

Abstract
Soon after its discovery, the attempts to develop anti-AIDS therapeutics focused on the retroviral protease (PR) — an enzyme used by lentiviruses to process the precursor polypeptide into mature viral proteins. An urgent need for the three-dimensional structure of PR to guide rational drug design prompted efforts to produce milligram quantities of this enzyme. However, only minute amounts of PR were present in the HIV-1 and HIV-2 viruses, and initial attempts to express this protein in bacteria were not successful. This review describes X-ray crystallographic studies of the retroviral proteases carried out at NCI-Frederick in the late 1980s and early 1990s and puts into perspective the crucial role that the total protein chemical synthesis played in unraveling the structure, mechanism of action, and inhibition of HIV-1 PR. Notably, the first fully correct structure of HIV-1 PR and the first cocrystal structure of its complex with an inhibitor (a substrate-derived, reduced isostere hexapeptide MVT-101) were determined using chemically synthesized protein. Most importantly, these sets of coordinates were made freely available to the research community and were used worldwide to solve X-ray structures of HIV-1 PR complexes with an array of inhibitors and set in motion a variety of theoretical studies. Publication of the structure of chemically synthesized HIV-1 PR complexed with MVT-101 preceded only by six years the approval of the first PR inhibitor as an anti-AIDS drug.
Keywords: Retroviral protease, Crystal structure, Chemical protein synthesis, HIV-1 PR, MVT-101, Substrate-based inhibitor
INTRODUCTION
The urgent need for finding clinical agents suitable for the treatment of AIDS has prompted unprecedented progress in both biochemical and structural studies on proteins essential to the retroviral life cycle. The observation that retroviruses encode a specific proteolytic enzyme vital for their replication indicated that a protease from HIV-1 would be a promising target for the design of anti-AIDS therapeutics. The genetic locus and primary structure of HIV-1 PR had been known since 1985; however, its molecular characterization was hampered by difficulties in obtaining enough purified protein (for review see Ref. 1). In September 1987, Pearl and Taylor published a hypothetical model of the fold of HIV-1 PR backbone. The model was based on the prediction that the viral PR corresponds to a single domain of the eukaryotic (“pepsin-like”) aspartic protease and functions in a dimeric form (Ref. 2 and references therein). This hypothesis certainly required confirmation by experimental results, and also a detailed knowledge of the tertiary structure, including side chain locations, was needed for design of specific inhibitors.
At the dawn of the structural biology era, the emerging new technologies, routinely used at present, were not yet fully developed. The NMR technique to determine three-dimensional structures of macromolecules was not yet in use. Very few groups had access to synchrotron facilities, and X-ray diffraction studies usually required quite a number of large crystals. In the case of a new structure, the phasing problem had to be overcome with the help of heavy atom derivatives. In the absence of crystallization robots, obtaining crystals and finding useful heavy-atom derivatives required a considerable amount of pure protein and were time-consuming. Nonetheless, the structure determination of about a 100 amino acid-long protein (124 and 99 for RSV and HIV-1 PR, respectively) containing cysteine was not a formidable task even 20 years ago. The main obstacle for the structural studies of PR to progress was the lack of adequate amounts of the protein. Recombinant methods and chemical synthesis of the large polypeptides found in proteins were in their infancy at that time, and the major part of material used for biophysical studies of proteins was extracted from cell cultures or organisms. In the case of HIV-1, it would require an accumulation of large volumes of a highly concentrated, dangerous pathogen. Scarcity of the protease present in HIV viruses and initial difficulties in cloning the enzyme in bacteria, prompted researchers to try chemical synthesis.3–6 However, obtaining milligram quantities of a folded homogenous protein of that size by the stepwise solid phase peptide synthesis7 posed a major challenge at that time. For these reasons, several groups turned to the closely related avian viruses, which produced ≈20 times more of their PR, and were not toxic to humans. The first crystal structure of retroviral PR was reported by Miller et al. for the enzyme from Rous sarcoma virus (RSV).8 A practically unlimited amount of protein supplied by Jonathan Leis from Case Western University allowed for obtaining crystals and their derivatives necessary for an unambiguous tracing of the polypeptide chain in the electron density maps. Shortly thereafter, a 3 Å resolution structure of recombinant HIV-1 PR was reported by the Merck group,9 which was the first one to succeed in large-scale purification and crystallization of the HIV-1 PR expressed in bacteria (see Ref. 1 and references therein). Although the structure revealed essential features of the catalytic apparatus and the presumed substrate binding cleft at the interface between the two subunits, it differed in important aspects from the X-ray structure of RSV PR. In addition, no side chain locations were publicly made available. Because of the importance of knowing the detailed atomic model of HIV protease for drug design, collaboration was initiated between the chemical protein synthesis group of Stephen Kent at the California Institute of Technology and the Crystallography Laboratory at NCI-Frederick, with an aim to solve the molecular structure of HIV-1 PR, both in an unliganded form and in a complex with substrate-based inhibitors.
