

Abstract
Phosphorylation of proteins is a key posttranslational modification in cellular signaling, regulating many aspects of cellular responses. We used a quantitative, integrated, phosphoproteomics approach to characterize the cellular responses of the yeast Saccharomyces cerevisiae to the fatty acid oleic acid, a molecule with broad human health implications and a potent inducer of peroxisomes. A combination of cryolysis and urea solubilization was used to minimize the opportunity for reorientation of the phosphoproteome, and hydrophilic interaction liquid chromatography and IMAC chemistries were used to fractionate and enrich for phosphopeptides. Using these approaches, numerous phosphorylated peptides specific to oleate-induced and glucose-repressed conditions were identified and mapped to known signaling pathways. These include several transcription factors, two of which, Pip2p and Cst6p, must be phosphorylated for the normal transcriptional response of fatty acid-responsive loci encoding peroxisomal proteins. The phosphoproteome data were integrated with results from genome-wide assays studying the effects of signaling molecule deletions and known protein-protein interactions to generate a putative fatty acid-responsive signaling network. In this network, the most highly connected nodes are those with the largest effects on cellular responses to oleic acid. These properties are consistent with a scale-free topology, demonstrating that scale-free properties are conserved in condition-specific networks.
In the face of dynamic cellular environments, cells must detect and compute signals they receive and execute an integrated and coordinated response involving multiple transcriptional and morphological programs. The rapid transduction of signals to the nucleus is accomplished in large part by altering the posttranslational states and thus activities of proteins that form relay networks for signal transmission. Phosphorylation is a ubiquitous posttranslational modification occurring on serine, threonine, and tyrosine aminoacyl residues that provides a common mechanism through which protein activity states are altered. Reversible phosphorylation is involved in virtually all cellular processes in eukaryotes, modulating enzymatic activities, protein subcellular distributions, protein half-lives, and protein-protein interactions (1, 2). The study of the phosphorylome is, therefore, a central component of systems approaches to understanding cellular processes.
We are interested in cellular responses to nutrient changes that induce peroxisomes. Peroxisomes are ubiquitous intracellular organelles responsible for many metabolic activities, most notably fatty acid (FA)1 β-oxidation. In yeast, genes encoding abundant peroxisomal proteins are repressed in cells grown in glucose and highly induced in response to FA. The induction process involves global reorganization of transcriptional networks and activities (3, 4), peroxisome proliferation (5) (a process that highlights the dynamic nature of the peroxisomal proteome (6, 7)), and a host of additional coordinated cellular responses such as alterations to the cell cycle, cytoskeleton, membrane dynamics, and chromatin. Signaling molecules are key regulators of these complex and coordinate responses. We previously used cell-based reporter studies of gene deletions to delineate a core FA response signaling network governing the induction of peroxisomes in yeast (8). In an effort to both expand the network beyond the focus on peroxisomes and to understand information flow within the FA response network, we took a quantitative phosphoproteomics approach to characterize the differences between the phosphorylated portions of the proteomes of glucose-grown (peroxisome-repressed) and oleic acid-treated (peroxisome-induced) cells.
Phosphoproteomes are a subset of proteomes with given phosphorylation events occurring at low stoichiometry (9). Mass spectrometry-based approaches to comprehensively identify phosphoproteomes have generally relied upon the reduction of sample complexity through fractionation, either gel-based (9, 10) or chromatographic (11–13), and enrichment strategies, generally through the use of immobilized metal affinity chromatography (IMAC) (14), titanium dioxide (TiO2) (15, 16), or phosphoramidate chemistry (17, 18). Although analysis of the phosphorylome remains a significant challenge, these approaches and improvements to mass spectrometers have significantly increased the number of identifications of phosphorylated peptides and their respective proteins. Known phosphorylation events are now on the order of several thousand (19).
Phosphorylation is a dynamic process with phosphorylation states changing rapidly on the order of minutes or even seconds (20–22). As mass spectrometry-based experiments rely on ex vivo approaches, careful consideration must be given to the cellular environment during the cell disruption procedures. Ideally, the presence of a stimulus should be maintained until the cells are processed to a point at which reorientation of the phosphoproteome is unable to occur. Given the rapidity with which phosphorylation states can reorient, we developed a cryolysis-based disruption, urea solubilization methodology to minimize the opportunity for kinase or phosphatase activity and maintain the condition-specific phosphorylation status of the proteome. We also combined stable isotope labeling with amino acids in cell culture with orthogonal peptide fractionation procedures to identify proteins that are significantly responsive to cell exposure to FAs. Using cryolysis, hydrophilic interaction-based chromatography, and a combination of LC-MS2 and multistage activation (MSA) approaches, we identified a global data set of proteins that are differentially phosphorylated upon transition from a glucose- to a FA-stimulated state.
