

Abstract
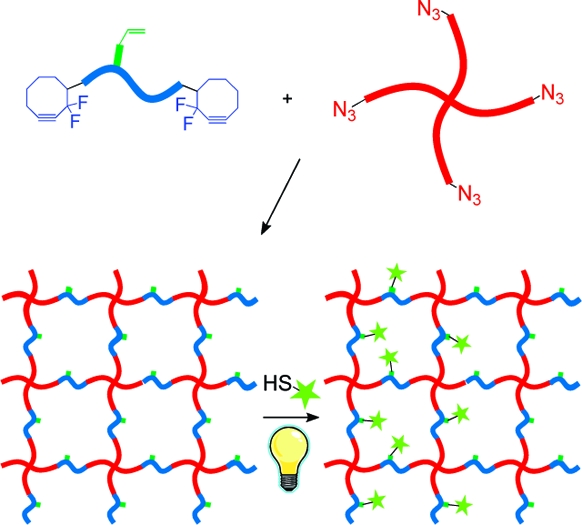
Click chemistry offers highly selective and orthogonal reactions that proceed rapidly and under a variety of mild conditions with the opportunity to create highly defined and multifunctional materials. This work illustrates a strategy where step-growth networks are formed rapidly via a copper-free, azide−alkyne click chemistry between tetrafunctional poly(ethylene glycol) molecules and difunctionalized synthetic polypeptides. The molecular weight of the polymer precursors (10, 15, or 20 kDa PEG) and the stoichiometry of reactive end group functionalities (1.5:1 to 1:1.5) provide control over the material cross-linking density, enabling elastic materials with tunable moduli (G′ = 1000−6000 Pa). A sequential photochemically activated thiol-ene chemistry allows subsequent functionalization of the network through reaction with pendant alkene moieties on the peptide. Because the thiol-ene reaction is light-driven, the degree of modification is directly related to the dosage of light delivered to the system (0−6 J cm−2). We exploit this feature to create complex biochemical gradients of multiple peptides with well-defined magnitude and slope throughout the three-dimensional (3D) network. Since both reactions can occur in the presence of cells, this material ultimately enables independent and in situ tuning of biochemical and biomechanical properties of biomaterial networks, suggesting an avenue to direct cell function throughout specific regions within a 3D material.
Short abstract
Multifunctionalized macromers react via a copper-free click chemistry to form an idealized 3D hydrogel. Subsequently, thiol-containing biomolecules are spatially patterned within the material with precise control over the amount and location of functionalization. Both the network formation and subsequent patterning reactions are fully cytocompatible, allowing these systems to be used to study individual cell behavior at user-defined locations throughout the material.
1. Introduction
There is a growing interest in developing and utilizing peptide-functionalized poly(ethylene glycol) (PEG) hydrogels for 3D cell culture applications.1−3 Peptides are readily designed to incorporate integrin-binding epitopes (e.g., the fibronectin-derived RGD sequence),4−7 enzymatically degradable sequences (e.g., the collagenase-sensitive sequence GPQG↓ILGQ),8−10 growth-factor sequestering properties (e.g., the basic fibroblast growth factor-binding KRTGQYKL sequence),11−13 as well as additional bioactivity14−17 into otherwise bioinert hydrogel material formulations. The inclusion of peptides within cell-laden hydrogel matrices enables tailorability of a cell’s microenvironment and potential control over the biological fate of encapsulated cells. Their ease of synthesis and purification render peptides as viable options for material functionalization. Additionally, peptides can be systematically engineered and modified with a variety of non-natural amino acids,(18) ultimately enabling a greater level of synthetic biological control than their complex biomacromolecular counterparts.
Taking lead from a variety of reactions originally developed for organic synthesis, many techniques have been utilized to incorporate peptides into hydrogel materials. Most commonly, monoacrylated PEG-N-hydroxysuccinimide (NHS) is coupled to the N-terminal α-amino group to yield an acrylated peptide, which can be copolymerized within PEG acrylate-based polymer networks.19−22 The photoinitiated solution polymerization of these molecules yields hydrogel systems that are useful for cell encapsulation, but are quite heterogeneous and polydisperse in their cross-linking density.(23) Alternatively, the Michael-type addition reaction has been used to generate step-growth networks which exhibit more uniform cross-linking densities, and do not require postsynthetic modification of the peptide sequence. In this reaction, cysteine-containing peptides (which include pendant thiols) are coupled to PEG with electron-poor olefin end groups (such as an acrylate or vinyl sulfone) under slightly basic conditions.24−26 More recently, a photoinitiated thiol-ene hydrogel platform, which combines the advantages of step-growth and photoinitiated polymerization has been used to form enzymatically degradable peptide-functionalized PEG gels, similar in structure to the Michael-addition reaction.(27) Further, the spatial and temporal regulation of the photopolymerization affords an additional level of control over network formation. Finally, the mixed-mode polymerization of acrylates in the presence of thiols enables nonstoichiometric amounts of cysteine-containing peptides to be directly incorporated into materials.(28) The combination of chain and step growth mechanisms give rise to networks with unique structure, where polyacrylate kinetic chains are truncated with single peptides, but the amount of peptide functionalization directly influences network structure and resulting gel mechanics. Although the interest in and application of peptide-functionalized PEG gels continues to grow, we sought to use a combination of orthogonal reactions to first form peptide-functionalized hydrogels with defined structures and mechanical properties and then to modify subsequently these materials with user-defined biochemical functionalities without altering the underlying network structure.