THE CRYSTAL STRUCTURE OF RSV-PR
The first batch of the avian enzyme (ASLV-PR) was produced for crystallographic studies in 1985; however, the protein did not crystallize, despite the two year-long effort of two groups (Peter Strop, personal communication). Single crystals suitable for X-ray diffractions studies (Figure 1(a)) were obtained with protein purified from viral particles from an RSV Pr-C strain. Trigonal crystals containing two molecules of the protease in the crystallographic asymmetric unit were grown under conditions in which the protease remains active (pH 5.4).10 The structure was solved by the multiple isomorphous replacement method (MIR).8 Several heavy-atom derivatives were easily obtained by soaking the crystals in uranyl acetate, mercury, and platinum-containing compounds. The electron density map (Figure 1(b)) based on MIR phases from four derivatives showed clearly the molecular boundary of the dimer and several characteristic sequence features, including the active site. With the exception of eight residues in the flap region (see below), which were disordered in the crystal, whole polypeptide chains corresponding to the two monomers were defined by contiguous electron density.
Figure 1.
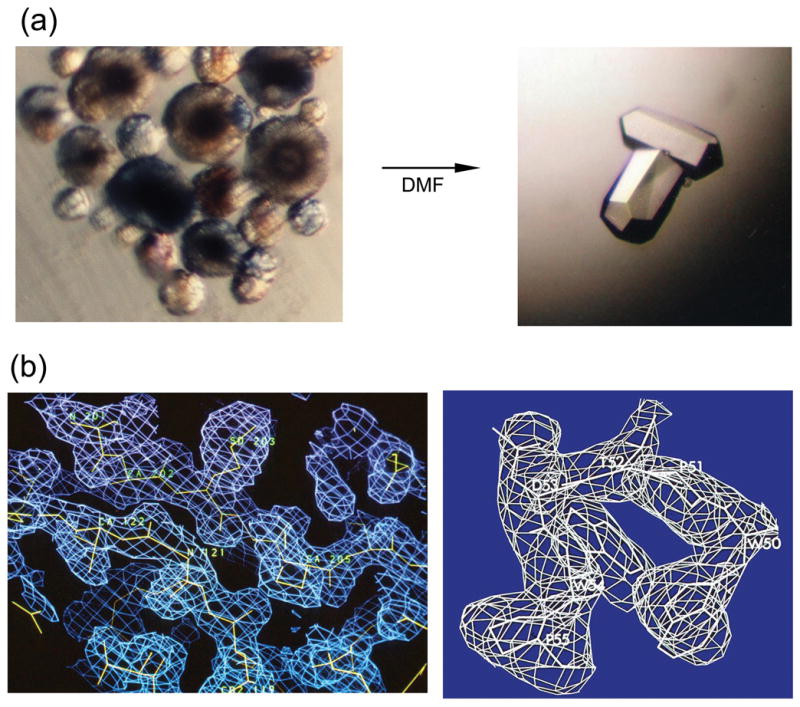
Determination of the RSV-PR crystal structure. (a) Crystallization: clusters of improper microcrystals, which grew from 10% ammonium sulfate; pH 5.4 and single crystals obtained upon the “magic” addition of dimethyl formamide (DMF). Crystals belong to the space group P3121 with the unit cell parameters a = b = 88.95 Å, c = 78.9 Å (b) Electron density maps (calculated using MIR phases improved by solvent flattening) are shown for the dimer interface region (left) and for the WPTDWP loop (right), which served as a starting point for sequence fitting. A combined lack of closure refinement of the best four derivatives (uranyl acetate, mercury acetate, methyl mercury acetate, and mersalyl) with anomalous scattering for the uranyl derivative included, gave a total figure of merit of 0.67 (>0.5 for the last resolution shell) for 3-Å data.
The crystal structure (Figure 2(a)) fully confirmed the predictions that the subunit of a retroviral PR homodimer corresponds to a single domain of the eukaryotic (“pepsin-like”) aspartic protease, so that the general topology of the molecule could be described by analogous nomenclature conventions.11 Two subunits interact to form a symmetric dimer. Each subunit donates the catalytic triad — Asp-Ser-Gly — to the active site that closely resembles the highly conserved active sites of monomeric, bilobal cellular proteases such as pepsin. Instead of one flap (a long flexible β-hairpin loop from the N-terminal domain) closing over the active site in pepsins, the homodimeric retroviral enzyme has two poorly ordered flaps. In fact, eight residues from the tips of each flap were not located after refinement of the structure at 2 Å resolution.12 The dimer interface — which corresponds to the inter-domain junction of pepsin-like proteases formed by six strands — is composed of the N- and C- termini from both monomers, which are intertwined to form a four-stranded antiparallel β-sheet. Surprisingly, the crystal structure of HIV-1 PR, which was reported a week later,9 showed differences in main chain connectivity and in the secondary structure in this region. The first five N-terminal residues were reported to be disordered and a helix which forms part of the highly conserved motif of aspartic proteases was missing, leading to a quite different intersubunit β-sheet topology in contrast to that found in the RSV PR structure. However, a model constructed by Weber et al., based on the initial RSV-PR coordinates, showed that the shorter HIV-1 PR polypeptide chain can have a similar core structure as the RSV enzyme.13
Figure 2.