Integration of this phosphoproteomics data set with data from an analysis of signaling molecules regulating peroxisome induction during the FA response (8) and known interactions from the literature allowed for the identification of numerous novel phosphorylated forms of signaling and transcription factors and the derivation of an expanded FA-responsive signaling network with properties indicative of scale-free topology. Interestingly, the most highly connected nodes influence the response to FAs to the greatest extent. Taken together, these analyses provide a comprehensive view of the network specifically responsive to the exposure of cells to FAs and further demonstrate the conservation of common network architecture within signaling networks with the feature of a few hubs exerting the largest effects persisting at condition- or phenotype-specific levels.
EXPERIMENTAL PROCEDURES
Cell Growth and Media
A single colony of BY4742 arg4Δ lys1Δ cells was grown overnight in 100 ml of rich medium to an A600 of 1.0 and then seeded into two 1-liter cultures of a minimal yeast medium (0.17% yeast nitrogen base without ammonium sulfate or amino acids, 0.5% ammonium sulfate) containing a full complement of amino acids and supplemented with 20 mg/liter isotopically normal or heavy arginine (13C6,15N4; Isotec) and lysine (13C6,15N2; Isotec). The cells were grown for 18 h to an A600 of 1.8. The isotopically heavy sample was pelleted and flash frozen in liquid nitrogen. The normal sample was pelleted and reseeded into an oleic acid-containing medium (isotopically normal arginine and lysine, 0.2% oleic acid (Sigma), 0.5% Tween 40), stimulated for another 85 min, pelleted, and flash frozen. The total time between the exposure of the cells to oleic acid and the flash freezing was 92 min. This yields an isotopically normal (with respect to arginine and lysine) oleic acid-stimulated sample and an isotopically heavy glucose-grown reference sample.
Cell Lysis, Isolation, and Fractionation of Peptides
1 ml of buffer (phosphate-buffered saline (PBS; Invitrogen), 10% glycerol, enzyme inhibitors (SigmaFAST Protease Inhibitor Tablets, Sigma), and HALT Phosphatase inhibitors (Thermo Scientific)) was added to the frozen pellets in liquid nitrogen. The frozen cell/buffer pellets were subjected to cryolysis using a Retcsh PM100 Planetary Ball Mill grinder (three runs of 3 min each with immersion in liquid nitrogen between each run).
The resulting ground sample was solubilized by brief sonication (2 × 10-s pulses) with a probe tip sonicator into 3 volumes of 8 m urea, 0.4 m ammonium bicarbonate (6 m urea final concentration). The lysate was cleared by centrifugation at 13,000 × g for 5 min, and the supernatant was reduced by incubation in 10 mm DTT at 37 °C for 1 h and then alkylated by incubation in 10 mm iodoacetamide in the dark at room temperature for 1 h. To validate incorporation of the heavy isotopes, 100 μl of the reduced and alkylated lysate were digested with trypsin, desalted with C18 Ultramicrospin columns (The Nest Group), and analyzed by mass spectrometry. Incorporation of isotopically heavy arginine and lysine was verified by a 10- and 8-Da shift in the respective m/z spectra using the QTIPS methodology (23). The incorporation for each amino acid was 98%. 11.5 mg of each protein sample (glucose and oleate) were combined and diluted 4-fold with distilled H2O, and proteins were digested with trypsin (Promega) for 16 h at 37 °C.
The trypsin-digested samples were acidified by the dropwise addition of 10% trifluoroacetic acid (TFA; Sigma) to a pH of 3. A white precipitate formed and was removed by centrifugation at 4,000 × g for 20 min. The cleared supernatant was passed through disposable Whatmann 2.7-μm-pore glass fiber filters. The supernatant was divided into four parts (∼5 mg/aliquot), and the samples were desalted by loading onto C18 columns (Waters Sep-Pak Vac 500 mg) (loaded in 0.1% TFA; eluted in 60% acetonitrile (ACN; Sigma), 0.1% TFA) and then dried. Fractionation was done successively with 5 mg of peptides on a TOSOH TSK-Gel Amide-80 4.6-mm × 25-cm analytical column with a guard column under the following conditions: Solvent A, 98% ACN, 0.1% TFA; Solvent B, 2% ACN, 0.1% TFA; load in 90% A over 20 min, 90–85% A over 5 min, 85–60% A over 40 min, 60–0% A over 5 min, and 0–90% A over 5 min. The flow rate was 1 ml/min, and fractions were collected at 2-min intervals beginning at the 85–60% A gradient. Fractions were pooled as follows and dried: Pool 1, Fractions 2, 3, 4, and 5; Pool 2, Fractions 6 and 7; Pool 3, Fraction 8; Pool 4, Fraction 9; Pool 5, Fraction 10; Pool 6, Fraction 11; Pool 7, Fraction 12; Pool 8, Fractions 13, 14, and 15; Pool 9, Fractions 16, 17, 18, 19, and 20; and Pool 10, Fractions 21, 22, 23, 24, 25, 26, and 27.
