimerization, and so binding to GAG and its receptors.
In this report, we have studied the role of the CXC motif for structure and function by creating CC versions of CXCL8 (termed as CC-CXCL8) by deleting the intervening residue X (Gln) of the CXC motif. We found that the tertiary and quaternary structures of CC-CXCL8 are similar to WT, except for local structural perturbation around the site of deletion. The CC-variant shows only very marginal binding affinity to CXCR1 and CXCR2 (Ki >1 μm versus ∼1 nm for WT) but most interestingly, selectively activates CXCR1 and not CXCR2. It is inactive for CXCR2 as measured from in vitro intracellular Ca2+ release and in vivo neutrophil recruitment, and is active for CXCR1 in the Ca2+ release assay. Compared with WT, CC-CXCL8 binds CXCR1 N-domain with only 5-fold lower affinity, indicating that the low affinity to the intact CXCR1 must be due to its weak binding at Site-II. Weak receptor binding is nevertheless sufficient for activating CXCR1 indicating that the structural basis and molecular mechanisms underlying receptor binding and receptor activation are separable functions, and that binding affinity cannot be simply correlated to function. We propose that the CXC motif is an integral component of the structural scaffold, and functions as a conformational switch mediating coupling interactions between N-loop (Site-I) and N-terminal residues (Site-II); these coupling interactions are critical for high affinity binding to both receptors but play differential roles for activation and downstream signaling events.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of CC-CXCL8
CC-CXCL8 variants were created by deleting the intervening glutamine residue (bold and underlined) in the CXC motif (in gray) in the CXCL8 sequence-SAKELRCQCIKTYSK∼. Two versions of CC-CXCL8, the full length CC-CXCL8 (residues 1–72) and a truncated monomeric form (residues 1–66), were generated. We have shown previously that the CXCL8(1–66) deletion mutant is a monomer, and that the structure and activity of this CXCL8(1–66) deletion mutant is similar to that of the trapped full-length monomer (25, 26). The full length CC-CXCL8 was expressed and purified as discussed previously (27). In the case of the CC-CXCL8(1–66) monomer construct, the fusion protein was trapped in inclusion bodies, so the cell pellet was first solubilized with lysis buffer containing 4.5 m GdnHCl with constant stirring at room temperature for 1 h followed by centrifugation at 15,000 rpm. The supernatant containing the solubilized protein was loaded on to the Ni-NTA column, and washed successively with the buffer containing 2, 1, and 0 m GdnHCl to allow on-column refolding. The refolded protein was eluted, cleaved, and purified as described previously (27). 15N-labeled proteins were produced essentially as described, except that cells were grown in minimal medium containing 15NH4Cl as the sole nitrogen source.
Receptor Binding Assays
HL-60 cells expressing CXCR1 or CXCR2 were suspended at a concentration of ∼107 cells/ml in phosphate-buffered saline (PBS) containing 0.1% (w/v) bovine serum albumin and 20 mm HEPES (pH 7.4) and incubated at 4 °C for 4–6 h in the presence of 0.4 nm 125I-CXCL8 and increasing concentrations of unlabeled CXCL8 or CC-CXCL8. The radioactivity in the pellet was measured in a γ-counter, and the Ki was calculated as described previously (28). Binding affinities are from at least two independent experiments, and each experiment was performed at least two times.
Intracellular Ca2+ Mobilization
Neutrophils were suspended in phosphate-buffered saline (PBS) at a density of ∼106 cells/ml and loaded with 5 μm Indo-1/AM for 30 min at 37 °C in the dark. Neutrophils were subsequently washed with ice-cold PBS and then resuspended at the same cell density for Ca2+ measurements as described (28). HL-60 cells expressing CXCR1 or CXCR2 were suspended in PBS buffer at a density of ∼107 cells/ml, loaded with Indo-1/AM for 30 min at 37 °C in the dark, and were subsequently washed with ice-cold buffer and resuspended at the same cell density in a buffer containing 1 mm probenecid. Neutrophils and HL-60 cells were placed in a continuously stirred cuvette maintained at 37°C in a RF5301PC spectrofluorometer (Shimadzu) and stimulated with monomeric CXCL8 or monomeric CC-CXCL8. Fluorescence intensity was measured using an excitation wavelength of 330 nm and an emission wavelength of 405 nm. Ca2+ release in human neutrophils was also measured in the presence of 1 μm CXCR2 inhibitor SB225002 (29).