Cartoon representations of the crystal structures discussed in the text. (a) Unliganded RSV-PR; PDB entry 2RSP. Note disordered ends of both flaps. (b) Unliganded HIV-1 PR obtained by total chemical synthesis; PDB entry 3HVP. Flaps are in “semi-open” conformation. (c) Aspartic protease from Rhizopus chinesis bound to a reduced isostere inhibitor; PDB entry 3APR. One flap closes in “flat” orientation over inhibitor. (d) synthetic HIV-1 PR bound to JG-365 inhibitor; PDB entry 7HVP. Both flaps close in “edge on” conformation over the inhibitor.
The RSV-PR was the first structure of a retroviral PR, and it was solved solely by MIR. With the exception of the chemically synthesized HIV-1 PR (see below), all subsequent structures of retroviral proteases were determined with the help of the existing models.
CRYSTAL STRUCTURES OF CHEMICALLY SYNTHESIZED HIV1-PR
Getting the things right: crystal structure of the free HIV1-PR
The amino acid sequence of chemically synthesized HIV-1 protease used for crystallographic studies (for an early review see Ref. 14) corresponded to the SF2 isolate, with the two Cys residues replaced by L-α-Amino-n-butyric acid (Aba) (see Figure 3(a)). The synthetic enzyme was prepared by Jens Schneider and displayed the same specific proteolytic activity, and had a turnover number similar to the enzyme derived from bacterial expression.15 Single crystals were grown in similar conditions and were isomorphous with those previously reported for the recombinant protein.9 The crystal structure was first solved by molecular replacement using a model based on the RSV PR crystal structure13 and was independently validated by the MIR method (Figure 3(c)). It is worth noting that useful heavy atom derivatives of HIV-1 PR crystals were difficult to obtain. Uranyl acetate (also lead nitrate used by Merck group for the same purpose) was bound in the active site located on the crystallographic two-fold axis and provided only limited phase information. Furthermore, the enzyme was highly active and autolysis, which occurred in the absence of an inhibitor, precluded formation of large, well diffracting crystals. The electron density map calculated using MIR phases based on two derivatives was of medium quality. Whereas most of the chain was found in the contiguous density, a number of breaks were present and chain tracing would have been very difficult in the absence of the information provided by the molecular replacement solution.
Figure 3.

Crystallization and MIR solution of synthetic HIV-1 PR. (a) A sample that made the difference. The initial sample of synthetic enzyme contained just 0.2 mg — not quite enough for extensive screening of the crystallization conditions — however, several small crystals were obtained using a modified protocol described by McKeever et al.47 On the news that synthetic material crystallized, within two weeks, Kent’s group set a precedent by producing ≈1mg of folded protein. (b) Single crystals of chemically synthesized HIV-1 PR. Tetragonal space group, unit cell parameters a = 50.2 Å, c = 106.6 Å. (c) MIR electron density maps based on two derivatives, uranyl acetate and K2PtCNS, showing (left) residues 1 to 10, and (right) residues 86 to 94, which were found in a helical conformation.
The final model at 2.8 Å resolution completely confirmed the conserved folding for retroviral proteases and their relationship with fungal and mammalian aspartic proteases.16 The HIV1-PR homodimer interface is composed of four well-ordered β strands from both the amino and carboxyl termini, and residues 86 to 94 have a helical conformation — in agreement with the RSV PR structure and the predicted models.2,13 Both retroviral enzyme molecules contain a mixed β-pleated sheet (Figure 2) of two intertwined motifs of four strands related by a pseudo dyad, the active site triad located on the wide loop and the buried “fireman’s grip” between the two identical subunits. Shorter than in the RSV-PR flaps, the residues 43–56 are well ordered but seem to be immobilized away from the active site by the intermolecular contacts in the crystal lattice. Importantly, the arrangement of N- and C-terminal strands, which interact to produce an active dimer, observed in both enzymes, indicated the intermolecular mechanism for the release of the PR from the precursor protein. These results also showed for the first time that a chemically synthesized protein can fold correctly without being exposed to a ribosome and can crystallize isomorphously with the protein obtained from living cells.