Enrichment of Phosphopeptides
Phosphopeptides were enriched using PHOS-SelectTM Iron Affinity Gel (Sigma). Pellets were resuspended in 500 μl of a 250 mm acetic acid, 30% ACN load solution and mixed using a Vortex mixer for 5 min. 15 μl of a 50% slurry (gel to load solution) were added to each sample, and the samples were incubated for 30 min with end-over-end rotation. Samples were washed four times with 500 μl of load solution and once with distilled H2O. Peptides were eluted with 2–3 min washes of 50 mm Na2HPO4, pH 8.4 acidified to pH 3 with 5 μl of 100% formic acid; flash frozen in liquid nitrogen; and dried. Phosphopeptides were resuspended in 200 μl of 0.1% TFA and cleaned on C18 columns. Eluted samples were dried and resuspended in 20 μl of 0.1% formic acid. 2 μl were used per mass spectrometric analysis.
Mass Spectrometry
Mass spectrometry was performed on a Thermo Electron Orbitrap mass spectrometer equipped with an electrospray ionization source and an Agilent HP1100 liquid chromatography system. Samples were loaded onto a precolumn (packed with 2 cm of 200-Å-pore size Magic C18AQ resin (Michrom Bioresources)) in Solvent 1 (0.1% formic acid, 2% ACN) and eluted using a 60- or 210-min gradient of an increasing percentage of Solvent 2 (100% ACN). The 77-min gradient was as follows: 2–4% over 10 min, 4–25% over 50 min, 25–35% over 10 min, 35–80% over 5 min, and 80–2% over 2 min. The 240-min gradient was as follows: 2–10% over 10 min, 10–25% over 170 min, 25–35% over 40 min, 35–80% over 10 min, and 80–2% over 10 min. Peptides were eluted onto a 75-μm × 10-cm fused silica capillary column packed with 100-Å-pore size Magic C18AQ resin. Flow rates were held constant at 0.350 μl/min.
The mass spectrometer was run in data-dependent acquisition mode with switching automatically between MS and MS2 or MS, MS2, and MSA modes with MSA mode triggered by neutral loss peaks of 98, 46, 32.66, and 24.5 m/z. Each sample was run four times with a 77-min gradient (twice with MS2 and twice with MSA) and twice with 240-min gradients (one each of MS2 and MSA).
Processing of Spectral Data
XCalibur raw files were converted to mzXML format using ReAdW (v4.2.0) using the readw profile and default parameters. Peptide assignments were done using a combination of search engines. Spectral searches were done for tryptic fragments using X!Tandem (24) (v2007.07.01.3 with the k-score plug-in (25)) and Mascot (26) (v2.2). Searches were done against a non-redundant Saccharomyces cerevisiae reference protein database (the forward and reverse protein sequences from the union of the Saccharomyces Genome Database, Ensembl, NCBI, and GenBankTM databases plus keratin and trypsin) containing 13,616 entries. Parent tolerance was ±0.25 Da and the fragment mass tolerance used was ±0.4 Da for X!Tandem searches and ±0.8 Da for Mascot searches with a maximum of three missed cleavages. This large parent mass tolerance was used in conjunction with the accurate mass model in PeptideProphet for high confidence peptide identifications to boost the probability of peptides that have a low mass error; the probability of peptides that have a high mass error will be subsequently reduced (27). Modifications included in the searches were as follows: static modification of cysteine (at 57.021464 Da) and variable modifications of methionine (at 15.994915 Da); serine, threonine, and tyrosine (at 79.96633 Da); lysine (at 8.014199 Da); and arginine (at 10.008269 Da). Individual search results were processed using the Trans Proteomic Pipeline (28). Search results were validated using PeptideProphet (v3.0) (29) with the use of accurate mass modeling (27). Determination of automated statistical analysis of peptide (ASAP) ratios (30), which integrate the area under the peaks of an ion chromatogram, was used to determine relative quantities of phosphopeptides from the glucose-grown and oleic acid-incubated cultures. Scores of −1 and 0 (no information) were excluded from the final quantitative analyses. Results falling outside of two standard deviations were confirmed by visual inspection. Statistical accuracy of the ASAP method is described elsewhere (30). As this analysis identified hundreds of quantitative differences, we relied upon data integration and network construction to validate the biological significance of several of the phosphoproteins.
A novel computational method, termed iProphet,2 was developed and used to integrate results from multiple search algorithms. iProphet takes as input PeptideProphet spectrum-level results from multiple LC-MS/MS runs and then computes a new probability at the level of a unique peptide sequence (or protein sequence (54)). This framework allows for the combination of results from multiple search tools and takes into account other supporting factors, including the number of sibling experiments identifying the same peptide ions, the number of replicate ion identifications, sibling ions, and sibling modification states. A model of iProphet performance with respect to the number of correct identifications versus error is shown in supplemental Fig. S1. Only those peptides with phosphorylated serines, threonines, and tyrosines were considered further. An iProphet probability of 0.58 was used as the cutoff for phosphopeptides. This cutoff returned 15,123 spectra containing 148 decoy peptides for a false discovery rate (FDR) of 1% (FDR = (148/(15,271 − 148) = 0.0099) for phosphopeptides. The selection of this cutoff was based on the accuracy to error plotted for all spectral results shown in supplemental Fig. S1 that returned an FDR of 0.002% for all peptides (both phosphorylated and non-phosphorylated) identified.