Neutrophil Recruitment in a Mouse Model
8 to 10-week-old female BALB/c mice were purchased from Harlan (Houston, TX) and housed under pathogen-free conditions in the animal research facility, in accordance with the National Institutes of Health and University guidelines for animal care. Under light anesthesia, mice were inoculated intranasally with 10 μg of WT CXCL8 and CC-CXCL8 variant in Dulbecco's phosphate-buffered saline (D-PBS). Mice were sacrificed, and neutrophil levels in the broncheo-alveolar lavage fluid (BALF) were determined as described previously (30).
Ultracentrifugation Measurements
Sedimentation equilibrium experiments were performed using a Beckman-Coulter model XLA analytical ultracentrifuge (Beckman-Coulter Inc., Fullerton, CA) with An-60 titanium rotors using absorbance optics at 280 nm. Experiments were carried using two different loading concentrations of 0.3 and 1.0 mg/ml at rotor speeds 25,000, 30,000, and 42,000 rpm. Sedimentation data were fitted to the monomer-dimer equilibrium using the Microcal Origin 4.1 software provided by the manufacturer.
NMR Structural Characterization
All NMR spectra were acquired at 30 °C or 40 °C using Varian Unity Plus 600 or INOVA 800 MHz spectrometers equipped with field gradient accessories. The chemical shifts were assigned from 2D-NOESY, 2D-TOCSY, 1H-15N HSQC, and 15N-edited NOESY experiments using standard pulse sequences (31, 32). The mixing time for the NOESY and TOCSY experiments were 150 and 80 ms, respectively. The protein concentrations were ∼0.5 mm in 50 mm sodium acetate buffer containing 1 mm sodium azide, 1 mm sodium 2,2-dimethyl-2-silapentane sulfonate (DSS), pH 6.0 in 90% H2O/10% H2O (v/v). All spectra were processed and analyzed as described previously (33).
Molecular Dynamics Simulations
The CXCL8(1–66) molecular model was generated using the CXCL8 monomer structure (PDB ID: 1IKM) (26). The N-methyl was replaced with NH for Leu-25, the last 6 amino acids (67–72) were deleted, and N-terminal residues Ser-1, Ala-2, and Lys-3 were added using Pymol (34). The CC-CXCL8(1–66) model was generated by deleting Gln-8, residue corresponding to X of the CXC motif. MD simulations in explicit solvent was carried out using Amber 9 for 13 ns for both CXCL8(1–66) and CC-CXCL8(1–66) variants as described previously (35, 36).
CC-CXCL8 Binding to CXCR1 N-domain
The binding of CC-CXCL8 variants to the CXCR1 N-domain was characterized by NMR spectroscopy. Unlabeled CXCR1-N-domain peptide prepared in the same buffer was titrated in aliquots into 15N-labeled CC-CXCL8, and binding was followed by changes in two-dimensional 15N-1H HSQC spectral characteristics (33). The final peptide:protein molar ratios for CC-CXCL8 and CC-CXCL8(1–66) were 11.4 and 8.3, respectively. Apparent binding constants were determined by fitting the binding-induced chemical shift changes as a function of peptide:protein molar ratios as described previously (33).
RESULTS
Receptor Binding Affinities
We measured the binding affinities of CC-CXCL8 and CXCL8 monomers to the CXCR1 and CXCR2 receptors individually expressed in HL-60 cells (Fig. 2). WT CXCL8 bound both CXCR1 and CXCR2 receptors with Ki of ∼1 nm, whereas CC-CXCL8, even at μm concentrations, could only partially displace 125I-CXCL8 bound to CXCR1 and could not displace any of 125I-CXCL8 bound to CXCR2 (Ki > 1 μm). These data indicate that the CXC motif is absolutely essential for high affinity binding to both CXCL8 receptors.
FIGURE 2.