Rao et al.17 compared the structural and evolutionary relationships between the retroviral RSV and HIV-1 protease homodimers against six bilobal fungal and mammalian aspartic proteases. In this study, the conserved parts corresponded to the structural regions related by the twofold axis present in both sets. The retroviral PR monomer exhibits the same degree of structural equivalence to the N- and C- domains of the eukaryotic enzymes. Two of the three highly conserved amino acid stretches of the retroviral enzyme in the Ψ loop–α helix motif bear moderate sequence similarity with the corresponding eukaryotic enzymes. The third one in the flap region bears no such sequence similarity. The interdomain antiparallel β sheet in the cellular enzymes differs from the intersubunit β sheet in the number, arrangement, and directionality of the strands. This study strongly supports the evolutionary relatedness of the eukaryotic and retroviral aspartic PRs to a common ancestral domain or subunit. Further, the pseudo-dyad in retroviral PR monomers was shown to be a consequence of topology and folding, and does not seem to have much evolutionary significance.18
Crystallographic studies of chemically synthesized HIV-1 PR complexed with the reduced isostere hexapeptide inhibitor MVT-101
From the structure of the unliganded enzyme, it was not possible to deduce the exact mode of substrate binding. Particularly intriguing was the role of the two flaps present in the homodimeric enzyme molecules. In the cell-encoded, pepsin-like aspartic proteases the single flap closes down over substrate, desolvates it, and contributes to the formation of the specificity pockets involved in substrate recognition (for a review see Ref. 19). In the available crystals of unliganded HIV-1 PR, the crystallographic asymmetric unit contained only one monomer, and the homodimer thus had perfect twofold symmetry. It was anticipated that binding of the polar protein substrate may introduce asymmetry in the retroviral enzyme itself. To address these considerations, we sought to cocrystallize HIV-1 PR with a number of substrate-based inhibitors.20 For that purpose, ample amounts of HIV1-PR were produced by total chemical synthesis at Caltech using a vastly improved protocol. The first compound to study was a design based on the sequence of the CA/NC cleavage site of the viral gag-pol polyprotein, in which the scissile peptide bond had been replaced by a reduced analog. The hexapeptide inhibitor MVT-101 with the sequence N-acetyl-Thr-Ile-Nle-Ψ[CH2-NH]-Nle-Gln-Arg·amide (Nle, norleucine) was synthesized in the laboratory of Garland Marshall at the Washington University School of Medicine. A mixture of the synthetic enzyme and a 10-fold molar excess of the inhibitor at pH 5.4 was used for screening the crystallization conditions. Orthorhombic cocrystals (Figure 4(a)) contained a protein dimer with one bound inhibitor molecule in the asymmetric unit. The structure was solved by molecular replacement, using the structure of unliganded HIV-1 PR as the model and was initially reported at 2.3 Å resolution.21 Despite the symmetric nature of the unliganded enzyme, the asymmetric inhibitor was fitted to the difference electron density map in only one orientation. However, further refinement of data collected at 2.0 Å resolution revealed biderectionality of this hexapeptide in the crystal lattice.22 In fact, following the static-disorder refinement, the orientation of the hexapeptide in a direction opposite to the one initially reported was assigned 70% of occupancy, and this orientation of the MVT-101 was recently reported as a unique one.23 Modeling of MVT-101 in two alternative orientations allowed for corrections of several inaccuracies in the original model. Nonetheless, the first structure revealed the mode of inhibitor binding, confirmed later by other structures of complexes with a variety of inhibitors.24–26
Figure 4.

Crystallographic studies of MVT-101:HIV-1 PR complex. (a) Crystals grew at room temperature from 60% ammonium sulfate at the space group P212121; unit cell parameters a = 51.7, b = 59.2 Å, c = 62.45 Å. (b) Two alternative orientations of MVT-101 hexapeptide; the major orientation (70% occupancy) is shown in red. Upper panel shows initial 2.3 Å (|Fo| - |Fc|) electron density map contoured at 1.25 σ level. Lower panel shows (2 |Fo| – |Fc|) electron density map, contoured at the 1.0 σ level, calculated after the final refinement using data extending to 2 Å resolution. (c) Hydrogen bond interactions of the MVT-101 backbone (green) in the active site.
The hexapeptide binds in an extended conformation, but its backbone is slightly bent, so that main chain–main chain interactions occur with both flaps and the body of the enzyme. Six direct hydrogen bonds are formed between the main chain carbonyls and NH groups, which include Gly27, Asp29, and the carbonyl of Gly48 from both subunits (Figure (4c)). No change in relative orientation of the two subunits of the dimer is necessary to maintain these interactions with the bound peptide, which binds in a pseudo-symmetrical way. On the other hand, in comparison with the structure of the unliganded enzyme, the (Ψ, ϕ) pair of dihedral angles of Gly27 changes from (−72°, −82°) to (−92°, 15°) and (−102°, 19°) in subunits 1 and 2, respectively. As a result, the carbonyls of Gly27 in both subunits turn approximately 90° toward the inhibitor, allowing for the formation of hydrogen bonds with amide NHs from P2′ and P1 sites. Also, planes of the carboxyl groups of Asp29 and Asp29′ are rotated about 90°, relative to their positions in the free enzyme. As the flaps fold over the inhibitor, a peptide bond in the flap of one monomer turns 180°, providing an attractive interaction between the tips of the flaps.
The mode of inhibitor binding differs from that observed for pepsin-like aspartic proteases in several aspects. Whereas the single flap of the cell-encoded aspartyl proteases closes down “flat” over the scissile bond, i.e., with the plane of the reverse turn at the tip nearly parallel to the plane of the inhibitor peptide (see Figure 2), the two flaps of the HIV-1 PR both close over the substrate-derived inhibitor, “edge on” (i.e., at ≈90°) to the plane of the substrate polypeptide chain. Moreover, direct hydrogen bonds between the amide NH of peptide bonds near the tip of the flap to the substrate that were observed in pepsin-like proteases, in the case of dimeric retroviral enzyme, are donated from both flaps and are mediated by a specific, tetrahedrally coordinated internal water molecule (Wat301, originally numbered 511).