The recent advent of click chemistry, an organic paradigm for robust, highly efficient, highly specific reactions29,30 has led to novel means to create peptide-functionalized biomaterials.(31) The orthogonal nature of these reactions ultimately allows for peptide introduction into materials both initially, as well as postgelation without altering the initial network structure.(32) The quintessential click reaction, the copper(I)-catalyzed 1,3-dipolar Huisgen cycloaddition between azides and alkynes (CuAAC),(33) has been utilized for peptide ligation,34,35 modification,36,37 macrocyclization,38,39 and polymer grafting.40,41 Unfortunately, this reaction’s catalyst, as with the majority of those reactions commanding “click” status, is cytotoxic and is unable to be performed in the presence of cells, thereby precluding its usage in the formation of peptide-functionalized materials for cell encapsulation. Recently, a novel cyclooctyne molecule (DIFO3) was developed whose ring strain and electron-withdrawing fluorine substituents give rise to an activated alkyne that reacts quickly with azides in the absence of a catalyst.42−44 This strain-promoted azide-alkyne cycloaddition (SPAAC) has enabled the application of the Huisgen reaction in living organisms to label glycosylation patterns in zebrafish and mice.(45) Similarly, the photoinduced thiol-ene reaction, a radical-mediated addition of a thiol across an olefin, has recently gained attention as an emerging click reaction.46,47 In addition to being fully bioorthogonal and biocompatible, this reaction is advantageous in that it can be controlled spatially and temporally with the presentation of light.(27) The cytocompatible nature of these two click reactions render them particularly well-suited for biomaterial synthesis and functionalization.
Beyond simply creating peptide-functionalized hydrogels, we are interested in developing and exploiting these constructs to study 3D cell behavior dynamically. Recently, we introduced a two-component hydrogel system that is capable of directing and detecting 3D cell behavior at user-defined locations within the material.(48) This system, comprising a four-arm PEG tetraazide and a bis(cyclooctyne)-peptide, utilizes two orthogonal click reactions; one for hydrogel formation (SPAAC) and another for biochemical patterning (thiol-ene). Ultimately, this formulation leads to an ideal, step growth network that enables control of the material cross-linking density and the ability to introduce pendant biochemical functionalities through the photochemical thiol-ene reaction. Exploiting these chemistries, this manuscript sought to understand how the molecular weight and (non)stoichiometric reaction of the precursor molecules could be used to control the final material mechanical properties. Subsequently, we quantified aspects of the thiol-ene photocoupling reaction and demonstrate the advantages of the photochemical reaction in forming complex chemical gradients of multiple peptides within the same material with precise control over location and concentration. We expect that these unique gels will prove particularly beneficial in elucidating the unique role of mechanotransduction versus biochemical signaling on cell function in a well-defined 3D environment.
2. Results and Discussion
2.1. Mechanical Tunability of Click-Based Hydrogels
2.1.1. Design of the Base Hydrogel Network
To illustrate the utility that is afforded by multiple bioorthogonal chemistries, we have developed a two-component system that enables hydrogel formation with tunable mechanics. Here, a four-arm PEG tetraazide is reacted with a bis(DIFO3) difunctional polypeptide cross-linker in an aqueous environment to form a multifunctional hydrogel (Scheme 1). PEG is chosen because of its biocompatibility, hydrophilicity, and resistance to nonspecific protein adsorption. Additionally, the biophysical properties of the gel are readily tuned by altering the length of the PEG arms as well as the stoichiometric ratio of azide:alkyne on the PEG and peptide cross-linker, respectively. Biological functionality is introduced into the matrix backbone via the choice of the synthetic cross-linking peptide sequence. Here, we utilize a functionalized matrix metalloproteinase cleavable sequence (GPQG↓ILGQ) that is susceptible to secreted enzymes, enabling cell-dictated local network degradation and material reformation.(49) Enzymatically degradable hydrogels enable cells to spread and migrate throughout the material and yield a significantly more viable population as compared to their nondegradable counterparts.8,9 Furthermore, by utilizing a synthetic peptide cross-linker with a pendant functional group, we are able to introduce additional chemical moieties into the material at later time points or in spatially defined regions.
Scheme 1. Click-Functionalized Macromolecular Precursors React via the Strain-Promoted [3 + 2] Huisgen Cycloaddition to Form an End-Linked Hydrogel Network; By Varying the Molecular Weight of the PEG, Well-Defined Networks of Differing Cross-Linking Density Are Formed.