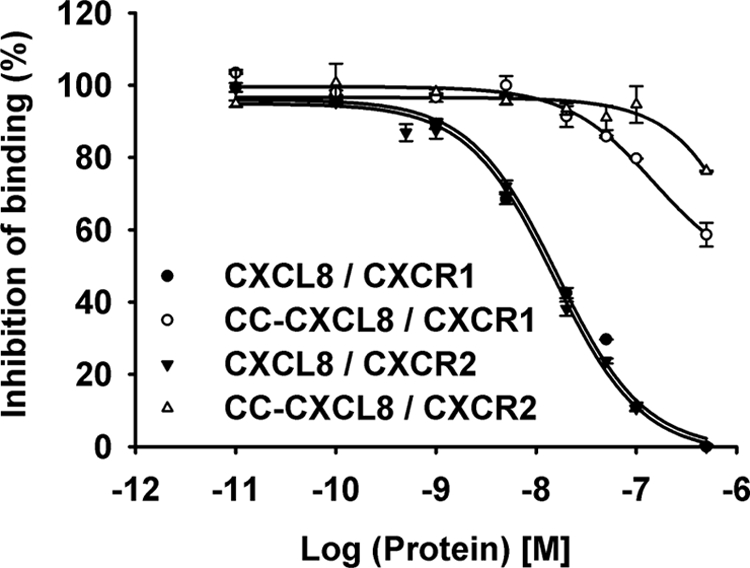
Binding affinity of CC-CXCL8 for CXCR1 and CXCR2 receptors. Binding affinities were calculated by measuring the inhibition of binding of 125I-CXCL8 to CXCR1 (○, ●) or CXCR2 (▵, ▾) receptors individually expressed in HL-60 cells by CC-CXCL8 (open symbols) and CXCL8 (closed symbols).
Receptor Activation
We studied the activities of CC-CXCL8 and CXCL8 monomers by measuring intracellular Ca2+ release in neutrophils and in HL60 cells expressing either CXCR1 or CXCR2. CC-CXCL8 could trigger robust Ca2+ response in neutrophils at 100 nm and higher concentrations (Fig. 3A). As neutrophils co-express both CXCR1 and CXCR2, activation could be either due to CXCR1, CXCR2, or both. To address this question, Ca2+ release was measured in the presence of CXCR2 inhibitor SB225002. CXCL8 is active indicating signaling via CXCR1, CXCL1 is not active as it signals only via CXCR2, and CC-CXCL8 is active indicating it must be due to signaling via CXCR1 (Fig. 3, panels B–G).
FIGURE 3.

Activity of CC-CXCL8 in neutrophils. Panel A, dose-response of intracellular Ca2+ release in neutrophils. Human neutrophils loaded with Indo-1 were stimulated with different CC-CXCL8 concentrations (10–1000 nm) and the intracellular Ca2+ release was measured by fluorescence measurements. Panels B–G, effect of CXCR2 inhibitor SB225002. Human neutrophils loaded with Indo-1 were tested for Ca2+ release in the absence and presence of SB225002 for 100 nm CXCL8 (panels B and C), 100 nm CXCL1 (panels D and E), and 200 nm CC-CXCL8 (panels F and G).
Results from HL-60 cells individually expressing either CXCR1 or CXCR2 receptors confirm that CC-CXCL8 activates CXCR1 but not CXCR2 receptors (Fig. 4). However, its activity (EC50 ∼300 nm) is not as robust as observed for CXCL8 (EC50 2 nm) indicating that it is a weak CXCR1 agonist. CC-CXCL8 failed to elicit any response for CXCR2 even at 1 μm concentration. Unlike humans, mouse neutrophils express only CXCR2, and CC-CXCL8 failed to recruit any neutrophils in the mouse lung model (Fig. 4D). CC-CXCL8 also was completely inactive in a mouse peritoneum model (data not shown). These data collectively indicate that the CXC motif is absolutely essential for activation of CXCR2 but not of CXCR1 receptor.
FIGURE 4.

Activity of CC-CXCL8 in HL-60 cells individually expressing CXCR1 and CXCR2. Panel A, HL-60 cells expressing CXCR1 loaded with Indo-1 were stimulated with CXCL8 or CC-CXCL8 and the Ca2+ release was monitored by fluorescence spectroscopy. Panels B and C, HL-60 cells expressing CXCR2 were loaded with Indo-1 and stimulated with 100 nm CXCL8 or 200 nm CC-CXCL8. Panel D, neutrophil recruitment in a mouse lung model. BAL samples from mice treated with CC-CXCL8, WT CXCL8, or PBS control were collected 6 h post-inoculation. Each data set represents average over 4–6 animals/group.
Structural Characterization of CC-CXCL8
The structural consequence of converting the CXC to CC motif in CC-CXCL8 dimer and monomer was characterized by solution NMR spectroscopy. NMR data showed that structural scaffolds of both CC-CXCL8 dimer and monomer are essentially identical to that of CXCL8. NMR structures are calculated largely on the basis of nuclear Overhauser effect (NOE) data, which provide information of the proton-proton distances in the context of the folded structure. Compared with the WT dimer, essentially the same set of intramolecular and intermolecular NOEs were observed in the CC-CXCL8 dimer. Regions of the CC-CXCL8 NOESY spectrum showing both intramolecular and intermolecular NOEs are shown in Fig. 5, panels A–D.