Processing of iProphet Results (MATLAB)
The details of the MATLAB algorithm used to process the peptides are provided in supplemental PhosPepAlign.m. Briefly, the algorithm counts the number of phosphorylation sites indicated on the peptide and then groups the peptides based on the number of phosphorylation sites, sequence overlap, and mutual PeptideProphet-level assignment. This program also returns the number of phosphorylation sites and the mean ASAP scores for each group.
ASCORE Analysis
A custom version (31) of the ASCORE algorithm (32) was implemented in the Python programming language to compute the probability of phosphorylation localizing to a specific residue. Peptides with scores greater than 19 (99% probability of being correct) were visually inspected. In instances of multiple possible assignments, the peptides identified most frequently with the highest probabilities and best localization scores were selected (supplemental Table S1-II).
Motif Analysis
All unambiguously assigned peptides were centered on the position of the phosphorylated aminoacyl residue, and a greedy search was used to identify highly enriched up- and downstream residues at specific positions compared with the proteome-wide frequency of each amino acid residue. In the first pass, simple “primary” patterns were identified in which a phosphorylated residue was paired with a highly enriched residue (or group) at a specific location (supplemental Table S3, primary feature). In a second pass, these primary patterns were held constant, and secondary patterns were identified using the same strategy (supplemental Table S3, secondary feature). At the conclusion of the second pass, three-part substrate patterns were identified: a phosphorylated residue, the enriched amino acid residue or group from the first pass, and a further amino acid residue or group identified in the second pass. Likelihood was determined using the hypergeometric distribution of the frequency of amino acids in the yeast proteome and the PhosphoPep database (33). The motifs with significant p values (supplemental Table S3) were iteratively queried against the Human Protein Reference Database using the PhosphoMotif Finder (34), identifying homologous human kinase motifs (supplemental Table S3, sequence). To identify the putative corresponding S. cerevisiae homologs, the identified human kinases were used to search the HomoloGene Database (35) (http://www.ncbi.nlm.nih.gov/sites/entrez?db=homologene) and the Saccharomyces Genome Database (36) (http://www.yeastgenome.org/).
Site-directed Mutagenesis
PCR-based mutagenesis was performed as described (37). The primers used are listed in supplemental Table S6. Mutagenized genes (PIP2 and CST6) were sequenced to confirm the fidelity of the mutagenesis.
Quantitative PCR
Quantitative PCR was done as described previously (38). Cells were grown overnight in YPBD (0.3% yeast extract, 0.5% peptone, 0.5% potassium phosphate buffer, pH 6.0, 2% glucose) and then grown for 16 h in YPB-low glucose (0.3% yeast extract, 0.5% peptone, 0.5% potassium phosphate buffer, pH 6.0, 0.15% glucose). Cells were pelleted by centrifugation and resuspended in YPBO (0.3% yeast extract, 0.5% peptone, 0.5% potassium phosphate buffer, pH 6.0, 0.5% Tween 40, 0.2% oleic acid), and samples were collected at 0, 30, and 90 min. Samples were pelleted by centrifugation, and the resulting pellets were flash frozen in liquid nitrogen. RNA was extracted using hot phenol extraction (39), and total RNA was enriched with a Qiagen RNeasyTM kit. A RevertAidTM First Strand cDNA Synthesis kit (Fermentas) was used to generate cDNA, and quantitative PCR was carried out using a DynamoTM Flash SYBR Green quantitative PCR kit (NEB) on the ABI 7900 RT-PCR system. The data were normalized to expression of YFL039C (actin), which does not change in expression significantly during the FA response (this study and Ref. 39).
Cytoscape Network Construction
Known physical interactions between these proteins were downloaded from the Saccharomyces Genome Database (March 14, 2009). Each protein-protein interaction was reduced to a single interaction, and self-interactions were removed to reduce edge density. ASAP scores of glucose- and oleic acid-enriched proteins were converted to log10 scores. Proteins in which multiple phosphorylation events were detected were treated as single species, and the maximal log10 phosphorylation score was assigned to the protein. Proteins for which both glucose- and oleic acid-enriched peptides were detected were treated as one protein that was designated as “dual,” and no indication of enrichment of phosphorylation was indicated. The network is available as supplemental FA_Net_Saleem_MCP.cys, allowing the reader to interactively view the network using Cytoscape (freely available at http://www.cytoscape.org/).
Network Connectivity
The connectivity of the nodes in the FA network were transformed to a normalized score between 0 (no connections) and 1 (maximal connections), indicated by the red line in Fig. 7, top (connectivity norm). Similarly, functional genomics data (8), testing the effects of deletions of the same nodes on expression of an FA-responsive locus, were transformed to range from 1 (enhancement of gene expression) to ∼−0.7 (severe defect in expression), indicated by the blue line in Fig. 7, top (deletion effect norm). A series of normalized score values (supplemental Fig. S2) was tested to determine the significance of the correlation between the connectivity norm and the deletion effect norm. At a cutoff of 25% (0.25 of the normalized score in Fig. 7, top) from maximum connectivity, a p value of 7.6 × 10−9 for the enrichment of highly connected nodes, whose respective gene deletions perturb expression of FA-responsive loci, was obtained based on the cumulative hypergeometric distribution function.