When the PEG tetraazide is mixed with the difunctionalized peptide, an end-linked polymerization occurs spontaneously and yields an idealized network with minimal defects and local heterogeneities. The nearly ideal structure of this system has been previously confirmed with rheological and magic-angle spinning NMR experiments (48) and agrees with previous work with click-based networks.(50) Though the gel is formed in just a few minutes, the reaction continues for ∼1 h, at which point all previously detectable azide and cyclooctyne functional groups have been consumed. This formulation is fully cytocompatible and has been successfully used for the encapsulation of primary cells, as well as established cell lines, with high viability.(48)
2.1.2. Effect of PEG Size and Network Connectivity on Mechanical Properties
We first sought to determine the effects of different PEG molecular weights on the resulting hydrogel network structure and elasticity. For this, we synthesized 10, 15, and 20 kDa 4-arm PEG tetraazide, spanning a 2-fold range of average repeat units per arm (∼56, 85, and 113 respectively). The functionalized (>95%) PEGs were then combined with a base peptide [AcK(DIFO3)RRGGK(alloc)GGPQGILGQRRK(DIFO3)-NH2] in a 13.5 wt % aqueous solution containing a stoichiometrically balanced amount of azide and alkyne moieties. After formation, these gels were allowed to equilibrate overnight in phosphate buffered saline (PBS) upon which rheological frequency sweep experiments were conducted to determine the shear elastic moduli of the constructs (Figure 1a). In addition, equilibrium swelling ratios (q) were calculated as the mass of the swollen network divided by that of the dry system (Figure 1b).
Figure 1.

Final mechanical properties are dictated by gel formulation. (a) The swollen hydrogel shear elastic moduli are easily tuned by changing the molecular weight of the PEG cross-linker (◻ = 10 000 g/mol, ▲=15 000 g/mol, ○ = 20 000 g/mol). Alternatively, small stoichiometric imbalances between the gel-forming azide and cyclooctyne reactive groups also give rise to materials with a wide range of moduli. (b) Changes in gel formulation result in hydrogels with different equilibrium swelling ratios (in PBS). (c) The swelling ratio (▲) and equilibrium shear moduli (○) data collapse onto a single curve for all conditions when normalized to cross-linking density. Data reported as mean ± std.
The shear moduli and overall water content varied significantly between formulations and were found to be directly related to the swollen hydrogel cross-linking density (ρx) (Figure 1c). For these networks, the ideal cross-linking density during network formation (ρx,0) is equal to twice the concentration of PEG tetraazide multiplied by r, the ratio of azide to alkyne functionalities such that 0 ≤ r ≤ 1, because each arm of the PEG azide is attached to half an infinite chain.(51) The gels containing the lowest molecular weight PEG (10 kDa, ρx = 0.023 mol/L) resulted in the highest moduli (6200 ± 280 Pa) and lowest swelling (6.6 ± 0.7), whereas those with the highest molecular weight PEG (20 kDa, ρx = 0.0018 mol/L) gave rise to the lowest observed moduli (2500 ± 220 Pa) and highest swelling (17.9 ± 0.9).
Because small stoichiometric imbalances can lead to significant changes in ρx, we quantified the changes in the elastic modulus and q for systems of varying ratios of azide to alkyne. Specifically, we created gels for each of the three PEG molecular weights (MWs) and the base peptide, this time varying the overall ratio of azide to alkyne functionality (1.5:1, 1.25:1, 1:1.25, and 1:1.5), and measured the resulting equilibrium swelling ratio (Figure 1a) and shear moduli (Figure 1b). Here, we find that the overall network stiffness decreases rapidly as the formulation departs from 1:1 stoichiometry with relatively modest deviations from unity resulting in large changes in mechanical properties. With one of the reactive groups in excess, a network containing fewer overall cross-links is formed (as r < 1). Again, with a lower ρx in the system, we observe a lower elastic moduli and a higher swelling ratio (Figure 1c). This trend is consistent through all chosen MWs of PEG and all stoichiometries.
By comparing the obtained gel moduli and swelling ratios with that expected for a perfect system with a given ρx,0, the degree of network ideality can be estimated.(51) When two azide functional groups on the same PEG molecule react with both cyclooctyne moieties present on a given peptide, a primary cycle is formed. This defect fully removes the ability of the PEG molecule to form cross-links, rendering it to behave like a difunctional linker, and lowers the final moduli. Similarly, when the tetrafunctional PEG contains two primary cycles, it will no longer be connected to the network and becomes part of the sol fraction. For all formulations containing an equal ratio of azide to cyclooctyne functionality, we estimate the degree of network ideality (i.e., extent of cross-linking versus cyclization) to be >90% (derivations and results for all formulations are given in the see the Supporting Information, Table S1).
2.1.3. Effect of Peptide Sequence and Charge on Network Properties
To determine the effect of the cross-linker composition on the hydrogel mechanical properties, we synthesized various peptides and gels were formed with each PEG tetraazide. We first sought to determine whether the order of amino acids in our peptide sequence had a significant effect on the gels’ resulting properties. We created a scrambled peptide sequence [Ac−K(DIFO3)QGK(alloc)RIPGRRLGGRGQGK(DIFO3)-NH2], which contained all of the internal amino acids of the original sequence in a randomly rearranged order. We find that the equilibrated shear elastic moduli of these stoichiometrically balanced scrambled systems are statistically indistinguishable from those of the original peptide sequence (Figure 2a). Similarly, we were interested in determining the effect of overall peptide charge on moduli values. For this, we synthesized charge neutral [AcK(DIFO3)REGGK(alloc)GGPQGILGQERK(DIFO3)-NH2] and negatively charged [-4, AcK(DIFO3)EEGGK(alloc)GGPQGILGQEEK(DIFO3)-NH2] peptide variants to complement our positively charged (+4) base peptide. Here, we find that the resulting equilibrated moduli are not statistically different from one another for the 10 kDa PEG system (Figure 2b). This is likely due to a charge-shielding effect of the buffered PBS solution that the gel imbibes. Ultimately, this enables the sequence of the peptide cross-linker to be altered without significant changes in gel mechanics under conditions for cell culture.