FIGURE 5.

NMR structural characteristics of CC-CXCL8 dimer. Panel A, intramolecular NOEs from Phe-17 NH to Trp-57 side chain protons, and (panels B and C) intermolecular NOEs across the dimer interface from Val-27 to Leu-25′ and Glu-24′ and from Phe-65 to Val-27′ and Glu-29′ are shown (residues across the dimer interface are highlighted by the sign ‘). Panel D shows the proximity of these residues in the WT CXCL8 structure (PDB ID: 1IL8); tertiary interactions within the monomer unit are shown in the top panel, and the quaternary interactions across the dimer interface are shown in the bottom panel. Panel E, 1H-15N HSQC spectrum of the CC-CXCL8 dimer. The chemical shifts are similar to those observed for the WT CXCL8. Characteristic upfield- and downfield-shifted peaks are labeled. Diagonal plot of HN (panel F) and Hα (panel G) chemical shifts of CC-CXCL8 (y-axis) and WT CXCL8 (x-axis).
Chemical shifts are sensitive to secondary, tertiary, and quaternary structure, and are especially useful for less structured regions of the protein and also for regions for which there is a paucity of NOEs. We observe that the shifts of CC-CXCL8 and WT CXCL8 dimers are essentially identical, and small differences if any are confined to regions around the site of deletion, indicating that the structure of the CC-CXCL8 variant is similar to WT CXCL8 (Fig. 5, panels E–G).
Molecular Dynamics Simulations
Because NMR structures are calculated based on NOE and dihedral restraints, it represents only a snapshot of multiple conformations that a structure can adopt, and so does not provide insights into the relative motions between structural modules that are dynamic in nature. In silico molecular dynamics (MD) simulation studies are a powerful tool that can complement NMR structural data by capturing real time motions, thereby providing insights into dynamics of such systems. We performed a 13-ns MD simulation on both CXCL8 and CC-CXCL8 monomers, in order to understand the consequence of deleting Gln8 (residue X; CXC → CC) on the crosstalk between different structural modules that could influence Site-II binding and receptor activation.
Analysis of the MD trajectory showed that deletion of Gln 8 does not disrupt the tertiary scaffold of the protein (Fig. 6A). The RMSD for the backbone of the tertiary scaffold is only ∼0.6 Å between the CC-CXCL8 and CXCL8 monomer structures. On the other hand, the deletion does introduce local conformational rearrangement around both the disulfide bridges. The conformation of the Cys9-Cys50 disulfide and the Cα-Cα distance (6.6 ± 0.1 Å) over the course of the MD trajectory is similar to that observed in WT (Fig. 6B). In contrast, larger conformational rearrangements are observed around the Cys7-Cys34 disulfide bridge. The N terminus is pulled closer to the 30s loop which also undergoes an inward bend to accommodate this change. This rearrangement introduces constraint on the relative orientation and flexibility of the 30s loop and the N-terminal residues (Fig. 6A). Further, Cys7-Cys34 disulfide is shown to adopt a more compact conformation over the course of the simulation (Cα-Cα distance = 5.5 ± 0.3 Å versus 6.2 ± 0.2 Å in WT) that could restrict the N terminus from efficient Site-II binding (Fig. 6B).
FIGURE 6.

MD simulations of CC-CXCL8. Panel A, average structures of the most populated clusters of CC-CXCL8(1–66) and CXCL8(1–66) over a 13 ns MD simulation. Structures are juxtaposed and not superimposed for clarity. Note the difference in the relative orientation of N terminus with respect to the 30s loop and the rearrangement in the Cys7-Cys34 disulfide bridge conformation. Panel B, plots of Cys7-Cys34 and Cys9-Cys50 Cα-Cα distances of 700 structures from the above clusters of CXCL8(1–66) (black) and CC-CXCL8(1–66) (red).
Characterization of Site-I Interaction
Site-I binding interactions of CC-CXCL8 monomers and dimers were characterized by titrating unlabeled CXCR1 N-domain peptide to 15N-labeled CXCL8 using NMR spectroscopy. We initially discuss the binding characteristics of the CC-CXCL8 dimer and then discuss the binding of the monomer.
