

Abstract
The success of high-throughput proteomics hinges on the ability of computational methods to identify peptides from tandem mass spectra (MS/MS). However, a common limitation of most peptide identification approaches is the nearly ubiquitous assumption that each MS/MS spectrum is generated from a single peptide. We propose a new computational approach for the identification of mixture spectra generated from more than one peptide. Capitalizing on the growing availability of large libraries of single-peptide spectra (spectral libraries), our quantitative approach is able to identify up to 98% of all mixture spectra from equally abundant peptides and automatically adjust to varying abundance ratios of up to 10:1. Furthermore, we show how theoretical bounds on spectral similarity avoid the need to compare each experimental spectrum against all possible combinations of candidate peptides (achieving speedups of over five orders of magnitude) and demonstrate that mixture-spectra can be identified in a matter of seconds against proteome-scale spectral libraries. Although our approach was developed for and is demonstrated on peptide spectra, we argue that the generality of the methods allows for their direct application to other types of spectral libraries and mixture spectra.
The success of tandem MS (MS/MS1) approaches to peptide identification is partly due to advances in computational techniques allowing for the reliable interpretation of MS/MS spectra. Mainstream computational techniques mainly fall into two categories: database search approaches that score each spectrum against peptides in a sequence database (1–4) or de novo techniques that directly reconstruct the peptide sequence from each spectrum (5–8). The combination of these methods with advances in high-throughput MS/MS have promoted the accelerated growth of spectral libraries, collections of peptide MS/MS spectra the identification of which were validated by accepted statistical methods (9, 10) and often also manually confirmed by mass spectrometry experts. The similar concept of spectral archives was also recently proposed to denote spectral libraries including “interesting” nonidentified spectra (11) (i.e. recurring spectra with good de novo reconstructions but no database match). The growing availability of these large collections of MS/MS spectra has reignited the development of alternative peptide identification approaches based on spectral matching (12–14) and alignment (15–17) algorithms.
However, mainstream approaches were developed under the (often unstated) assumption that each MS/MS spectrum is generated from a single peptide. Although chromatographic procedures greatly contribute to making this a reasonable assumption, there are several situations where it is difficult or even impossible to separate pairs of peptides. Examples include certain permutations of the peptide sequence or post-translational modifications (see (18) for examples of co-eluting histone modification variants). In addition, innovative experimental setups have demonstrated the potential for increased throughput in peptide identification using mixture spectra; examples include data-independent acquisition (19) ion-mobility MS (20), and MSE strategies (21).
To alleviate the algorithmic bottleneck in such scenarios, we describe a computational approach, M-SPLIT (mixture-spectrum partitioning using library of identified tandem mass spectra), that is able to reliably and efficiently identify peptides from mixture spectra, which are generated from a pair of peptides. In brief, a mixture spectrum is modeled as linear combination of two single-peptide spectra, and peptide identification is done by searching against a spectral library. We show that efficient filtration and accurate branch-and-bound strategies can be used to avoid the huge computational cost of searching all possible pairs. Thus equipped, our approach is able to identify the correct matches by considering only a minuscule fraction of all possible matches. Beyond potentially enhancing the identification capabilities of current MS/MS acquisition setups, we argue that the availability of methods to reliably identify MS/MS spectra from mixtures of peptides could enable the collection of MS/MS data using accelerated chromatography setups to obtain the same or better peptide identification results in a fraction of the experimental time currently required for exhaustive peptide separation.
EXPERIMENTAL PROCEDURES
Problem Formulation
A mixture spectrum is defined as an MS/MS spectrum from two different peptides, and a spectral library is a collection of identified MS/MS spectra. Analogous to the identification of MS/MS spectra by comparison against a database of known protein sequences, our goal is to identify mixture spectra by comparison against a spectral library. More formally, we modeled a mixture spectrum M as M = A + αB, where A and B are MS/MS spectra from two different peptides and α, the mixture coefficient, indicates their relative abundance. Without loss of generality, we assume that A and B are scaled to Euclidean norm 1 and that 0 ≤ α ≤ 1 (i.e. A always corresponds to the higher abundance peptide). We can now formulate the following computational problem:
Mixture Spectrum Identification Problem (MSIP):
Input: A putative mixture spectrum M and a spectral library L
Output: A constant 0 ≤ α ≤ 1 and pair of spectra A, B ϵ L, maximizing similarity (M, A + αB)
Although there are several ways to define similarity between two peptide spectra (12, 14, 15, 22), the normalized dot product or cosine2 measure of spectral similarity is widely accepted to be robust and makes no special assumptions concerning peptide mass spectra (14). Moreover, as we show below and in the supplemental materials, cosine similarity has a number of useful mathematical properties that allow us to derive theoretical bounds to guide our approach.
Simulation of Mixture Spectra
Because there are currently no publicly available data with validated identifications of mixture MS/MS spectra, we created a dataset of simulated mixture spectra to develop and benchmark our approach. To this end, we used the human MS/MS spectral library from the National Institute of Standards and Technology (version 6/06) and grouped the spectra according to their identified peptide. This resulted in 27,966 groups in the library, each containing two or more spectra belonging to the same peptide. The spectral library was then divided into two sets: 1) a set X, which has exactly one spectrum per peptide, used to create the simulated mixture spectra and 2) a spectral library L containing all the remaining spectra, used for searching. All spectra in the library are first scaled to norm 1; because in a mixture the two peptides will most likely be present at different abundances, mixture spectra were created by randomly selecting two spectra AX and BX from X and linearly combining them using a predefined mixture coefficient α. In other words, a mixture spectrum is of the form M = AX + αBX, where M represents a simulated mixture spectrum and AX and BX represent two single-peptide spectra, 0 ≤ α ≤ 1. Below we benchmark our approach for αϵ{0.1, 0.2, 0.5, 1}.