Figure 2.

Network properties are largely independent of peptide cross-linker properties in buffered solutions (pH 7.4). (a) The amino acid sequence does not influence the shear elastic moduli of formed gels. (b) Similarly, the overall charge of the peptide does not influence the mechanical properties of the formed networks. Data reported as mean ± std.
2.2. Biochemical Patterning within Click-Based Hydrogels
2.2.1. Incorporating Biochemical Tunability into Base Hydrogel
An additional benefit of using a synthetic peptide cross-linker to form our hydrogel network is that we are able to dictate precisely its sequence to include a variety of biological functionalities, as well as to include non-natural amino acids that enable additional modification of the system postgelation independent of the network mechanics. Here, we chose to include a photoreactive allyl ester within the peptide sequence via the commercially available Fmoc-Lys(alloc)−OH amino acid, which enables relevant biomolecules to be covalently coupled pendant to the hydrogel backbone via the bioorthogonal thiol-ene click reaction (Scheme 2). Though allyloxycarbonyl (alloc) was originally designed as a selectively removable protecting group for peptide branch creation, it contains a vinyl group capable of undergoing the thiol-ene reaction with any thiol-containing molecule, including cysteine.32,52 This protecting group is fully stable to Fmoc deprotection and trifluoroacetic acid peptide cleavage from resin. The electron-rich alloc allyl ester is not susceptible to the nonspecific Michael-type addition, ensuring that chemical immobilization is confined to user-defined regions of interest within the material.(53)
Scheme 2. Post-Gelation, Cysteine-Containing Peptides Are Covalently Affixed Pendant to the Hydrogel Backbone via the Radical-Mediated Thiol-ene Reaction; Using a Photoinitiator, Chemical Radical Generation Is Confined to Areas of Light Exposure, Ultimately Enabling Spatial Patterning Control Throughout the Material.

The thiol-ene reaction is a radical-mediated addition of a thiol to an alkene, involving a sequential propagation of a thiyl radical with a vinyl functional group followed by a chain transfer of the radical to another thiol.(54) Thiyl radicals can be readily generated via the photocleavage of a hydrogen-abstracting initiator. Here, thiyl radicals are only generated in the presence of UV light, ultimately dictating when, where, and how much of the thiol-ene functionalization will occur. After functionalization is complete, unreacted thiols are allowed to diffuse out of the network, leaving the covalently patterned substrate behind (Scheme 2). Additionally, spatially controlled patterns can be formed in 3D using focused multiphoton laser light.(48)
2.2.2. Controlling Patterning Extent with Light Dosage and Initiator Concentration
Upon swelling cysteine-containing peptides and photoinitiator into the preformed hydrogel, UV light was used to induce thiol-ene functionalization within the network. By varying the amount of photoinitiator present (1.1, 2.2, 4.4, and 8.8 mM Irgacure 2959, 2-hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone), as well as the total dosage of light imposed on the system (0.6, 1.8, 3.0, and 6.0 J cm−2), we control the extent of ene conversion, ultimately dictating the patterning concentrations of peptide within our materials. Here, fluorescently labeled peptides (AF488-AhxRGDSC-NH2 and AF546-AhxPHSRNC-NH2) are patterned into the network and the system’s resulting fluorescence is used to quantify the resulting concentration range. For a given light dosage, we find that the degree of patterning scales roughly linearly with the photoinititiator concentration (Figure 3a). Similarly, for a given amount of photoinitiator, we find that the patterning concentration scales roughly linearly with the light dosage. Interestingly, the patterning concentrations for a given condition remains nearly constant throughout the full ∼1 mm thickness of the gel (Figure 3b). Additionally, network mechanical properties in the form of swelling ratios and moduli remain unchanged throughout the patterning process (see the Supporting Information, Figure S1).
Figure 3.

Patterning concentration depends on the photoinitiator concentration and the total light dosage delivered to the system. (a) By varying the amount of photoinitiator present, we are able to control the level of patterning concentration for a given dosage (▼ = 1.1 mM, ▲ = 2.2 mM, ◻ = 4.4 mM, ○ = 8.8 mM photoinitiator I2959). (b) As the light intensity remains near-constant throughout the thickness of the gel, patterning concentration is largely uniform throughout the entirety of the gel. Data reported as mean ± std. Scale bar = 200 μm.
For thiol-ene polymerizations, the rate of reaction (Rp) is given as:
where [C=C] is the ene concentration, [S·] is the thiyl radical concentration, and k is the reaction rate constant.(54) In this system, both the alkene (∼20 mM) and thiol (∼10 mM) functionalities are in significant excess with regards to the patterned concentrations obtained (∼1 mM). Because only a small portion of the total ene functionality is consumed (<5%) in any one of our patterning experiments, the [C=C] is taken as a largely unchanging constant, leaving Rp only as a function of [S·] (i.e., pseudo-first-order reaction). Additionally, the thiyl radical concentration directly depends on the rate of photoinitiation (Ri) given by
where f = initiator efficiency, ε = molar absorptivity of initiator, [I] = initiator concentration, I0 = light intensity, NAV = Avogadro’s number, h = Planck’s constant, and c = speed of light.(54) Because <2% of the total initiator is consumed in any one of our patterning experiments (see the Supporting Information, Figure S2), [I] can also be approximated as a constant, implying that overall degree of patterning is proportional to the light dosage and the initiator concentration. This result is consistent with our experimental findings. For example, for 4.4 mM I2959 a dosage of 6 J cm−2 results in 0.57 ± 0.03 mM patterning. Halving the dosage to 3 J cm−2 gives half the patterning (0.29 ± 0.02 mM) with the same I2959 concentration, while halving the amount of initiator to 2.2 mM I2959 with the same light conditions also results in half the patterning amount (0.30 ± 0.04 mM). Thus, the patterning concentration is observed to be roughly linear with light dosage and initiator concentration.