We have shown previously for both the WT CXCL8 dimer and CXCL8(1–66) monomer that binding induces significant chemical shift perturbations for residues corresponding to N-loop, 3rd β-strand, and C-terminal helix residues, and dissociation of the dimer-receptor to the monomer-receptor complex (33). For the CC-CXCL8 dimer, we observe perturbation of the same set of residues (data not shown), and also observe a new set of peaks in the spectra that correspond to the monomer-N-domain complex during the course of the titration, indicating binding-induced dissociation of the dimer-receptor complex (Fig. 7A). The chemical shift perturbation profiles of the CC-CXCL8(1–66) monomer were also similar to that observed for CXCL8(1–66) monomer, though the apparent binding affinities were ∼5 fold lower (Fig. 7, panels B and C). These observations indicate that the CXC motif does play a role, albeit minor, in mediating Site-I interaction, and that it plays a more prominent role in mediating Site-II interaction.
FIGURE 7.

Binding of CC-CXCL8 to CXCR1 N-domain. Panel A, binding induced dissociation of CC-CXCL8 dimer. A section of the 15N-1H HSQC spectra showing binding-induced chemical shift changes in the CC-CXCL8 dimer, and the appearance of bound monomer (labeled in red). Panel B, section of the 15N-1H HSQC spectra showing binding-induced chemical shift perturbations of the CC-CXCL8(1–66) monomer. Unbound peaks are in black and the final bound peaks are in red. Panel C, left and right, binding affinity measurements. Binding-induced chemical shift changes are shown for residues Ile-39 and Leu-49 of CC-CXCL8(1–66) (●) and CXCL8(1–66) (○). Average Kd calculated from a subset of CC-CXCL8(1–66) residues is ∼5-fold lower compared with CXCL8(1–66) monomer (63 ± 6 versus 12 μm).
Dimerization of CC-CXCL8
The monomer-dimer equilibrium constant (Kd) of WT CXCL8 has been characterized under a variety of solution conditions including at different pH values, buffers, and at various temperatures using sedimentation equilibrium methods (37–39). These studies have shown that the Kd of CXCL8 is ∼10 μm, and that the values are fairly insensitive to temperature, but show some differences as a function of pH. We measured the Kd of CC-CXCL8 in phosphate buffer at pH 7 and acetate buffer at pH 5 using sedimentation equilibrium. The Kd values for CC-CXCL8 variant were identical within experimental error to that observed for WT CXCL8, indicating that the CXC motif plays no role in influencing the dimerization affinity (Table 1).
TABLE 1.
Sedimentation equilibrium studies of WT CXCL8 and CC-CXCL8
| Protein | Buffer | Kd (μma) |
|---|---|---|
| WT CXCL8 | 50 mm phosphate, pH 7 | 12 ± 5b |
| 50 mm acetate, pH 5.0 | 15 ± 3 | |
| CC-CXCL8 | 50 mm phosphate, pH 7 | 6 ± 2 |
| 50 mm acetate, pH 5.0 | 9 ± 3 |
a The Kd was calculated from a global fit of the data collected at rotor speed of 25,000, 30,000, and 42, 000 rpm. The phosphate buffer also contains 50 mm NaCl.
b Data from Ref. 16.
DISCUSSION
Chemokines are classified as either CXC or CC on the basis of conserved N-terminal cysteines. The CXC/CC motif separates the functionally important N-terminal and N-loop domains, and also are an integral component of the structural framework by forming disulfides with the other two cysteines present in the protein core (Fig. 1). In general, CXC chemokines bind CXC receptors and CC chemokines bind CC receptors, but a subset of CXC chemokines that are agonists for the CXCR3 receptor also bind the CCR3 receptor not as an agonist but as an antagonist (40). Further, the human chemokine receptor DARC can bind both CXC and CC chemokines with high affinity, but DARC is not a signaling receptor and is believed to play a role in trafficking chemokines (41). Currently, nothing is known regarding what, if any, role(s) the CXC/CC motif plays in CXC/CC chemokine structure and receptor function.
Human CXCL8, a CXC chemokine, is one of the best-studied chemokines and the structural basis for its binding and receptor activation has been well characterized (5, 6). To understand the role of the CXC motif, we created and characterized structural and functional properties of CC variants of CXCL8 by deleting the intervening residue (CXC → CC). NMR studies show that deleting the intervening residue Gln-8 in the CXC motif does not affect either the global fold or quaternary structure, and MD simulations predict a change in the relative orientation of the N-terminal ELR and 30s loop. Switching CXC to CC motif in CXCL8 has no effect either on dimerization or GAG binding (supplemental materials). On the other hand, CC-CXCL8 binds poorly to both receptors but functions as an agonist for CXCR1 but not for CXCR2 receptor. CC-CXCL8, in comparison to WT, binds CXCR1 N-domain peptide with a slightly lower affinity indicating that the poor binding affinity to the intact receptor must be due its weak binding at Site-II.