Contacts between the side chains of the inhibitor and protein residues define six or seven specific binding pockets along the active site clef; however, only the four central ones are well defined and they do not include solvent molecules, other than Wat301. Binding of the inhibitor introduces substantial changes in the protein backbone, which leads to decreasing of the active site cavity. The overall movement of the subunits can be described as a hinge motion by 1.7 Å, with the hinge axis located in the intersubunit β-sheet interface. As expected, the largest movement involves flap regions on both monomers, where the change of positions for the tips of both flaps is as much as 7 Å.
These coordinates derived from X-ray structural analysis of the chemically synthesized PR enzyme complexed with the substrate-derived inhibitor were immediately used for a variety of purposes. The shape of the active site facilitated a search of Cambridge Data Bank for nonpeptide compounds that could potentially provide a good fit to the enzyme.27 The results of the multiple copy simultaneous search (MCSS) method applied to the construction of peptide ligands in the binding site of HIV-1 PR were first tested with the MVT-101:HIV-1 PR complex structure.28 These coordinates were also used in modeling studies of inhibitor binding to HIV-2 PR,29 and to determine the X-ray structures of several complexes with the recombinant enzyme.30 The structure was used for the molecular mechanics analysis of inhibitor binding to HIV-1 PR,31,32 and for a number of molecular dynamics simulation studies.33–35
HIV-1 PR prepared by total chemical synthesis in the Kent lab at Caltech was also used to determine the structures of two other complexes with substrate-derived inhibitors containing hydroxyethylamine (JG-36536 provided by J. Green and D.H. Rich; University of Wisconsin) and hydroxyethylene (U-85548e37 provided by R.L. Herikson, A.G. Tomasselli and T.K. Sawer; The Upjohn Company) isosteres. These were the first structures of the enzyme with canonical examples of the two most important classes of inhibitors of PR. These data have provided a unique opportunity for rational drug design, based on detailed knowledge of the three-dimensional structure of the target molecule, but details of such studies are beyond the scope of this article.
PROTEIN SYNTHESIS WITHOUT BORDERS
A new synthetic technology — the chemical ligation method — was soon developed by Kent’s group, which not only allowed the routine production of proteins with high purity regardless of their size, but also permitted to introduce specific modifications of individual functional groups.38 This was achieved by chemical ligation of large unprotected peptide segments by means of unique, mutually reactive functionalities. The resulting ligated protein had a non-peptide bond at the site of reaction between two peptides. This creative innovation made possible the precise replacement of a single atom in the protein backbone, and brought the studies of structure-activity relationship to a completely new level. This method was applied to produce fully active HIV-1 PR (a native peptide bond was replaced by a thioester bond between residues Gly51–Gly52) and to prepare backbone-engineered HIV-1 PR analogues to study the mechanism of enzymatic catalysis (for review see Ref. 39).
The crystal structure of ligated all-D HIV-1 PR
One of the first proteins prepared by the chemical ligation method was “mirror image” HIV-1 PR composed of D-amino acids.40 This was done to show that a polypeptide composed entirely of D-amino acids (and glycines) is able to properly fold to form a functional molecule and to investigate the chiral specificity of the peptide substrate. Also, co-crystals of both enantiomers of the enzyme, if obtained in the centrosymmetric space group, would provide very high-quality data for structural studies and render it amenable for easy solution of the phase problem. The D-enantiomer of HIV-1 PR prepared by chemical ligation, ([COS]51-52)2HIV-1 PR), had the same chemical specificity as HIV-1 protease, but displayed reciprocal chiral specificity, i.e., the D-HIV-1 PR cleaved D-peptide substrates but would not act on L-peptide substrates. The same rule applied toward inhibitors.
Unfortunately, a racemic mixture of HIV-1 PR did not crystallize. On the other hand, co-crystals of the D-protease with a D-hexapeptide MVT-101 inhibitor were obtained (see Figure 5(a)).39,41 The structure was solved by molecular replacement, using the coordinates of protein dimer from the structure of the complex of native backbone L-HIV-1 PR and MVT101 inhibitor. The structure was refined to an R-factor of 0.188 in the resolution range of 10–2.5 Å, following the same refinement protocol as used for the L-complex. A comparison of the structure with that of the natural-backbone synthetic L-amino acid enzyme showed that the two molecules were in all respects the mirror images of each other, including the centers of asymmetry not directly determined by the chirality of Cα atoms in the polypeptide backbone. These results vividly demonstrated that no chiral influences other than those inherent to the covalent structure of a polypeptide chain are required for correct three-dimensional folding to form a protein structure.
Figure 5.

Crystal structure of the D-enantiomer of backbone engineered HIV-1 PR prepared by total chemical synthesis complexed to D-MVT101 inhibitor. (a) Cocrystals were obtained from 50% ammonium sulfate, pH 5.4, in the space group P212121 a = 67.5, b = 92.8, c = 29.4 Å. There were two monomers of protein and one inhibitor in the crystallographic asymmetric unit. (b) Stereo view of Cα tracing of D HIV-1 PR. Bound D MVT-101 inhibitor is shown as green sticks. Coordinates were deposited to HIV Structural Database;48 accession code: NCI2009.