Fig. 7.

Network analysis. Top, the most highly connected nodes are the nodes that tend to exert the greatest effect on the expression of FA-responsive loci. The x axis indicates proteins from the network for which there are data on the role of the protein in expression of FA-responsive loci. The y axis is the normalized score for connectivity with 1 indicating the most highly connected node in the network. The red line indicates node connectivity (connectivity norm), whereas the blue line (deletion effect norm) indicates the role of the network protein as a negative effector (positive score on the y axis) or positive effector (negative score on the y axis) of the FA response. Middle, most nodes in the network have few connections. The x axis indicates the number of connections (k); the y axis indicates the number of nodes with a given number of connections (P(k)). Bottom, the degree of distribution of the FA network follows the power law. The x axis is the log10 of connectivity (k); the y axis represents the log10 of the number of nodes in a given bin (P(k)). Each red dot represents a given number of connections (k) from the x axis of the middle panel. This connectivity shows that this network has topology consistent with scale-free networks.
Microarray Analysis
A 100-ml culture seeded at an A600 of ∼0.05 into minimal yeast medium (described above) + 2% glucose was grown at 30 °C for 8 h. One-half (50 ml) of the culture was harvested at an A600 of 0.5 by centrifugation and washed once with induction medium (minimal yeast medium with 0.2% oleate, 0.5% Tween 40) prewarmed to 30 °C, and cells were transferred to 50 ml of prewarmed induction medium. This culture was induced for 90 min, and then the cells were harvested by centrifugation and flash frozen. The remaining 50 ml of the original culture were grown to an A600 of 1 and harvested as for the induced sample. RNA was extracted and purified as described above. Labeled cRNA was prepared with the Agilent Quick AmpTM two-color kit using 0.5 μg of input RNA. Arrays were Agilent 8 × 15K (AMAID). Analysis was performed as described previously (39). The strains tested were BY4742, hog1Δ, and ste20Δ. Deletion strains were obtained from the commercially available yeast deletion library.
RESULTS
Experimental Strategy and Rationale
Our previous study identified signaling molecules that are required for the normal induction of loci encoding peroxisomal matrix proteins in response to FA stimulation (8); however, that study did not address the nature of changes in the phosphoproteome that accompany FA exposure. We therefore sought to complement these initial efforts by developing a robust methodology to quantitatively interrogate phosphoproteomes and apply this approach to enumerate changes of the phosphorylated proteome corresponding to a shift from glucose- (2% glucose in YPB) to oleic acid (0.2% oleic acid, 0.5% Tween 40 in YPB)-grown S. cerevisiae. Our work flow is shown in Fig. 1.
Fig. 1.
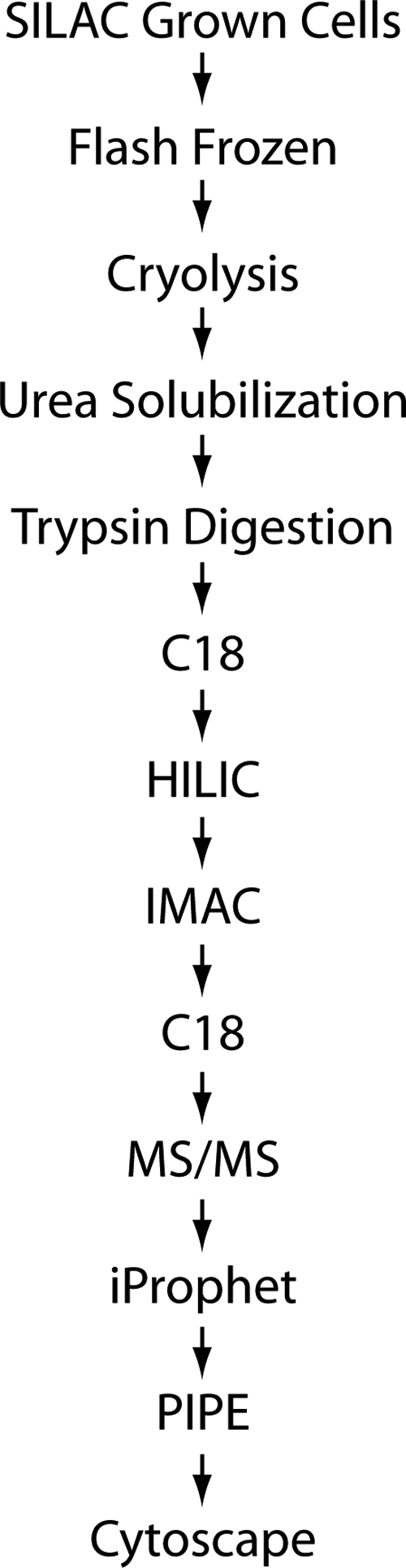
Work flow for isolation and integration of phosphopeptides into signaling networks. SILAC, stable isotope labeling with amino acids in cell culture.