Therefore, the CXC-motif is critical for the conformational change(s) that is/are essential for optimal binding at Site-II. Weak binding affinities to both the receptors also implies that the extent and/or duration of engagement by the N-terminal ELR residues of receptor residues at Site-II are less stringent for CXCR1 activity but more stringent for CXCR2 activity. Results from this study have provided critical insights into the molecular basis of the relationship among receptor binding affinity, receptor activation, and receptor specificity. In particular these data indicate that binding affinities and receptor activation are not correlated and are separable functions, and that the role of CXC motif is subtle, and receptor dependent. A previous study that measured binding affinities and activities of CXCL8 and CXCL1 for various CXCR1/CXCR2 chimera also came to the conclusion that binding affinity and agonist potency are separable functions (42).
On the basis of our data, we now propose a two-site multistep model of how CXCL8 binds and activates its receptor (Fig. 8). We propose that the N-terminal, N-loop, and the disulfides are best described as structural/functional modules, and that the CXC spacer functions as a conformational switch/trigger allowing optimal coupling interactions between these modules for binding affinity and receptor activation. In the first step, the N-loop residues form an encounter complex with the receptor N-domain leading to Site-I binding (panel A). The disulfides and the CXC motif functioning as a spacer mediate this initial binding. This initial binding triggers structural/dynamic changes throughout the ligand, especially of the 30s loop and N-terminal residues. These conformational changes orient the N-terminal ELR residues for optimal binding to the receptor residues, leading to Site-II binding (panel B). In the final step, ligand N-terminal residues engage site-II receptor residues, triggering conformational changes in the receptor resulting in downstream signaling events. In the absence of the spacer X (CXC → CC), CC-CXCL8 can bind to the receptor N-domain (Site-I) but its ability to communicate with Site-II is compromised. Nonetheless, CC-CXCL8 can still function as a selective agonist for CXCR1, but is completely incompetent to activate CXCR2 (panels C and D).
FIGURE 8.

A schematic of the two-site multistep binding of CXCL8 to its receptor. The structural scaffold of CXCL8 is shown in black, the 30s loop in pink, disulfides in blue, and the N-terminal residues are in black (panel A). The figures are not to scale and are purely for illustrative purposes only. The initial binding interactions at Site-I results in structural/dynamic changes that are essential for subsequent binding of N-terminal ELR residues to receptor transmembrane/extracellular loop residues and receptor activation (Site-II, panel B). The absence of the spacer X (CXC to CC) nevertheless allows binding and activation at Site-II for CXCR1 (panel C) but not CXCR2 (panel D).
It is very likely that the CC motif plays a similar important role in CC chemokines, and we propose that carrying out the reciprocal experiment of inserting a residue in the CC motif of a CC chemokine could substantially impact its structural and functional properties. Chemokine structures consistently reveal that the regions of the ligand that interact with the receptor and the CXC/CC motif is structurally not rigid but plastic, and suggest that the information for receptor specificity and promiscuity is encoded in the functional residues in the background of the CXC/CC motif structural scaffold. We propose that the CXC/CC motifs function as conformational switches, and that their ability to mediate coupling interactions between N-terminal and N-loop binding domains and between these domains and the protein core play important roles in determining binding affinities, receptor and ligand selectivity, and for various receptor signaling activities.
Supplementary Material
Acknowledgments
We thank Dr. Lavanya Rajagopalan for heparin binding experiments, Aishwarya Ravindran for protein purification, Pavani Gangavarapu for the peritoneum animal model data, and Christopher Chin for sedimentation equilibrium measurements.
This work was supported, in whole or in part, by National Institutes of Health Grant R01AI069152, the John Sealy Endowment Fund (to K. R.), R01GM081798 (to J. N.), and a McLaughlin Fellowship (to S. T. D.). J. M. S. is on leave from the Instituto de Fisiologia, Universidad Austral de Chile, Valdivia, Chile.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- GAG
- glycosaminoglycans
- HL-60
- human promyelocytic leukemia cells
- HSQC
- heteronuclear single quantum correlation
- NOESY
- nuclear Overhauser enhancement spectroscopy
- MD
- molecular dynamics
- PDB
- Protein Data Bank.