Main Method
Although the MSIP formulation is simple, the rapidly growing size of target spectral libraries (already on the order of 105-106 spectra) makes searching all possible pairs of spectra a prohibitive approach (1011 comparisons per query spectrum). We note that although one can prefilter the target spectral library to consider only combinations of spectra with the same precursor mass as the query spectrum, such an approach would currently not provide a realistic estimate of performance on quickly growing proteome-scale spectral libraries. By not enforcing any parent mass filters on our performance estimates, we argue that the approach proposed here should seamlessly scale to much larger spectral libraries and be directly applicable to complex searches (e.g. metaproteomics studies). We propose two ways to avoid the quadratic penalty of searching all pairs. First, we use an efficient projected-cosine filter to eliminate a large fraction of spectra in the library. After filtering, we use a branch-and-bound search strategy to find the best-matching pairs by considering only a subset of all possible pairs. The overall strategy is detailed in Fig. 1.
Fig. 1.

Pseudocode for matching strategy.
Filtering with Projected-Cosine
Although cosine is generally a good measure of spectrum similarity, a mixture spectrum M derived from peptides A and B may have limited similarity to the corresponding single-peptide spectra; e.g. the presence of B in the mixture results in many unmatched peaks between M and A. We address this with a projected-cosine similarity, a modified cosine function that only considers a peak in M if the corresponding peak in A is not zero. More precisely, for two vectors A and M, the projection of M on A (M(p)A) is defined as:
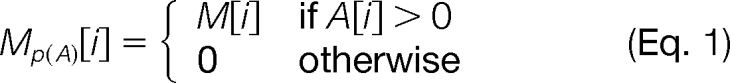 |
The projected cosine between M and A is then simply the cosine of the M(p)A and A:
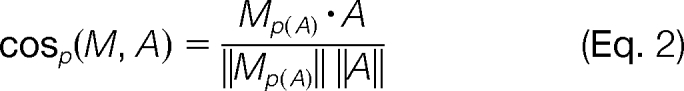 |
Given a spectrum M, the filtering step consists of computing the projected-cosine similarity between M and all spectra in L and retaining the top most similar matches. The filtering efficiency of projected-cosine similarity is determined by the highest (i.e. worst) rank of a correct match of M to the library L. Note that a correct match in L has the same peptide as M; single-peptide spectra have one correct match, and mixture spectra have two correct matches. As shown in Fig. 2a, the resulting ranks of correct matches indicate that projected-cosine is an efficient filter that, in most cases, retains the correct matches at ranks less than 500 in a library of approximately 27,966 spectra. In fact, for 95% of cases, the correct pair of peptides in a mixture spectrum M can be identified by considering only the top 100 library spectra with highest projected cosine similarity to M (for α ≥ 0.2).
Fig. 2.

Effectiveness of filtering and branch-and-bound strategies. a, cumulative distributions of maximum rank for correct matches to the spectral library. Spectra in the library are first sorted according to decreasing projected-cosine similarity to the mixture spectrum (library containing 27,966 spectra). The rank of correct matches are then determined. Correct matches are spectra identified as one of the peptides in the mixture. Because each mixture spectrum has two correct matches (i.e. it is generated from two peptides), we take the maximum (i.e. worst) rank of the two matches. b, effectiveness of the branch-and-bound strategy. To avoid considering all pairs of spectra in the library, we derive a branch-and-bound search strategy to eliminate a large fraction of all possible pairs. The number of evaluated pairs of spectra is shown. Because the total number of possible pairs is 3.9 × 108 and our approach never evaluates more than 15,000 pairs, this self-adjusting strategy achieves speedups of at least 2 × 104. c, combining the projected-cosine filter (a) with branch-and-bound search (b). We first filter the spectral library with projected-cosine and retain only the top 500 candidates; the branch-and-bound search strategy is then applied to further reduce number of pairs of spectra that needed to be evaluated. The curves for α = 0.1 clearly shows that projected-cosine is a effective pruning filter; note that prefiltering the library with cosine results in the evaluation of more pairs of spectra. The combined filters typically achieve speedups of approximately 6 orders of magnitude (≈(3.9 × 108)/500 = 7.8 × 105 speedups).