2.2.3. Generation of Peptide Gradients Throughout Hydrogel
Having established a relationship between light dosage and patterning concentration, we were interested in utilizing this information to create well-defined complex biochemical gradients within a hydrogel. Hydrogels were exposed to a linear gradient of light exposure generated by a moving photomask (see the Supporting Information, Scheme S1),(55) resulting in a linear gradient of AF488-AhxRGDSC-NH2 patterning concentrations (Figure 4a). By varying the rate of photomask coverage (0.05, 0.1, or 0.2 cm min−1), the chemical gradient slope was precisely tuned (Figure 4b). As only a fraction of the available alkene functionalities are consumed in any given patterning process, the thiol-ene process can be repeated to pattern multiple epitopes within a single sample. This was exploited to introduce sequential and independent gradients of arbitrarily chosen slope of multiple peptides (AF488-AhxRGDSC-NH2 and AF546-AhxPHSRNC-NH2) within the same material (Figure 4c). By shuttering the light as it is delivered to the system, stepped gradients are easily created (Figure 4d). 3D biochemical gradients are of particular note to those interested in studying fundamental biological processes in vitro, including induced cell migration and alignment with RGD(56) and directed neurite extension with nerve growth factor gradients.(57)
Figure 4.

Biochemical gradients are created throughout the hydrogel network. (a) By subjecting the hydrogel to a linear gradient of light exposure, well-defined gradients of fluorescent patterning agents are created across relatively large distances. (b) The magnitude of these gradients is readily controlled by tuning the slope of the light gradient. (c) Multiple peptide gradients are patterned within the same material. (d) Using a stepped gradient of light exposure, a stepped patterning concentration is obtained. Scale bar = 200 μm.
3. Conclusions
In conclusion, we developed a versatile click-based PEG/peptide hydrogel system whose biomechanical properties are precisely tuned by altering the cross-linking density and network connectivity of the system. This was accomplished by (1) changing the molecular weight of the PEG cross-linker and (2) off-stoichiometric network polymerization. In classic cell culture buffers, the sequence and charge of the peptide were found to have little effect on the mechanical properties of the gel. By incorporating a photoreactive alkene functionality pendant to the hydrogel backbone, we were able to exploit the thiol-ene click reaction for spatially controlled, full-thickness network functionalization of cysteine-containing peptides. We demonstrate that the degree of patterning within the network is directly proportional to the dosage of light imposed on the system as well as to the initiator concentration. Exploiting this finding, we used gradients of light to synthesize well-defined linear chemical gradients of defined slope throughout a gel. This process is fully additive, and gradients of multiple peptides are easily achieved. Though not explicitly demonstrated in this work, both network formation and subsequent patterning reactions are fully cytocompatible. As such, this system can be utilized to elucidate systematically the effects of individual chemical cues in addition to gradients of multiple cues on individual cell behavior, as well as to spatially direct 3D stem cell differentiation.
4. Experimental Section
Synthesis of Poly(ethylene glycol) Tetraazide
4-arm PEG (Mn ≈ 10, 15, or 20 kDa) (0.5 mmol, Jenkem) was dried overnight in a vacuum oven and dissolved at 0 °C in a 20% mixture of pyridine in dichloromethane (DCM) under argon. A solution of methanesulfonyl chloride (1.15 g, 10 mmol, 5x/OH, Sigma) dissolved in minimal DCM was added over 10 min via syringe pump and allowed to react overnight. This solution was concentrated, washed with saturated Na2HCO3 (Fisher), dried over MgSO4 (Fisher), and precipitated in ice-cold diethyl ether (Fisher). 1H NMR (500 MHz, DMSO-d6, δ): 4.30 (m, 8H; 4 x MsOCH2), 3.67 (m, 8H; 4 × MsOCH2CH2), 3.57−3.47 (m, [CH2CH2O]n), 3.18 (s, 12H; 4 × CH3SOO−). The activated mesylate was dissolved in anhydrous dimethylformamide containing sodium azide (650 mg, 10 mmol, 5x/Ms, Fluka) and stirred overnight at 80 °C under argon. The reaction was filtered through Celite and concentrated. The product was dissolved in deionized water and dialyzed for 48 h (MWCO ∼ 2 kDa, SpectraPor). The solution was lyophilized to yield the desired product. The functionality was determined as >95% for all products. 1H NMR (500 MHz, CDCl3, δ): 3.77−3.50 (m, [CH2CH2O]n), 3.39 (m, 8H; 4 × CH2N3) (see the Supporting Information, Figure S3).