REFERENCES
- 1.Luster A. D. (2002) Curr. Opin. Immunol. 14, 129–135 [DOI] [PubMed] [Google Scholar]
- 2.Moser B., Wolf M., Walz A., Loetscher P. (2004) Trends Immunol. 25, 75–84 [DOI] [PubMed] [Google Scholar]
- 3.Zou Y. R., Kottmann A. H., Kuroda M., Taniuchi I., Littman D. R. (1998) Nature 393, 595–599 [DOI] [PubMed] [Google Scholar]
- 4.Belperio J. A., Keane M. P., Arenberg D. A., Addison C. L., Ehlert J. E., Burdick M. D., Strieter R. M. (2000) J. Leukoc. Biol. 68, 1–8 [PubMed] [Google Scholar]
- 5.Rajagopalan L., Rajarathnam K. (2006) Biosci. Rep. 26, 325–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark-Lewis I., Dewald B., Loetscher M., Moser B., Baggiolini M. (1994) J. Biol. Chem. 269, 16075–16081 [PubMed] [Google Scholar]
- 7.Jarnagin K., Grunberger D., Mulkins M., Wong B., Hemmerich S., Paavola C., Bloom A., Bhakta S., Diehl F., Freedman R., McCarley D., Polsky I., Ping-Tsou A., Kosaka A., Handel T. M. (1999) Biochemistry 38, 16167–16177 [DOI] [PubMed] [Google Scholar]
- 8.Kofuku Y., Yoshiura C., Ueda T., Terasawa H., Hirai T., Tominaga S., Hirose M., Maeda Y., Takahashi H., Terashima Y., Matsushima K., Shimada I. (2009) J. Biol. Chem. 284, 35240–35250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xanthou G., Williams T. J., Pease J. E. (2003) Eur. J. Immunol. 33, 2927–2936 [DOI] [PubMed] [Google Scholar]
- 10.Fernandez E. J., Lolis E. (2002) Annu. Rev. Pharmacol. Toxicol. 42, 469–499 [DOI] [PubMed] [Google Scholar]
- 11.Swaminathan G. J., Holloway D. E., Colvin R. A., Campanella G. K., Papageorgiou A. C., Luster A. D., Acharya K. R. (2003) Structure 11, 521–532 [DOI] [PubMed] [Google Scholar]
- 12.Baldwin E. T., Weber I. T., St Charles R., Xuan J. C., Appella E., Yamada M., Matsushima K., Edwards B. F., Clore G. M., Gronenborn A. M., Wlodawer A. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 502–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajarathnam K., Li Y., Rohrer T., Gentz R. (2001) J. Biol. Chem. 276, 4909–4916 [DOI] [PubMed] [Google Scholar]
- 14.Clore G. M., Appella E., Yamada M., Matsushima K., Gronenborn A. M. (1990) Biochemistry 29, 1689–1696 [DOI] [PubMed] [Google Scholar]
- 15.Crump M. P., Gong J. H., Loetscher P., Rajarathnam K., Amara A., Arenzana-Seisdedos F., Virelizier J. L., Baggiolini M., Sykes B. D., Clark-Lewis I. (1997) EMBO J. 16, 6996–7007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajagopalan L., Chin C. C., Rajarathnam K. (2007) Biophys. J. 93, 2129–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajarathnam K., Sykes B. D., Dewald B., Baggiolini M., Clark-Lewis I. (1999) Biochemistry 38, 7653–7658 [DOI] [PubMed] [Google Scholar]
- 18.Jin H., Shen X., Baggett B. R., Kong X., LiWang P. J. (2007) J. Biol. Chem. 282, 27976–27983 [DOI] [PubMed] [Google Scholar]
- 19.Rajarathnam K., Prado G. N., Fernando H., Clark-Lewis I., Navarro J. (2006) Biochemistry 45, 7882–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nasser M. W., Raghuwanshi S. K., Grant D. J., Jala V. R., Rajarathnam K., Richardson R. M. (2009) J. Immunol. 183, 3425–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veldkamp C. T., Seibert C., Peterson F. C., De la Cruz N. B., Haugner J. C., 3rd, Basnet H., Sakmar T. P., Volkman B. F. (2008) Sci. Signal. 1, ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Proudfoot A. E., Handel T. M., Johnson Z., Lau E. K., LiWang P., Clark-Lewis I., Borlat F., Wells T. N., Kosco-Vilbois M. H. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 1885–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoogewerf A. J., Kuschert G. S., Proudfoot A. E., Borlat F., Clark-Lewis I., Power C. A., Wells T. N. (1997) Biochemistry 36, 13570–13578 [DOI] [PubMed] [Google Scholar]