Searching with Branch-and-Bound
To better describe the concepts behind the branch and bound search strategy, let us assume for the moment that a mixture spectrum M is obtained from two single-peptide spectra with same abundance (i.e. α = 1; see supplemental materials for analysis when α < 1). Therefore, for any pair of spectra (A, B) we have the following relation for our objective function:
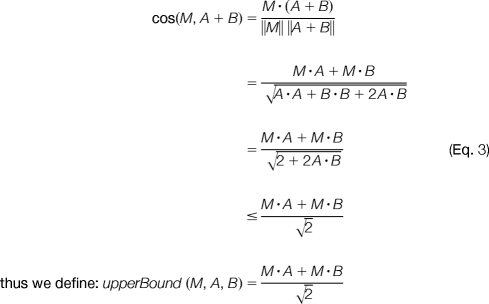 |
Assume that at certain a stage of our search, the best solution we have seen so far is: A* + B* and, without loss of generality, let us also assume cos(M, A*) ≥ cos(M, B*). By the above equations, we do not need to pair A* with any spectrum C such that upperBound(M, A*, C) < cos(M, A* + B*) because upperBound(M, A*, C) is never less than cos(M, A* + C). Moreover, a spectrum D with cos(M, D) ≤ cos(M, C) necessarily implies that upperBound(M, A* + D), < upperBound(M, A* + C) thus implying that the pair (A, D) can be excluded from consideration. This leads to the following search strategy: 1) sort spectra in the library according to their cosine similarity to the query spectrum M; 2) set A to the spectrum with highest cos(M, A) in the library; 3) pair A with remaining spectra C ϵ L until we find a spectrum that has upperBound(M, A, C) < cos(M, A* + B*); 4) delete A from the library, and repeat from step 2.
We determine the efficiency of this method by counting the number of pairs that are evaluated before the algorithm terminates with the optimal answer. As shown in Fig. 2b, in most cases, we consider only hundreds to thousands of combinations, approximately 5 orders of magnitude less than the total number of possible pairs (≈3.9 × 108). To take advantage of both the projected-cosine filter and the branch-and-bound strategy, we first filter the library with projected-cosine to retain only the top 500 candidates and then apply the branch-and-bound strategy to limit the number of evaluated pairs. As shown in Fig. 2c, only a few hundred pairs of spectra need to considered before M-SPLIT finds the optimal answer. We also note that projected-cosine is a better filter than cosine; as shown in Fig. 2c for α = 0.1, prefiltering the library with cosine results in more pairs of spectra being matched to each query spectrum (yellow line). A full comparison of the two filters is provided in supplemental materials.
Estimating the Mixture Coefficient α
When trying to identify a mixture spectrum M = A + αB, the mixture coefficient α is generally not known in advance. Because an incorrect α will distort the cosine similarity between M and its correct library matches, it is important to estimate it correctly. To distinguish the true and estimated values of α, we denote the estimated mixture coefficient as α̂ and compare two methods to compute α̂. In the residual-spectrum approach, we first identify the dominant component in the mixture (A) and construct a residual spectrum R by removing from the mixture spectrum all common peaks between A and M. It can be shown that α̂ is directly related to the magnitude of the residual spectrum (‖R‖) and can be estimated by solving the following equation:
 |
In the optimal-cosine approach, α̂ is chosen to maximize the cosine similarity between M and A + α̂B. By taking the derivative of the cosine similarity function with respect to α̂, setting it to zero, and solving for α̂ (details provided in supplemental materials), we get
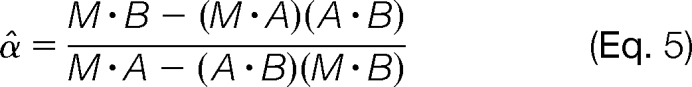 |
The performance of both methods is shown in Table I and supplemental Fig. S1. Although the performance of the residual-spectrum method is reasonable when α is large, the error becomes quite substantial when α is small. By contrast, the optimal-cosine method is robust in the presence of noise and delivers comparable performance across different values of α.
Table I. Mean and S.D. of the log-2 ratios of estimated (α̂ and true (α) mixture coefficients.
Although both approaches are roughly equivalent when α ≥ 0.5, optimal-cosine estimation performs substantially better on the more difficult cases of smaller mixture coefficients (see supplemental Fig. 2 for the complete distributions of log-2 ratios).
| True α | Residual-spectrum approach |
Optimal-cosine approach |
||
|---|---|---|---|---|
| Mean | S.D. | Mean | S.D. | |
| 1.0 | −0.1312 | 0.4278 | −0.0393 | 0.4646 |
| 0.5 | 0.051 | 0.4449 | −0.0103 | 0.4770 |
| 0.2 | 0.4592 | 0.6767 | 0.0816 | 0.5264 |
| 0.1 | 1.0139 | 0.9021 | −0.0014 | 0.5317 |
Classifications of Spectral Library Matches
As with regular database search of MS/MS spectra from isolated peptides, a spectral library search will always identify some top-scoring pair for any given query. To assess whether a match is significant, we consider three possible outcomes when searching a given query spectrum S:
No match: S does not match any spectrum in the library
Single-peptide match: S matches one peptide in the library
Mixture match: S is identified as a pair of peptides in the library
Let A* + α̂ B* be the best pair of spectra in the library returned by M-SPLIT; we distinguish between the possible outcomes using P and Δ defined as follows: P = Max(cos(S, A*), cos(S, B*)) and Δ = cos(S, A* + α̂B*) − P.
Intuitively, if S is from a peptide not present in the library, both A* and B* should have low cosine similarity to S. It follows that P should be low in the no-match case but relatively high in the other two cases. In addition, in mixture matches, the term B* should increase the similarity to S by a significant amount, as determined by Δ. We thus determine the outcome of a particular match by a simple two-step process: 1) a match is classified as no match if P is below a certain threshold; 2) distinguish single-peptide and mixture matches by checking whether Δ is below or above a chosen threshold, respectively.