Synthesis of Difluorinated Cyclooctyne (DIFO3)
DIFO3 was synthesized following a published synthetic route.(44) Briefly, 1,3-cyclooctanedione was difluorinated followed by a Wittig reaction to introduce an orthoester linker handle. The remaining ketone was converted to its vinyl triflate, and cyclooctyne was generated via a lithium diisopropylamide-mediated elimination. Finally, the orthoester was deprotected to yield the DIFO3 carboxylic acid product. 1H NMR (500 MHz, CDCl3, δ): 11.72 (br s, 1H), 2.82−2.69 (m, 2H), 2.43−2.22 (m 3H), 2.19−2.05 (m, 2H), 1.87−1.74 (m, 2H), 1.70−1.61 (m, 1H), 1.45−1.35 (m, 1H); 13C NMR (101 MHz, CDCl3, δ): 178.53 (s), 119.10 (dd, J = 237.7, 239.4), 110.71 (t, J = 11.1 Hz) 84.62 (dd, J = 41.6 Hz, 46.9), 52.60 (t, J = 24.3 Hz), 33.80 (d, J = 3.1 Hz), 32.84 (d, J = 4.5 Hz), 32.65 (d, J = 1.9 Hz), 27.87 (s), 20.45 (s); 19F NMR (376 MHz, CDCl3, δ): −95.84 (d, J = 259.9; 1F), −102.05 (ddt, J = 7.0, 21.1, 260.2; 1F); HRMS (ESI+): calculated for C10H12F2LiO2+ [M + 7Li]+, 209.0960; found 209.0965 (Δ = +2.5 ppm). NMR spectra are found in the Supporting Information, Figures S4 and S5.
Peptide Synthesis and Modification
Peptide sequences (Ac-KRRGGK(alloc)GGPQGILGQRRK-NH2, regular; Ac-KQGK(alloc)RIPGRRLGGRGQGK-NH2, scrambled; Ac-KREGGK(alloc)GGPQGILGQERK-NH2, neutral; Ac-KEEGGK(alloc)GGPQGILGQEEK-NH2, negative) were synthesized (Protein Technologies, Inc. Tribute Peptide Synthesizer) though standard Fmoc solid-phase methodology and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate/N-hydroxybenzotriazole (HBTU/HOBt) activation. Resin was treated with trifluoroacetic acid/triisopropylsilane/water/phenol (94:2.5:2.5:1) for 3 h and precipitated and washed (3x) in ice-cold diethyl ether. DIFO3 was coupled to the ε-amino groups of the terminal lysines via standard 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) coupling chemistry. Peptides were purified using a semipreparative reversed-phase high-performance liquid chromatography (RP-HPLC) (Waters Delta Prep 4000) using a 70 min linear gradient (5 to 95%) of acetonitrile and 0.1% trifluoroacetic acid (Sigma). Peptide purity was confirmed by analytical RP-HPLC and matrix-assisted laser desorption, ionization time-of-light (MALDI-TOF) mass spectrometry (Applied Biosystems DE Voyager) using α-cyano-4-hydroxycinnamic acid matrix (Sigma). Regular calculated ([M+H]+ 2442.793); observed ([M+H]+ 2441.815); scrambled calculated ([M+H]+ 2442.793); observed ([M+H]+ 2443.159); neutral calculated ([M+H]+ 2388.649); observed ([M+H]+ 2388.568); negative calculated ([M+Na]+ 2357.471); observed ([M+H]+ 2357.205). MALDI spectra are found in the Supporting Information, Figures S7−S10.
Synthesis of Fluorescently Labeled Patterning Agents
H-AhxRGDSC-NH2 and H-AhxPHSRNC-NH2 (0.25 mmol) were synthesized via standard Fmoc solid-phase methodology and HBTU/HOBt activation. Fluorescent activated esters [Alexa Fluor 488 carboxylic acid, 2,3,5,6-tetrafluorophenyl ester (1 mg, Invitrogen) for H-AhxRGDSC-NH2 and Alexa Fluor 546 carboxylic acid, succinimidyl ester (1 mg, Invitrogen) for AhxPHSRNC-NH2] were dissolved in NMP with a catalytic amount of DIEA and stirred with respective resins overnight at room temperature. Resins were treated with trifluoroacetic acid/triisopropylsilane/water/dithiothreitol (94.5:2.5:2.5:0.5) for 1 h and precipitated and washed (3×) using ice-cold diethyl ether. These products, denoted as AF488-AhxRGDSC-NH2 and AF546-PHSRNC-NH2, were used with no further purification.
Hydrogel Formation
Four-arm PEG tetraazide was reacted with bis(DIFO3) difunctionalized polypeptides in an aqueous environment at room temperature. Component concentrations were based on a 13.5 wt % total macromer content for a variety of differing stoichiometric ratios of azide to alkyne functionality (1.5:1, 1.25:1, 1:1, 1:1.25, 1:1.5). Gels were allowed to swell in PBS overnight prior to patterning or mechanical testing.
Rheological Characterization of Hydrogel Equilibrium Properties
Dynamic rheometry was performed with parallel plate geometry on an ARES 4400 rotational rheometer (TA Instruments). Frequency sweep experiments (10% strain, ω = 1 − 100 rad s−1) were conducted on equilibrated hydrogels of known geometry (7 mm diameter × 1 mm tall disks). Gel moduli were determined within the linear viscoelastic regime for each sample.