Despite the overall similarity, the crystal structure of ([COS]51-52)2HIV-1 PR:MVT-101 complex revealed some important differences to the native backbone enzyme in the flap region. Instead of a single tetrahedrally coordinated water molecule, two distinct water molecules were observed to mediate hydrogen bonds between the Gly49-Ile50 amide bond and inhibitor P2 and P1′ carbonyl groups. These two water molecules were characterized by high thermal vibration factors and only partial (≈50%) occupancy. In addition, whereas the N–Cα dihedral angle (ϕ) for Ile50 was observed to be −65° in both subunits in the original L-HIV-1 PR:MVT-101 complex,21,22 in the structure of ([COS]51-52)2HIV-1 PR:MVT-101 complex, the Ile50 N–Cα dihedral angle was −110° in subunit 1 and −95° in subunit 2. These changes in the flap geometry were most probably induced by the replacement of the Gly52 NH moiety by the sulfur atom of the thioester isostere, which affected the Gly49-Ile50 N-H vector and thus water 301 binding. This was the first observation to cast doubt on the role of “crystallographic” water 301 in HIV-1 PR enzymatic activity and provided support for the hypothesis that the retroviral enzyme may make use of only one flap.41–43 Detailed analysis of the flap structures in three complexes of HIV-1 PR, prepared by total chemical synthesis, with inhibitors designed to mimic different states on the reaction coordinate of peptide bond hydrolysis was recently investigated by pulse-EPR spectroscopy by Torbeev et al.44
PERSPECTIVE
The importance of total chemical protein synthesis for the field of protein science has been widely recognized. However, its potential for biological research is best illustrated by the impact on the structural and functional studies of the HIV-1 PR. As described here, chemically synthesized protein was used for the determination of the first correct structure of the free HIV-1 PR enzyme and the first structure of the HIV-1 PR complex with an inhibitor, as well as of two other important complexes. The coordinates of the synthetic HIV-1 PR:MVT-101 complex were deposited to Protein Data Bank in April 1990, and for the two most critical years were the only ones available free to researchers working worldwide on the design of specific PR inhibitors. By 1994, papers reporting the structure of chemically synthesized HIV-1 PR16 and its complex with the MVT-101 inhibitor21 were cited 409 and 268 times, respectively—reflecting wide usage of these coordinates for solving countless other structures of complexes of recombinant PR with a variety of inhibitors, as well as for a number of theoretical studies. Ligated D- HIV-1 PR obtained by total chemical synthesis was used to explore the world of D-proteins, for the first time experimentally illustrating the reciprocal chiral specificity of mirror image enzyme molecules.
These early successes of an application of total chemical synthesis were, however, just a prelude to a completely novel approach to study physico-chemical and biological properties of macromolecules. Most notably, by providing the means of producing protein analogs by replacing a single backbone atom at any desired location, the new chemical methods invented by Kent and his colleagues opened endless possibilities to investigate the action of proteins. The significance of this breakthrough has recently become evident by its role in the studies of the mechanism of HIV-1 PR catalysis. Backbone-engineered analogs of HIV-1 PR were used to investigate the contribution of flap-substrate hydrogen bonds to the enzymatic activity of the retroviral enzyme and to obtain atomic-resolution structures of HIV-1 PR with inhibitors that provided invaluable insights into the catalytic apparatus.23,45,46 These insights are currently being used to design novel classes of inhibitors, which would be able to prevent emergence of mutated drug-resistant viruses. These pioneering studies have now been carried on for more than 15 years and will likely benefit efforts to stop AIDS epidemics for many years to come.