To determine the actual threshold used in this process, we constructed two negative control datasets. One consisted of 5,000 mixture spectra (with α = 1.0) in which the peptides used to create the mixture spectra are deleted from the library. The second dataset consists of 5,000 single-peptide spectra. These two datasets were combined with another mixture dataset and searched against the library for the best pairs of matches. As shown in Fig. 3, when the peptides are not present in the library (no-match case), P has relatively low values (green dots) and can thus distinguish these from single or mixture-match cases by placing a threshold on P (see Fig. 4 left for precision/recall curves). In distinguishing single-peptide from mixture matches, Fig. 3 shows that Δ is higher for mixture matches than for single-peptide matches. However, Fig. 3 also shows that this threshold depends on α. To build a general model, we first choose the threshold for cases where αϵ{0.1, 0.2, 0.5, 1.0} and use linear regression to obtain the relationship between Δ and α. During our experiments, we also found low-complexity spectra (i.e. spectra dominate by only a few peaks) can lead to artificially high P or Δ, we computed a measure similar to dot-bias used in Ref. 14 and use this to filter out any significant matches that may be due to low-complexity spectra (see supplemental materials for details).
Fig. 3.

Comparison of spectral library search outcomes. Searching a query spectrum S against a spectral library has three possible outcomes: 1) no match when S matches no spectrum in the library (green dots); 2) single-peptide match when S matches only one peptide in the library (red dots); and 3) mixture match when S is identified as a pair of peptides in the library (pink, blue, cyan dots represent mixture matches when α = 1.0, 0.5, 0.1, respectively). As illustrated by the colored sets, M-SPLIT can distinguish no match from the rest by thresholding p = max(cos(M, A), cos(M, B)), shown on the x axis. Likewise, single-peptide and mixture matches can be distinguished by thresholding Δ = cos(M, A + B) − P, (shown on the y axis as the distance from the main diagonal line).
Fig. 4.

Classification of spectral library matches. Left, precision/recall curves when distinguishing No-match from single-peptide and mixture matches; decisions are made by checking whether p = max(cos(M, A), cos(M, B)) is above a predetermined threshold. Right, precision/recall curves when distinguishing single-peptide from mixture matches; decisions are made by checking whether Δ = cos(M, A + B) − P is above a predetermined threshold.
RESULTS
Our running hypothesis is that a mixture spectrum can be identified by matching it to a linear combination of single-peptide spectra. To test this hypothesis, we simulated a series of different mixture spectra (as described under “Experimental Procedures”) and verified whether the resulting mixture matches correctly identified the peptides used to construct each simulated mixture. As shown in Table II, the performance of our approach varies with α but is able to select the correct peptides in 90–99% of all cases. As expected, as α decreases, it becomes more difficult to identify both peptides in the mixture spectra because the signal-to-noise ratio substantially decreases for the low-abundance peptide. In addition, the accuracy decreases faster at a ratio of 1:0.1, suggesting that this may be the lowest α that can be handled without substantially decreasing sensitivity. Of course, high MS/MS mass accuracy should seamlessly elevate the performance of this approach to lower values of α.
Table II. Selecting the correct pair of peptides from the spectral library; each row indicates the percentage of cases in which the top-ranking pair is correct.
M-SPLIT is compared with an iterative approach in which one first identifies the spectrum with the top-scoring projected-cosine, removes shared peaks between the top-scoring spectrum and the query spectrum, and finally searches the library a second time to identify the second peptide in the mixture. As shown here, the iterative approach is generally worse than M-SPLIT and especially error-prone for low values of mixture coefficients, consistent with our observations on estimation of mixture coefficients. For smaller values of α, M-SPLIT gains an advantage by simultaneously considering both peptides in the mixture.
| Mixture coefficient (α) | M-SPLIT | Iterative approach |
|---|---|---|
| 1:1 | 99.4 | 98.4 |
| 1:0.5 | 98.7 | 98.3 |
| 1:0.3 | 96.8 | 96.4 |
| 1:0.1 | 89.6 | 77.1 |
Because of multiple factors in MS/MS data acquisition, it is possible that not all peaks in a single-peptide spectrum will appear in a mixture spectrum containing the same peptide. It is reasonable to assume that high-intensity peaks in the single-peptide spectrum will be detectable, whereas low-intensity peaks may not be observed. We simulate this scenario by applying a window filter where a peak is kept if it has rank less than or equal to N in a window of W Da around its mass. We show that our method is robust against missing peaks using different values of W and N (see supplemental materials). This is consistent with previous studies showing that one does not need all peaks in a spectrum for single-peptide identification purposes: in X!Hunter (12), the authors speed up the computation by showing that it is generally enough to retain only the top 20 peaks per spectrum.
Having observed that the highest abundance peptide in a mixture can be identified as the top ranking match using projected-cosine, one could reason that if the peaks from this peptide are removed from the mixture spectrum, we are left with a non-mixture spectrum. This leads to an iterative strategy to identify peptides in mixture spectra: first, identify the spectrum with top-scoring projected-cosine, remove shared peaks between the top-scoring spectrum and the mixture spectrum, and search the library a second time to identify the second peptide in the mixture. The accuracy of the iterative method is compared with that of M-SPLIT in Table II and observed to be worse. Note that this is consistent with our results on estimation of α: as α gets smaller, it is important to consider both components in the mixture for accurate identification and quantification of both peptides.