Biochemical Patterning
Hydrogels were swollen in PBS (pH 7.4) containing Irgacure 2959 (1.1, 2.2, 4.4, or 8.8 mM, I2959, Ciba) and 10 mM patterning agent (AF488-AhxRGDSC-NH2 or AF546-PHSRNC-NH2) for one hour. Using conventional photolithographic techniques, gels were exposed to collimated UV light (365 nm wavelength at 10 mW cm−2) for various times (1, 3, 5, or 10 min). After patterning is complete, the gel is washed for approximately two hours with fresh PBS to remove any unbound material, yielding the final patterned hydrogel. For gradient patterned hydrogels, a light gradient (365 nm at 10 mW cm−2 for 0 − 12 min of exposure time) was achieved by irradiating the hydrogel containing 2.2 mM I2959 and 10 mM patterning agent while a unidirectional moving photomask continuously covers the hydrogel at a predetermined rate (0.5, 1.0, or 2.0 cm min−1).(55) For step-gradient formation, the light was shuttered on and off while the photomask continued to cover the sample at a fixed rate. Patterning concentrations were determined using confocal microscopy by comparing the fluorescence intensity at a point 200 μm below gel surface against a standard curve relating fluorescence intensity to that of gels swollen in known concentrations of the patterning agent (see the Supporting Information, Figure S11). Light dosage was calculated as the product of the intensity and the exposure time.
Acknowledgments
The authors thank Dr. April Kloxin and Dr. Benjamin Fairbanks for discussion on photopatterning, Radhika Rawat for initial assistance with gradient generation, Mark Tibbitt for communication on network nonidealities, and Alex Aimetti for useful discussion on peptide synthesis. Fellowship assistance to C.A.D. was awarded by the U.S. Department of Education’s Graduate Assistantships in Areas of National Need program and the National Institutes of Health (T32 GM-065103).
Supporting Information Available
Network ideality calculations, network mechanical properties postpatterning, photoinitiator consumption, and NMR and MALDI-TOF characterization (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tibbitt M. W.; Anseth K. S. Biotechnol. Bioeng. 2009, 103, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing M. C.; Anseth K. S. Science 2007, 316, 1133. [DOI] [PubMed] [Google Scholar]
- Lutolf M. P.; Raeber G. P.; Zisch A. H.; Tirelli N.; Hubbell J. A. Adv. Mater. 2003, 15, 888. [Google Scholar]
- Ruoslahti E.; Pierschbacher M. D. Science 1987, 238, 491. [DOI] [PubMed] [Google Scholar]
- Burdick J. A.; Anseth K. S. Biomaterials 2002, 23, 4315. [DOI] [PubMed] [Google Scholar]
- Hern D. L.; Hubbell J. A. J. Biomed. Mater. Res. 1998, 39, 266. [DOI] [PubMed] [Google Scholar]
- Schmedlen K. H.; Masters K. S.; West J. L. Biomaterials 2002, 23, 4325. [DOI] [PubMed] [Google Scholar]
- Lutolf M. P.; Lauer-Fields J. L.; Schmoekel H. G.; Metters A. T.; Weber F. E.; Fields G. B.; Hubbell J. A. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutolf M. P.; Hubbell J. A. Nat. Biotechnol. 2005, 23, 47. [DOI] [PubMed] [Google Scholar]
- Mann B. K.; Gobin A. S.; Tsai A. T.; Schmedlen R. H.; West J. L. Biomaterials 2001, 22, 3045. [DOI] [PubMed] [Google Scholar]
- Lin C. C.; Anseth K. S. Adv. Funct. Mater. 2009, 19, 2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C. C.; Metters A. T.; Anseth K. S. Biomaterials 2009, 30, 4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayalia P.; Mooney D. J. Adv. Mater. 2009, 21, 3269. [DOI] [PubMed] [Google Scholar]
- Mann B. K.; Schmedlen R. H.; West J. L. Biomaterials 2001, 22, 439. [DOI] [PubMed] [Google Scholar]
- Zisch A. H.; Lutolf M. P.; Ehrbar M.; Raeber G. P.; Rizzi S. C.; Davies N.; Schmokel H.; Bezuidenhout D.; Djonov V.; Zilla P.; Hubbell J. A. Faseb Journal 2003, 17, 2260. [DOI] [PubMed] [Google Scholar]
- Aimetti A. A.; Tibbitt M. W.; Anseth K. S. Biomacromolecules 2009, 10, 1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H.; Moon J. J.; Miller J. S.; West J. L. Biomaterials 2007, 28, 3163. [DOI] [PubMed] [Google Scholar]
- Patch J. A.; Barron A. E. Curr. Opin. Chem. Biol. 2002, 6, 872. [DOI] [PubMed] [Google Scholar]
- Roberts M. J.; Bentley M. D.; Harris J. M. Adv. Drug Delivery Rev. 2002, 54, 459. [DOI] [PubMed] [Google Scholar]