After cryolysis and urea solubilization of the cells, the proteome was digested to peptides, and the peptides were fractionated using hydrophilic interaction liquid chromatography (HILIC). HILIC-based fractionation works in a manner orthogonal to reverse-phase chromatography and has been used effectively in the enrichment of phosphorylated peptides (11, 12). Peptides with net hydrophobic properties elute first from the HILIC column followed by peptides with net hydrophilic properties. The charged nature of phosphate groups tends to increase the hydrophilic properties and the retention time of phosphorylated peptides on the column.
The largest percentages of phosphopeptides were delayed in their elution from the column and appeared near the end of the chromatographic trace (Fig. 2), although phosphopeptides were detected in other fractions as well. HILIC fractions were pooled for enrichment by IMAC based on the overall peptide abundance estimated from the chromatographic trace (Fig. 2).
Fig. 2.

Fractionation and analysis of phosphoproteome. A, HILIC fractionation of tryptic peptides retains phosphopeptides until later in the elution profile. Chromatographic traces at 214 (black) and 280 nm (gray) and the gradient profile are shown with the percentage of Solvent A (“Experimental Procedures”) indicated above the gradient. Pools of the HILIC-based fractionation for IMAC-based enrichment of phosphopeptides are shown on the x axis. Combined Fractions 8, 9, and 10 have the highest percentage of phosphopeptides. B, Venn diagram of the number of non-redundant peptides identified by MS2- and MSA-based mass spectrometry. The majority of the phosphoproteome can be identified by MS2 analysis alone. C, number of phosphorylation sites (N Phos) per peptide. The majority of the peptides identified were doubly phosphorylated (60%).
Mass spectrometric data were searched with both X!Tandem (24) and Mascot (26) and processed with the Trans-Proteomic Pipeline (28) using the spectral intensities of the isotopically different amino acids to compute quantitative differences between ion pairs in the glucose-grown and oleic acid-incubated cells (30). These spectral search data were integrated using iProphet,2 which uses PeptideProphet spectrum-level results from multiple MS runs to compute a new probability at the level of a unique peptide sequence (supplemental Fig. S1). Using this approach, 13,941 phosphorylated peptides were identified, and the relative abundances were quantified at an FDR of 1% (using a decoy database to establish the FDR).
Identification of Phosphopeptides
Phosphorylated peptides were identified by mass spectrometric analyses of both MS2 experiments and MSA analyses. Once redundant peptides were removed from the entire data set, 3,898 unique species of phosphorylated peptides remained. Peptides with the same primary sequence and different assignments of phosphorylated residues were counted as unique species and are included in supplemental Table S1A. Phosphopeptides that were ambiguously assigned to proteins were removed, leaving a total of 3,788 non-redundant phosphopeptides that were confidently assigned to proteins. The bulk of the identifications came from MS2 data (Fig. 2B), but a unique subset of the phosphoproteome was identified only through the use of MSA-based spectrometry. MSA analysis increased the number of peptide identifications by 12%. To account for instances where the same peptide would be counted multiple times due to missed cleavages, peptides that overlapped significantly (>50%), mapped to the same protein, and contained the same number of phosphates were grouped together (supplemental PhosPepAlign.m), yielding a total of 1,324 non-redundant peptides (supplemental Table S1B) that mapped unambiguously to 697 proteins (supplemental Table S1C). Most peptides were multiply phosphorylated peptides: 414 peptides (32%) with single phosphorylation sites were identified compared with 770 that were doubly phosphorylated (60%) and 109 that were triply phosphorylated (8%) (Fig. 2C and supplemental Table S1B, inset). A small number of peptides containing more than three phosphates were also identified (supplemental spectra). To assign the specific site of phosphorylation, a modified version of ASCORE was applied to the search results. This resulted in the unambiguous assignment of 892 phosphorylated residues for 527 peptides identified at a 99% confidence cutoff (supplemental Table S1-II).
The phosphopeptide data set was queried against the PhosphoPep yeast database (33) (www.phosphopep.org/index.php) (supplemental Table S2-I). Based on primary sequence comparisons, the majority (92%) of the proteins identified in this study were present in the PhosphoPep database, validating the approach; however, several new phosphopeptides and phosphoproteins were also identified (supplemental Table S2-II). Among the previously identified 179 phosphoproteins in the data set, 213 novel phosphorylation sites were identified; in addition, 54 novel phosphoproteins were identified (59 phosphorylation sites).
In an effort to understand kinase-substrate relationships in this data set, phosphopeptides were analyzed for enriching motifs by primary sequence analysis. These motifs were matched to known motifs using PhosphoMotif Finder (34). Each query motif and the possible submotifs identified are listed in supplemental Table S3. As reported in other studies (10, 40), these motifs included substrates for casein kinases I and II, G-protein-coupled receptor kinase I, and PKA/PKC. The S. cerevisiae homologs for kinases for which motifs were identified are listed in Table I.