Peptide Identification with Compressed Chromatography
Whereas the simulation experiments demonstrate the ability of M-SPLIT to reliably identify mixture spectra against large spectral libraries, we further validated our method on experimental data. The dataset consists of six bovine proteins (apotransferrin, carbonic anhydrase, catalase, glutamate dehydrogenase, lactoperoxidase, and serum albumin) from Michrom Bioresources, Inc. (Auburn, CA). 500 pmol of each protein were mixed in an equimolar ratio in a 50:50 mix of acetonitrile and water, reduced, alkylated, and trypsinized. This same sample was analyzed under two different chromatographic time scales: one dataset was obtained with an 80-min chromatography (Long dataset), whereas the other dataset was obtained with a Short 3-min chromatography (Short dataset). MS data were acquired on an LTQ-Orbitrap XL (Thermo Fisher Scientific) operating on an acquisition cycle of two consecutive survey scans (first in the linear ion trap, second in the Orbitrap at 60,000 resolution) followed by MS/MS scans at unit resolution (linear ion trap, centroid mode, AGC on). We note that although the high-resolution survey scans readily provide accurate precursor masses, these particular settings assign MS/MS precursor masses based on the low-resolution survey scans, thus allowing us to verifiably test the performance of our approach as if operating in the (still) most common data acquisition mode. Peak lists in RAW files were converted to mzXML using ReAdW. Excluding the initial load and final wash periods, we obtain 251 MS/MS spectra in the Short dataset that could possibly be mapped to spectra in the Long dataset. Under these chromatographic conditions, we assumed that each spectrum in the Long dataset comes from only one peptide and used these as our library of single-peptide spectra. Conversely, because the Short dataset was obtained from the same sample with much less chromatography time, we assumed that some spectra might contain pairs of peptides that had been separated in the Long run; the Short dataset was thus used as our set of query spectra against the spectral library defined by the Long dataset.
The Long dataset was annotated using InsPecT (4) to search SwissProt (ver.15.9) with parent mass tolerance of 2 Da and fragment mass tolerance of 0.5 Da; a 5% false discovery rate was enforced using a standard target/decoy strategy (10), and no modifications were allowed. We note that although 5% FDR is generally too high for peptide identification purposes, our main utilization of search results was in grouping repeated spectra from the same peptide. To further increase the coverage of peptide identification, we grouped spectra in the library by assigning two spectra to the same group if their parent masses were within tolerance (2 Da, 0.05 Da if precursor masses are corrected using the high accuracy survey scans) and their cosine similarity is high. Then if any spectrum in a group is annotated by InsPecT, annotations are transferred to every member in the group. To reduce potential errors, annotations are transferred only when coherent across all identified spectra in the same group; otherwise, all spectra in that group are considered unidentified. Spectra in the Short dataset were annotated in two different ways: 1) M-SPLIT with parameters determined from the simulation experiments and 2) by InsPecT using the same search parameters used for the Long dataset. The results are shown in Fig. 5 and Table 3. Of 251 MS/MS spectra, M-SPLIT returned a total of 187 matches and InsPecT returned 22 IDs. As a first level of validation, we ran M-SPLIT without a parent mass filter and used parent mass as an a posteriori independent test to estimate the accuracy and sensitivity of our approach. The lack of a parent mass filter also allowed us to estimate the performance of M-SPLIT on a much larger spectral library (e.g. proteome-scale spectral library) in which searches would be conducted only against spectra with matching parent masses, thus resulting in a comparable number of candidate matches. We manually compared the MS1 isotopic profile of the query spectra to the MS1 isotopic profile of the top match(es) returned by M-SPLIT and verified whether these were the same. Two isotopic profiles were considered the same if both indicated the same peptide charge and if isotopic peaks had a difference in m/z of less than 0.05 Da. We also manually visualized both the MS1 and MS/MS spectra of mixture match cases to verify that the matches are valid (details for all mixture matches from the Short dataset are provided in supplemental materials). The estimated accuracy for both single-peptide and mixture matches are shown in Table III, Part A.
Fig. 5.

Classification of spectral library matches between Short (3-min) and Long (80-min) chromatography runs of the same sample. We assumed that each MS/MS spectrum in the Long dataset comes from only one peptide and used these as our library of single-peptide spectra. On the other hand, because the Short dataset was obtained from the same sample with compressed chromatography, we would expect that some MS/MS spectra might contain pairs of peptides that were separated in the Long run and thus use this as our set of query spectra. Each spectrum in the Short dataset was searched against the Long dataset for the best pair and labeled as mixture, single-peptide and no match, shown here as purple, green, and blue dots, respectively.
Table III. M-SPLIT results on the compressed-chromatography (Short) data set.
Of 251 spectra, 186 have a match to the spectral library obtained from an 80-minute run of the same sample (Long dataset). In Part A, precision was estimated by comparing the MS1 isotopic profile of each query spectrum and the top matches returned by M-SPLIT in the Long dataset. Two isotopic profiles are considered matched if they indicate the same peptide charge, have correlated intensities and isotopic peaks have m/z difference = 0.05 Da. In Part B, M-SPLIT matches are divided into four categories according to whether the spectra were identified by InsPecT. In Part C, the 64 spectra that did not match to the Long data set were further investigated manually. For most cases (54 of 64), this was due to missing data in the Long data set; either there was no MS/MS spectra for the corresponding MS1 precursor or no matching MS1 precursor was found. In Part D, the number of unique peptides identified by M-SPLIT and InsPecT is reported.
| Category |
Precision |
|---|---|
| A. All M-SPLIT matches | |
| Single-peptide matches | 97% (174/179) |
| Mixture matches | 87% (7/8) |
| B. Identified M-SPLIT matches | ||
|---|---|---|
| Identified by InsPecT |
Counts | |
| Long dataset | Short dataset | |
| No | No | 95 |
| Yes | No | 73 |
| No | Yes | 8 |
| Yes | Yes | 11 |
| C. Spectra in the Short dataset not matched to the Long dataset | |
|---|---|
| Category | Counts |
| MS1 not found | 17 |
| MS2 not found | 37 |
| MS1 and MS2 found | 10 |
| D. Unique peptide identifications | ||
|---|---|---|
| Method | No. of peptides identified |
|
| Long dataset | Short dataset | |
| InsPecT | 211 | 14 |
| M-SPLIT | NA | 43 |
NA, not available.