- Gobin A. S.; West J. L. Faseb J. 2002, 16, 751. [DOI] [PubMed] [Google Scholar]
- DeLong S. A.; Moon J. J.; West J. L. Biomaterials 2005, 26, 3227. [DOI] [PubMed] [Google Scholar]
- Weber L. M.; Hayda K. N.; Haskins K.; Anseth K. S. Biomaterials 2007, 28, 3004. [DOI] [PubMed] [Google Scholar]
- Lin-Gibson S.; Jones R. L.; Washburn N. R.; Horkay F. Macromolecules 2005, 38, 2897. [Google Scholar]
- Lutolf M. P.; Tirelli N.; Cerritelli S.; Cavalli L.; Hubbell J. A. Bioconjugate Chem. 2001, 12, 1051. [DOI] [PubMed] [Google Scholar]
- Park Y.; Lutolf M. P.; Hubbell J. A.; Hunziker E. B.; Wong M. Tissue Eng. 2004, 10, 515. [DOI] [PubMed] [Google Scholar]
- Miller J. S.; Shen C. J.; Legant W. R.; Baranski J. D.; Blakely B. L.; Chen C. S.. Biomaterials 2010, 31, 3736− 3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbanks B. D.; Schwartz M. P.; Halevi A. E.; Nuttelman C. R.; Bowman C. N.; Anseth K. S. Adv. Mater. 2009, 21, 5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas C. N.; Anseth K. S. Macromolecules 2008, 41, 6019. [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Angew. Chem., Int. Ed. 2001, 40, 2004. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Sharpless K. B. Drug Discovery Today 2003, 8, 1128. [DOI] [PubMed] [Google Scholar]
- Hawker C. J.; Wooley K. L. Science 2005, 309, 1200. [DOI] [PubMed] [Google Scholar]
- Polizzotti B. D.; Fairbanks B. D.; Anseth K. S. Biomacromolecules 2008, 9, 1084. [DOI] [PubMed] [Google Scholar]
- Huisgen R. Angew. Chem., Int. Ed. 1963, 75, 604. [Google Scholar]
- Tornoe C. W.; Christensen C.; Meldal M. J. Org. Chem. 2002, 67, 3057. [DOI] [PubMed] [Google Scholar]
- Franke R.; Doll C.; Eichler J. Tetrahedron Lett. 2005, 46, 4479. [Google Scholar]
- Moses J. E.; Moorhouse A. D. Chem. Soc. Rev. 2007, 36, 1249. [DOI] [PubMed] [Google Scholar]
- Binder W. H.; Sachsenhofer R. Macromol. Rapid Commun. 2007, 28, 15. [Google Scholar]
- Bock V. D.; Perciaccante R.; Jansen T. P.; Hiemstra H.; van Maarseveen J. H. Org. Lett. 2006, 8, 919. [DOI] [PubMed] [Google Scholar]
- Jagasia R.; Holub J. M.; Bollinger M.; Kirshenbaum K.; Finn M. G. J. Org. Chem. 2009, 74, 2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish B.; Breitenkamp R. B.; Emrick T. J. Am. Chem. Soc. 2005, 127, 7404. [DOI] [PubMed] [Google Scholar]
- Engler A. C.; Lee H. I.; Hammond P. T. Angew. Chem., Int. Ed. 2009, 48, 9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. J. Am. Chem. Soc. 2004, 126, 15046. [DOI] [PubMed] [Google Scholar]
- Laughlin S. T.; Baskin J. M.; Amacher S. L.; Bertozzi C. R. Science 2008, 320, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codelli J. A.; Baskin J. M.; Agard N. J.; Berozzi C. R. J. Am. Chem. Soc. 2008, 130, 11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P. V.; Prescher J. A.; Sletten E. M.; Baskin J. M.; Miller I. A.; Agard N. J.; Lo A.; Bertozzi C. R. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyle C. E.; Lee T. Y.; Roper T. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 5301. [Google Scholar]
- Dondoni A. Angew. Chem., Int. Ed. 2008, 47, 8995. [DOI] [PubMed] [Google Scholar]
- DeForest C. A.; Polizzotti B. D.; Anseth K. S. Nat. Mater. 2009, 8, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H.; Fields G. B. Biopolymers 1996, 40, 399. [DOI] [PubMed] [Google Scholar]
- Malkoch M.; Vestberg R.; Gupta N.; Mespouille L.; Dubois P.; Mason A. F.; Hedrick J. L.; Liao Q.; Frank C. W.; Kingsbury K.; Hawker C. J. Chem. Commun. 2006, 2774. [DOI] [PubMed] [Google Scholar]
- Rydholm A. E.; Reddy S. K.; Anseth K. S.; Bowman C. N. Polymer 2007, 48, 4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T. Y.; Smith Z.; Reddy S. K.; Cramer N. B.; Bowman C. N. Macromolecules 2007, 40, 1466. [Google Scholar]
- Mather B. D.; Viswanathan K.; Miller K. M.; Long T. E. Prog. Polym. Sci. 2006, 31, 487. [Google Scholar]
- Cramer N. B.; Reddy S. K.; O’Brien A. K.; Bowman C. N. Macromolecules 2003, 36, 7964. [Google Scholar]
- Johnson P. M.; Reynolds T. B.; Stansbury J. W.; Bowman C. N. Polymer 2005, 46, 3300. [Google Scholar]
- DeLong S. A.; Gobin A. S.; West J. L. J. Controlled Release 2005, 109, 139. [DOI] [PubMed] [Google Scholar]
- Yu X. J.; Dillon G. P.; Bellamkonda R. V. Tissue Eng. 1999, 5, 291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.