Table I. Kinases for which motifs are significantly enriched in this phosphopeptide data set.
Statistical information for the identified motifs is provided in supplemental Table S3. The highly enriching motifs were queried against the Human Protein Reference Database using the PhosphoMotif Finder (34). Putative yeast homologs of the human kinases were identified through the HomoloGene Database (35) and searches of the Saccharomyces Genome Database (36).
| Feature of motif described in literature | Instances | S. cerevisiae homolog |
|---|---|---|
| Casein kinase II substrate motif | 155 | Yck1p, Yck2p, Yck3p, Cka1p, Cka2p, Ckb1p, Ckb2p |
| Casein kinase I substrate motif | 77 | Hrr25p |
| G-protein-coupled receptor kinase 1 substrate motif | 57 | Gpr1p, Gpa2p, Gpb1p, Gpb2p |
| MAPKAPK2 kinase substrate motif | 52 | MAPK |
| PKA kinase substrate motif | 48 | Tpk1p, Tpk2p, Tpk3p, Bcy1p |
| GSK3 kinase substrate motif | 26 | Mck1p, Rim11p, Mrk1p, Ygk3p |
| PKC kinase substrate motif | 25 | Pkc1p |
| GSK-3, ERK1, ERK2, CDK5 substrate motif | 22 | Mck1p, Rim11p, Mrk1p, Ygk3p, Fus3p, Kss1p, Pho85p |
| Calmodulin-dependent protein kinase II substrate motif | 8 | Cmk1p, Cmk2p |
| Unknown | 3 |
Quantitative Comparison of Carbon Source-specific Phosphoproteomes
Cells were grown in glucose-repressing or oleic acid-inducing medium in the presence of isotopically different amino acids (41), allowing the downstream quantification of phosphopeptides by ASAP ratio in the MS1 spectra of identified phosphopeptides (30). Peptides were considered enriched in glucose-repressing medium if they had an ASAP ratio (log2-based) less than 0.5 and were considered enriched in oleic acid-inducing medium if they had an ASAP ratio greater than 2. There were 467 peptides, corresponding to 350 proteins enriched in the oleate-inducing medium. 249 peptides, corresponding to 197 proteins, were enriched in the glucose-repressing condition (Fig. 3A). When both the oleic acid and glucose data sets were combined, 67 proteins showed dual enrichment; i.e. they contained at least one phosphopeptide that was enriched in oleic acid and at least one different phosphopeptide that was enriched in glucose (Fig. 3B).
Fig. 3.

Specific subsets of phosphoproteome enriched from cells incubated in oleic acid- or glucose-containing medium. A, comparison of the number of unique phosphopeptides, the number of phosphoproteins, and the number of unique phosphoproteins for oleic acid-enriched, glucose-enriched, and non-enriching phosphopeptides. B, Venn diagram showing the overlap between identified proteins with peptides increasing in phosphorylation in oleic acid and those proteins with peptides increasing in phosphorylation in glucose. C, subcellular localizations for proteins enriched in either oleic acid or glucose. D, functional annotations of the proteins as a kinase or phosphatase (Signaling), a transcription factor, or other. ER, endoplasmic reticulum.
The data set of 480 oleate-, glucose-, and dual enriched phosphoproteins was uploaded via the Firegoose interface (42) into the Protein Information and Property Explorer (43), and subcellular localizations were obtained for proteins in the data set (Fig. 3C). The data set was also queried for significantly enriching gene ontology annotations and showed enrichment for signaling/kinase activity (Fig. 3D and supplemental Table S4), regulation of actin, and the presence of plasma membrane proteins.
Within the oleate- or glucose-enriched phosphoprotein data set were a number of proteins that were previously identified as nodes in the core FA response signaling network governing the induction of peroxisomes (8). For example, phosphorylated forms of the transcription factors Adr1p and Pip2, which are known to be key transcriptional regulators of the FA response (3, 4), were enriched in oleic acid-induced cells. Enrichment of the glucose repression signaling molecule (Yck3p) was also detected in oleate-inducing medium. Interestingly, of six known negative effectors of derepression, three showed increased phosphorylation in oleic acid-induced cells (Hsl1p, Ypk1p, and Apl5p). Nine signaling molecules thought to act as positive effectors of oleic acid-induced expression were also identified: four with increased phosphorylation in oleic acid (Inp52p, Ste20p, Tps3p, and Ypk2p), four with increased phosphorylation in glucose-repressed conditions (Cdc19p, Gin4p, Lcb4p, and Tor1p), and Reg1p, which showed dual enrichment (different phosphorylated sites in glucose versus oleic acid). Signaling molecules known to be involved in the morphology of peroxisomes were also differentially phosphorylated. Pbs2p exhibited increased phosphorylation in glucose, whereas Rck1p, Ssk1p, and Yck1p exhibited increased phosphorylation in oleate-inducing medium; Akl5p showed dual enrichment.