Of all 251 MS/MS spectra in the Short dataset, 64 did not match any spectra in the Long dataset (Table III, Part B). After manual investigation of the Long dataset, it turned out that for most cases (54/64), M-SPLIT did not find a match because either the corresponding peptide was missing (i.e. no corresponding MS1 isotopic profile was found) or it was not selected for MS/MS (MS2 not found). Hence, these unannotated spectra are likely a limitation in the library derived from the Long dataset and not a shortcoming of M-SPLIT, which correctly classifies them as no-match cases. Considering the remaining 10 cases as false negatives leads to a ≈94% (186/196) estimate of M-SPLIT's sensitivity. Note that these numbers, although not identical, are close to those seen in our simulated dataset and thus indicate that our simulation is able to capture important aspects of mixture spectra in real chromatographic settings.
To quantify the peptide identification gain in M-SPLIT, we further compared the number of spectra and unique peptides identified in the Short dataset with those obtained by InsPecT. Although this comparison violates the common assumption of database search methods (i.e. that each spectrum comes from a single peptide), it nevertheless mimics the typical setup in MS/MS experiments and allows us to estimate the expected gains from using M-SPLIT. The 187 matches from the Short dataset to the Long dataset are divided into four groups in Table III, Part B, according to their InsPecT annotations. Although InsPecT was able to annotate only 22 spectra in the Short dataset, M-SPLIT was able to successfully annotate approximately four times as many spectra in the same dataset. When comparing the number of unique peptides identified in the Short dataset, InsPecT identified only ≈6% of the peptides identified in the Long run, whereas M-SPLIT matches recover approximately ≈20% of all identifications in the Long dataset, including IDs from mixture spectra.
Peptide Identification in Yeast
To illustrate the utility of our method in a typical scenario, we further tested M-SPLIT on a larger experimental yeast dataset (23), generously made publicly available in Tranche/ProteomeCommons (24) by researchers at the University of Vanderbilt. In brief, a tryptic digest of Saccharomyces cerevisiae was analyzed on an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific), and MS/MS spectra were acquired using a data-dependent scanning mode in which one full MS scan (m/z 300–2000) was acquired on the Orbitrap at a resolution of 60,000, followed by eight MS/MS scans collected on the LTQ (see Ref. 23 for full details). To retain the utility of accurate precursor masses for a posteriori validation of search results, InsPecT was run with 2.5-Da parent mass tolerance and 0.5-Da fragment mass tolerance on the Saccharomyces Genome Database (version 5/8/2009); a 1% false discovery rate was enforced using a target/decoy strategy, and no modifications were allowed. M-SPLIT was run with default parameters against the yeast spectral library from NIST (version 5/4/2009); a 3-Da parent mass filter was used to prefilter the library before the search. The results are summarized in Table IV. In short, InsPecT was able to identify a total of 19,297 spectra and 4,486 unique peptides. On the other hand, M-SPLIT was able to identify 28,993 single-peptide spectra, 1,505 mixture spectra, and a total of 6,089 unique peptides. Because the yeast dataset was acquired with high-accuracy survey scans, this information was further used to validate our annotations by comparing the theoretical m/z value of the peptides returned by InsPecT/M-SPLIT and the observed precursor m/z in the corresponding survey scans. An annotation is considered correct if the theoretical precursor m/z is within 10 ppm of the observed m/z; the estimated accuracies are summarized in Table IV. The comparison between M-SPLIT and InsPecT further reveals that their annotations are same in ≈99% of the cases for which both make an annotation, thus demonstrating the coherence of these two independent methods.
Table IV. M-SPLIT and InsPecT search results on the Yeast dataset [24].
Part A reports numbers of identified spectra (single-peptide and mixture) and unique peptides. Part B reports the precision of peptide identifications was estimated by comparing the theoretical precursor m/z of peptides returned by M-SPLIT or InsPecT and the observed precursor m/z values in the corresponding MS1 scan (isotopic profile). An identification is considered correct if the difference between theoretical and observed precursor m/z values is less than 10 ppm. For mixture spectra the overall precision is computed by dividing the number of correct peptide identifications by the total number of identifications (i.e. twice the number of mixture spectra). The precision for the second-peptide identifications is also shown (in parentheses); this precision is lower because the second peptide in the mixture is usually of low abundance (average α = 0.3) and thus harder to identify.
| Method | Spectrum identifications |
Unique peptides | ||
|---|---|---|---|---|
| Single-peptide | Mixture | Total | ||
| A. Spectrum and peptide identifications in the yeast dataset | ||||
| InsPecT | 19,297 | NA | 19,297 | 4,486 |
| M-SPLIT | 28,993 | 1,505 | 30,498 | 6,089 |
| B. Estimated precision in the yeast dataset | ||
|---|---|---|
| Method | Single-peptide matches | Mixture matches |
| InsPecT | 98% | NA |
| M-SPLIT | 98% | 95.7% (91.4%) |
NA, not available.