Acknowledgments
We are grateful to Stephen Kent for his inspiration, advice and discussions through the period of this fruitful collaboration. The success of the NCI team would not have been possible without the truly heroic efforts of Jens Schneider and other members of Kent’s group who in 1989 within a period of just two weeks prepared a milligram of pure chemically synthesized HIV-1 PR for crystallographic studies. I would like to thank Stephen Kent, Garland Marshall, and Alex Wlodawer for trusting the precious material in my hands. I also thank Mohana Rao for over 20 years of collaboration and friendship; Robert Harisson and Osnat Herzberg for encouragement and help during work on the MIR solution of RSV PR; and Steven Sheriff for help with the static-disorder refinement of the MVT-101 inhibitor. I am indebted to David Davies for supporting my scientific career at NIH and for invaluable advice during the preparation of this manuscript. This project was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Reference List
- 1.Blundell TL, Lapatto R, Wilderspin AF, Hemmings AM, Hobart PM, Danley DE, Whittle PJ. Trends in Biochemical Sciences. 1990;15:425–430. doi: 10.1016/0968-0004(90)90280-o. [DOI] [PubMed] [Google Scholar]
- 2.Pearl LH, Taylor WR. Nature. 1987;329:351–354. doi: 10.1038/329351a0. [DOI] [PubMed] [Google Scholar]
- 3.Kent SB. Annual Review of Biochemistry. 1988;57:957–989. doi: 10.1146/annurev.bi.57.070188.004521. [DOI] [PubMed] [Google Scholar]
- 4.Kent SB, Schneider J, Selk C, Chen Q. Chemical Synthesis Approach to the Molecular Biology of Retroviral Proteinases: Structure-Function Studies of the HIV-1 and HIV-2 Enzymes and their Substrates. In: Krausslich H, Oroszlan S, Wimmer W, editors. Current Communications in Molecular Biology: Viral Proteinases as Targets for Chemotherapy. Cold Spring Harbor Press; Cold Spring Harbor: 1989. pp. 223–230. [Google Scholar]
- 5.Copeland TD, Oroszlan S. Gene Anal Tech. 1988;5:109–115. doi: 10.1016/0735-0651(88)90010-6. [DOI] [PubMed] [Google Scholar]
- 6.Nutt RF, Brady SF, Darke PL, Ciccarone TM, Colton CD, Nutt EM, Rodkey JA, Bennett CD, Waxman LH, Sigal IS. Proc Natl Acad Sci USA. 1988;85:7129–7133. doi: 10.1073/pnas.85.19.7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merrifield RB. Journal of the American Chemical Society. 1963;85:2149–2154. [Google Scholar]
- 8.Miller M, Jaskólski M, Rao JKM, Leis J, Wlodawer A. Nature. 1989;337:576–579. doi: 10.1038/337576a0. [DOI] [PubMed] [Google Scholar]
- 9.Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, Herber WK, Sigal IS, Darke PL, Springer JP. Nature. 1989;337:615–620. doi: 10.1038/337615a0. [DOI] [PubMed] [Google Scholar]
- 10.Miller M, Leis J, Wlodawer A. Journal of Molecular Biology. 1988;204:211–212. doi: 10.1016/0022-2836(88)90610-9. [DOI] [PubMed] [Google Scholar]
- 11.Blundell TL, Jenkins J, Pearl L, Sewell T, Pedersen V. In: Aspartic Proteases and Their Inhibitors. Kostka V, editor. deGruyter; Berlin: 1985. pp. 151–161. [Google Scholar]
- 12.Jaskólski M, Miller M, Rao JKM, Leis J, Wlodawer A. Biochemistry. 1990;29:5889–5898. doi: 10.1021/bi00477a002. [DOI] [PubMed] [Google Scholar]
- 13.Weber IT, Miller M, Jaskólski M, Leis J, Skalka AM, Wlodawer A. Science. 1989;243:928–931. doi: 10.1126/science.2537531. [DOI] [PubMed] [Google Scholar]
- 14.Miller M, Swain AL, Jaskolski M, Sathyanarayana BK, Marshall GR, Rich DH, Kent SBH, Wlodawer A. X-ray analysis of HIV-1 protease and its complexes with inhibitors. In: Pearl L, editor. Retroviral Proteases: Control of Maturation and Morphogenesis. MacMillan Press; York, England: 1990. pp. 93–106. [Google Scholar]
- 15.Schneider J, Kent SB. Cell. 1988;54:363–368. doi: 10.1016/0092-8674(88)90199-7. [DOI] [PubMed] [Google Scholar]
- 16.Wlodawer A, Miller M, Jaskólski M, Sathyanarayana BK, Baldwin E, Weber IT, Selk LM, Clawson L, Schneider J, Kent SBH. Science. 1989;245:616–621. doi: 10.1126/science.2548279. [DOI] [PubMed] [Google Scholar]
- 17.Rao JKM, Erickson JW, Wlodawer A. Biochemistry. 1991;30:4663–4671. doi: 10.1021/bi00233a005. [DOI] [PubMed] [Google Scholar]
- 18.Rao JKM, Wlodawer A. Federation of European Biochemical Societies Letters. 1990;260:201–205. [Google Scholar]
- 19.Davies DR. Annual Review of Biophysics and Biophysical Chemistry. 1990;19:189–215. doi: 10.1146/annurev.bb.19.060190.001201. [DOI] [PubMed] [Google Scholar]
- 20.Rich DH, Green J, Toth MV, Marshall GR, Kent SB. Journal of Medicinal Chemistry. 1990;33:1285–1288. doi: 10.1021/jm00167a003. [DOI] [PubMed] [Google Scholar]