- 24.Frevert C. W., Kinsella M. G., Vathanaprida C., Goodman R. B., Baskin D. G., Proudfoot A., Wells T. N., Wight T. N., Martin T. R. (2003) Am. J. Respir. Cell Mol. Biol. 28, 464–472 [DOI] [PubMed] [Google Scholar]
- 25.Fernando H., Nagle G. T., Rajarathnam K. (2007) Febs. J. 274, 241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajarathnam K., Clark-Lewis I., Sykes B. D. (1995) Biochemistry 34, 12983–12990 [DOI] [PubMed] [Google Scholar]
- 27.Rajagopalan L., Rajarathnam K. (2004) J. Biol. Chem. 279, 30000–30008 [DOI] [PubMed] [Google Scholar]
- 28.Suetomi K., Lu Z., Heck T., Wood T. G., Prusak D. J., Dunn K. J., Navarro J. (1999) J. Biol. Chem. 274, 11768–11772 [DOI] [PubMed] [Google Scholar]
- 29.White J. R., Lee J. M., Young P. R., Hertzberg R. P., Jurewicz A. J., Chaikin M. A., Widdowson K., Foley J. J., Martin L. D., Griswold D. E., Sarau H. M. (1998) J. Biol. Chem. 273, 10095–10098 [DOI] [PubMed] [Google Scholar]
- 30.Das S. T., Rajagopalan L., Guerrero-Plata A., Sai J., Richmond A., Garofalo R. P., Rajarathnam K. (2010) PLoS ONE 5, e11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kay L., Keifer P., Saarinen T. (1992) JACS 114, 10663–10665 [Google Scholar]
- 32.Muhandiram D. R., Kay L. E. (1994) J. Mag. Reson., Ser. B 103, 203–216 [Google Scholar]
- 33.Ravindran A., Joseph P. R., Rajarathnam K. (2009) Biochemistry 48, 8795–8805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeLano W. L. (2002) The Pymol Users Manual DeLano Scientific LLC, Palo Alto, CA [Google Scholar]
- 35.Case D. A., Darden T. A., Cheatham T. E., III, Simmerling C. L., Wang J., Duke R. E., Luo R., Merz K. M., Pearlman D. A., Crowley M., Walker R. C., Zhang W., Wang B., Hayik S., Roitberg A., Seabra G., Wong K. F., Paesani F., Wu X., Brozell S., Tsui V., Gohlke H., Yang L., Tan C., Mongan J., Hornak V., Cui G., Beroza P., Mathews D. H., Schafmeister C., Ross W. S., Kollman P. A. (2006) AMBER 9, University of California, San Francisco [Google Scholar]
- 36.Lee M. C., Deng J., Briggs J. M., Duan Y. (2005) Biophys. J. 88, 3133–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burrows S. D., Doyle M. L., Murphy K. P., Franklin S. G., White J. R., Brooks I., McNulty D. E., Scott M. O., Knutson J. R., Porter D. (1994) Biochemistry 33, 12741–12745 [DOI] [PubMed] [Google Scholar]
- 38.Lowman H. B., Fairbrother W. J., Slagle P. H., Kabakoff R., Liu J., Shire S., Hébert C. A. (1997) Protein Sci. 6, 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kagiampakis I., Jin H., Kim S., Vannucci M., LiWang P. J., Tsai J. (2008) Biochemistry 47, 10637–10648 [DOI] [PubMed] [Google Scholar]
- 40.Loetscher P., Pellegrino A., Gong J. H., Mattioli I., Loetscher M., Bardi G., Baggiolini M., Clark-Lewis I. (2001) J. Biol. Chem. 276, 2986–2991 [DOI] [PubMed] [Google Scholar]
- 41.Pruenster M., Mudde L., Bombosi P., Dimitrova S., Zsak M., Middleton J., Richmond A., Graham G. J., Segerer S., Nibbs R. J., Rot A. (2009) Nat. Immunol. 10, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahuja S. K., Lee J. C., Murphy P. M. (1996) J. Biol. Chem. 271, 225–232 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.