M-SPLIT identifications indicate that mixture spectra consist of approximately 5% of all identifiable spectra in the yeast dataset, suggesting that these constitute a modest but significant fraction of identifiable spectra in typical proteomics experiments. It should be emphasized that even though the number of mixture spectra is not large, these result in more than one peptide identification per spectrum and thus carry more information than single-peptide spectra. In the yeast dataset, there are a total of 28,993 single-peptide spectra identified by M-SPLIT as 5,873 unique peptides. In addition, M-SPLIT further identifies 1,505 mixture spectra as 1,627 unique peptides, 239 of which are identified only in mixture spectra; a summary of the overlap between the two methods is shown in Fig. 6.
Fig. 6.

Peptide identifications in yeast dataset with M-SPLIT and InsPecT. Peptides identified by M-SPLIT and InsPecT are compared in a Venn diagram indicating the numbers of unique peptides in each category.
DISCUSSION
Despite the success of mainstream software for peptide identification from MS/MS spectra, the ubiquitous assumption that each spectrum arises from only one peptide is often not valid, making the interpretation difficult in such scenarios. To address this computational bottleneck, we propose the first spectral library-based approach (M-SPLIT) to the identification of mixture spectra generated from pairs of peptides. Theoretical bounds were derived to prune the search space using branch-and-bound techniques and further improved using a new projected-cosine metric. Thus, M-SPLIT dramatically reduces the search space by 6 orders of magnitude and is able to deliver results at an average of 2 s/spectrum (on a regular laptop with a Pentium Core2 Duo, 1.6 GHz, 2 GB RAM), even when searching against proteome-scale spectral libraries. Despite considering only a tiny fraction of the whole search space, our benchmarks on both simulated and experimental data consistently show that M-SPLIT has both high sensitivity (≈94%) and high accuracy (up to 98%).
In addition to accurate peptide identification, M-SPLIT robustly quantifies the relative abundance of coeluting peptides at the time of MS/MS acquisition, as determined by the fraction of MS/MS ion current assigned to each peptide. In principle, extending this approach to relative peptide abundance per run (e.g. in Data Independent Acquisition setups (25)) could be as simple as adding the estimated intensities over consecutive MS/MS scans followed by a posteriori computation of per-run relative abundance. It should be noted that, as in other label-free MS-based quantification approaches (26), there are MS-specific confounding factors that may result in distortion of the observed relative abundance (e.g. peptide-specific ionization efficiencies) and thus require follow-up experiments to validate the observed relative abundance.
We further note that M-SPLIT makes no assumptions about the type of query or library spectra. Although M-SPLIT was developed and tested on peptide MS/MS spectra, the current implementation is readily applicable to any type of spectra. In particular, it would be straightforward to extend any target spectral library to include spectra of common peptide and chemical contaminants and thus reduce their negative effect on peptide identifications (by matching experimental contaminant spectra to library contaminant spectra). However, as with other spectral library search approaches, M-SPLIT do assumes that experimental spectra are acquired under conditions prone to generate spectra comparable with those available in the spectral library. Although our yeast results demonstrate that standard NIST spectral libraries can be used to identify independently collected spectra from an unrelated research group, we note that all spectra were acquired using similar tandem mass spectrometry (CID/IonTrap) instruments and settings. By blindly identifying the best pairs in a given spectral library, M-SPLIT automatically classifies each query spectrum as a mixture match, single match, or no match; thus it is a general self-adjusting tool that can be used on experimental setups promoting the acquisition of either/both single-peptide or/and mixture spectra.
The development of mass spectrometry algorithms typically requires large datasets with validated identified spectra that are difficult to obtain. The unavailability of datasets with validated identifications of mixture spectra was a limiting factor that we addressed in two different ways: by generating large datasets of simulated mixture spectra and by acquiring MS/MS spectra from the same sample using different chromatographic time scales. The level of control afforded by the generation of simulated mixture spectra was instrumental in determining spectrum identifiability over a range of relative abundance of coeluted peptides. These results were then corroborated using an experimental dataset in which it was possible to provide exhaustive manual validation. As such, we were able to determine both the accuracy and sensitivity of our approach—a commonly difficult task because the set of true positives (and its complementary false negatives) is typically not known in advance. After our validation, we estimated that M-SPLIT delivers a false negative rate of only 5% at accuracy levels of up to 98%.
Focusing M-SPLIT on the identification of mixture spectra from pairs of peptides allowed us to derive theoretical bounds and filtration techniques that can be extended for spectra from more complex mixtures. In particular, the utility of the projected-cosine metric is likely to increase as mixture spectra become more complex. In addition, although M-SPLIT is already able to reliably annotate mixture spectra with inaccurate fragment masses (still the dominant MS/MS acquisition mode), its performance is very likely to further improve for high accuracy MS/MS data. Such data could seamlessly enable the identification of coeluted peptides at more disparate relative abundance ratios and would probably greatly simplify the extension to mixture spectra from more than two peptides.
Supplementary Material
Acknowledgments
We thank NIST, ProteomeCommons, and Vanderbilt University for the public availability of the mass spectrometry data used in this research and the reviewers for insightful suggestions.