- 21.Miller M, Schneider J, Sathyanarayana BK, Toth MV, Marshall GR, Clawson L, Selk L, Kent SBH, Wlodawer A. Science. 1989;246:1149–1152. doi: 10.1126/science.2686029. [DOI] [PubMed] [Google Scholar]
- 22.Miller M, Geller M, Gribskov M, Kent SB. Proteins: Structure, Function, and Genetics. 1997;27:184–194. doi: 10.1002/(sici)1097-0134(199702)27:2<184::aid-prot4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 23.Johnson EC, Malito E, Shen Y, Pentelute B, Rich D, Florian J, Tang WJ, Kent SB. J Mol Biol. 2007;373:573–586. doi: 10.1016/j.jmb.2007.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzgerald PMD. Current Opinion in Structural Biology. 1993;3:868–874. [Google Scholar]
- 25.Appelt K. Crystal structures of HIV-1 protease-inhibitor complexes. In: Anderson PS, Kenyon GL, Marshall GR, editors. Perspectives in Drug Discovery Design. ESCOM Science Publishers B.V; Leiden, The Netherlands: 1993. pp. 23–48. [Google Scholar]
- 26.Wlodawer A, Erickson JW. Annual Review of Biochemistry. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]
- 27.DesJarlais RL, Seibel GL, Kuntz ID, Furth PS, Alvarez JC, Ortiz de Montellano PR, DeCamp DL, Babe LM, Craik CS. Proceedings of the National Academy of Sciences USA. 1990;87:6644–6648. doi: 10.1073/pnas.87.17.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caflisch A, Miranker A, Karplus M. J Med Chem. 1993;36:2142–2167. doi: 10.1021/jm00067a013. [DOI] [PubMed] [Google Scholar]
- 29.Gustchina A, Weber IT, Wlodawer A. Molecular modeling of HIV-2 protease. In: Dunn BM, editor. Structure and Function of the Aspartic Proteinases. Plenum Press; New York: 1991. pp. 549–553. [Google Scholar]
- 30.Thanki N, Rao JKM, Foundling SI, Howe WJ, Moon JB, Hui JO, Tomasselli AG, Heinrikson RL, Thaisrivongs S, Wlodawer A. Protein Sci. 1992;1:1061–1072. doi: 10.1002/pro.5560010811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sansom CE, Wu J, Weber IT. Protein Engineering. 1992;5:659–667. doi: 10.1093/protein/5.7.659. [DOI] [PubMed] [Google Scholar]
- 32.Gustchina A, Sansom C, Prevost M, Richelle J, Wodak SY, Wlodawer A, Weber IT. Protein Engineering. 1994;7:309–317. doi: 10.1093/protein/7.3.309. [DOI] [PubMed] [Google Scholar]
- 33.Harte WE, Jr, Beveridge DL. Journal of the American Chemical Society. 1993;115:3883–3886. [Google Scholar]
- 34.Harte WE, Jr, Beveridge DL. Methods in Enzymology. 1994;241:178–95. 178–195. doi: 10.1016/0076-6879(94)41065-8. [DOI] [PubMed] [Google Scholar]
- 35.Geller M, Miller M, Swanson SM, Maizel J. Proteins: Structure, Function, and Genetics. 1997;27:195–203. [PubMed] [Google Scholar]
- 36.Swain AL, Miller MM, Green J, Rich DH, Schneider J, Kent SB, Wlodawer A. Proceedings of the National Academy of Sciences USA. 1990;87:8805–8809. doi: 10.1073/pnas.87.22.8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaskólski M, Tomasselli AG, Sawyer TK, Staples DG, Heinrikson RL, Schneider J, Kent SB, Wlodawer A. Biochemistry. 1991;30:1600–1609. doi: 10.1021/bi00220a023. [DOI] [PubMed] [Google Scholar]
- 38.Schnolzer M, Kent SB. Science. 1992;256:221–225. doi: 10.1126/science.1566069. [DOI] [PubMed] [Google Scholar]
- 39.Kent SB, Baca M, Elder J, Miller M, Milton R, Milton S, Rao JK, Schnolzer M. Breaking the shackles of the genetic code: engineering retroviral proteases through total chemical synthesis. In: Takahashi K, editor. Aspartic Proteinases. Vol. 362. Plenum Press; New York: 1995. pp. 425–438. [DOI] [PubMed] [Google Scholar]
- 40.Milton RC, Milton SC, Kent SB. Science. 1992;256:1445–1448. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- 41.Miller M, Baca M, Rao JKM, Kent SBH. J Mol Struct (Theochem) 1998;423:137–152. [Google Scholar]
- 42.Baca M, Kent SB. Proceedings of the National Academy of Sciences USA. 1993;90:11638–11642. doi: 10.1073/pnas.90.24.11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baca M, Kent SBH. Tetrahedron. 2000;56:9503–9513. [Google Scholar]
- 44.Torbeev VY, Raghuraman H, Mandal K, Senapati S, Perozo E, Kent SB. J Am Chem Soc. 2009;131:884–885. doi: 10.1021/ja806526z. [DOI] [PubMed] [Google Scholar]
- 45.Torbeev VY, Kent SB. Angew Chem Int Ed Engl. 2007;46:1667–1670. doi: 10.1002/anie.200604087. [DOI] [PubMed] [Google Scholar]
- 46.Torbeev VY, Mandal K, Terechko VA, Kent SB. Bioorg Med Chem Lett. 2008;18:4554–4557. doi: 10.1016/j.bmcl.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 47.McKeever BM, Navia MA, Fitzgerald PM, Springer JP, Leu CT, Heimbach JC, Herbert WK, Sigal IS, Darke PL. J Biol Chem. 1989;264:1919–1921. [PubMed] [Google Scholar]
- 48.Vondrasek J, Wlodawer A. Proteins. 2002;49:429–431. doi: 10.1002/prot.10246. [DOI] [PubMed] [Google Scholar]