* This work was supported in part by National Institutes of Health Grant 1-P41-RR024851 from the National Center for Research Resources.
 This article contains supplemental material and supplemental Fig. S1.
This article contains supplemental material and supplemental Fig. S1.
2 Because all spectra were scaled down to norm 1, normalized dot product simply reduces to estimating the cosine between two unit vectors. In addition, we reduce the disproportionate influence of high-intensity peaks by first applying the square-root transform to all peak intensities (13) (preprocessing details provided in Supplemental materials).
1 The abbreviations used are:
- MS/MS
- tandem mass spectrometry
- M-SPLIT
- mixture-spectrum partitioning using libraries of identified tandem mass spectra
- NIST
- National Institute of Standards and Technology.
REFERENCES
- 1.Eng J. K., McCormack A. L., Yates J. R. (1994) An approach to correlate tandem mass-spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 2.Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 3.Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 4.Tanner S., Shu H., Frank A., Wang L. C., Zandi E., Mumby M., Pevzner P. A., Bafna V. (2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 77, 4626–4639 [DOI] [PubMed] [Google Scholar]
- 5.Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 6.Frank A., Pevzner P. (2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 7.Fischer B., Roth V., Roos F., Grossmann J., Baginsky S., Widmayer P., Gruissem W., Buhmann J. M. (2005) NovoHMM: a hidden Markov model for de novo peptide sequencing. Anal. Chem. 77, 7265–7273 [DOI] [PubMed] [Google Scholar]
- 8.Mo L., Dutta D., Wan Y., Chen T. (2007) Msnovo: a dynamic programming algorithm for de novo peptide sequencing via tandem mass spectrometry. Anal. Chem. 79, 4870–4878 [DOI] [PubMed] [Google Scholar]
- 9.Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 10.Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 11.Frank A. M., Bandeira N., Shen Z., Tanner S., Briggs S. P., Smith R. D., Pevzner P. A. (2008) Clustering millions of tandem mass spectra. J. Proteome Res. 7, 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Craig R., Cortens J. C., Fenyo D., Beavis R. C. (2006) Using annotated peptide mass spectrum libraries for protein identification. J. Proteome Res. 5, 1843–1849 [DOI] [PubMed] [Google Scholar]
- 13.Frewen B. E., Merrihew G. E., Wu C. C., Noble W. S., MacCoss M. J. (2006) Analysis of peptide ms/ms spectra from large-scale proteomics experiments using spectrum libraries. Anal. Chem. 78, 5678–5684 [DOI] [PubMed] [Google Scholar]
- 14.Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 15.Bandeira N., Tang H., Bafna V., Pevzner P. (2004) Shotgun protein sequencing by tandem mass spectra assembly. Anal. Chem. 76, 7221–7233 [DOI] [PubMed] [Google Scholar]
- 16.Savitski M. M., Nielsen M. L., Zubarev R. A. (2006) Modificomb, a new proteomic tool for mapping substoichiometric post-translational modifications, finding novel types of modifications, and fingerprinting complex protein mixtures. Mol. Cell. Proteomics 5, 935–948 [DOI] [PubMed] [Google Scholar]
- 17.Bandeira N., Tsur D., Frank A., Pevzner P. A. (2007) Protein identification via spectral networks analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 6140–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phanstiel D., Brumbaugh J., Berggren W. T., Conard K., Feng X., Levenstein M. E., McAlister G. C., Thomson J. A., Coon J. J. (2008) Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 105, 4093–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venable J. D., Dong M. Q., Wohlschlegel J., Dillin A., Yates J. R., 3rd (2004) Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 1, 39–45 [DOI] [PubMed] [Google Scholar]
- 20.Masselon C., Pasa-Tolić L., Lee S. W., Li L., Anderson G. A., Harkewicz R., Smith R. D. (2003) Identification of tryptic peptides from large databases using multiplexed tandem mass spectrometry: simulations and experimental results. Proteomics 3, 1279–1286 [DOI] [PubMed] [Google Scholar]
- 21.Chakraborty A. B., Berger S. J., Gebler J. C. (2007) Use of an integrated MS-multiplexed MS/MS data acquisition strategy for high-coverage peptide mapping studies. Rapid Commun. Mass Spectrom. 21, 730–744 [DOI] [PubMed] [Google Scholar]
- 22.Atwater B. L., Stauffer D. B., McLafferty F. W., Peterson D. W. (1985) Reliability ranking and scaling improvements to the probability based matching system for unknown mass spectra. Anal. Chem. 57, 899–903 [Google Scholar]
- 23.Li J., Zimmerman L. J., Park B. H., Tabb D. L., Liebler D. C., Zhang B. (2009) Network-assisted protein identification and data interpretation in shotgun proteomics. Mol. Syst. Biol. 5, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falkner J. A., Falkner J. W., Andrews P. C. (2007) ProteomeCommons.org IO Framework: reading and writing multiple proteomics data formats. Bioinformatics 23, 262–263 [DOI] [PubMed] [Google Scholar]
- 25.Venable J. D., Yates J. R., 3rd (2004) Impact of ion trap tandem mass spectra variability on the identification of peptides. Anal. Chem. 76, 2928–2937 [DOI] [PubMed] [Google Scholar]
- 26.Mueller L. N., Brusniak M. Y., Mani D. R., Aebersold R. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 7, 51–61 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.