The phosphorylated proteins detected by this study were compared with known signaling pathway components identified in the literature and by querying against the Kyoto Encyclopedia of Genes and Genomes for relevant pathways (8, 39, 44, 45) (supplemental Table S5). Remarkably, combining these phosphoproteomics data with the results from the functional genomics study (8) showed involvement of numerous signaling pathways, which must be coordinated during the response. Some pathways such as glucose sensing, G-protein signaling, and quiescence pathways were particularly evident in the data with as much as 60% of the known components identified (Fig. 4).
Fig. 4.

Detection of known signaling pathway proteins. Pathways identified in the literature were compared with proteins that were detected by mass spectrometry-based phosphoproteomics analysis.
Mutational Analysis of Transcription Factor Phosphorylated Amino Acid Residues
To test novel predictions arising from the identification of phosphorylation sites in these data, we focused on two novel phosphorylated transcription factors, Pip2p and Cst6p. Pip2p is a well characterized central regulator of oleate-responsive loci but has not been previously reported to be phosphorylated, and models of the transcriptional response involving Pip2p do not consider its potential posttranslational modifications (3, 4). Phosphorylated Pip2p was enriched in oleate-induced conditions but barely above the statistical cutoff used. On the other hand, phosphorylated Cst6p was strongly enriched in the oleate-induced state and has not been previously implicated in the transcriptional response to oleate.
Pip2p was determined to be phosphorylated in oleate at position Ser-783. Therefore, a serine to alanine (S783A) mutation was introduced, and the induction of oleate-responsive loci was tested. By comparison with wild-type cells, the S783A mutant led to a sharp increase in the levels of transcription after 30 min of oleate induction (Fig. 5). By 90 min, this effect was much less dramatic (data not shown), leading to the hypothesis that phosphorylation of Pip2p at Ser-783 tempers the levels of transcription of oleate-responsive loci at the early stages of the response.
Fig. 5.

Quantitative PCR reveals a role for oleate-enriched phosphorylation of Pip2p and Cst6p. A, yeast cells carrying a S783A-mutagenized version of Pip2p were tested for expression of oleate-responsive loci POT1, CTA1, FOX2, and POX1. Shown are the levels of induction after 30 min in oleate-inducing medium. By 90 min, the differences between wild type and the Pip2p S783A strain were not significant (data not shown). B, at 90 min of oleate induction, increased expression of oleate-responsive loci (POT1, CTA1, FOX2, and POX1) is detected in the two Cst6p mutagenized strains. Cst6-1 is multiply mutated (S396A,S399A,T401A), whereas Cst6-2 is singly mutated (T401A). Error bars show the standard deviation.
A similar effect was observed with Cst6p. Cst6p is a phosphoprotein with several phosphorylation sites (33); however, the biological relevance of these phosphorylation states is not clear. A doubly phosphorylated Cst6p peptide (at positions Ser-399 and Thr-401 with Thr-401 being a novel phosphosite) was enriched in oleate. Both singly (T401A) and multiply (S396A,S399A,T401A) mutated species of Cst6p were generated, and the effect of these mutations on expression of peroxisomal, FA-responsive loci was examined. As shown in Fig. 5, mutant Cst6p cells also showed an increase in expression of POT1, CTA1, FOX2, and POX1 compared with wild type, suggesting that Cst6p is a novel transcriptional regulator of oleate-responsive loci and that its phosphorylation dampens the response.
Nodes within Fatty Acid-responsive Signaling Network Show High Correlation of Connectivity and Influence
A network was constructed from the interactions between the phosphoproteins identified in this study and the kinases and phosphatases implicated in the core response to FA-mediated peroxisome induction (8) (Fig. 6). This network represents the known components and potential interactions between the components that regulate the response of S. cerevisiae to glucose and oleic acid and includes the core transcription factors Adr1p, Pip2p, Oaf1p, and Oaf3p, which differentially bind fatty acid-induced loci (4). The majority of the nodes in the network show few connections with a small number of highly connected nodes (Fig. 7, top). The degree of distribution of the network follows the power law, which is indicative of a scale-free network (Fig. 7, middle). The connectivity of this network was compared with the effect of node deletions on the regulation of FA-responsive loci (Fig. 7, bottom). The nodes with the greatest effect (more than one standard deviation from wild type) on the expression of FA-responsive loci showed significant enrichment for high connectivity (p value = 7.6 × 10−9) (supplemental Fig. S2).
Fig. 6.

FA signaling network. Integrated network of enriching phosphoproteins, signaling proteins known to regulate responses to FAs, and regulatory proteins. Proteins are indicated by nodes, and protein-protein interactions are shown by edges between the nodes. Green nodes indicate enrichment in oleic acid, red nodes indicate enrichment in glucose, blue nodes indicate enrichment of different sites in glucose and oleic acid, and white nodes indicate no information on phosphorylation status. The node border indicates the role of the protein as a positive (red), negative (green), or neutral (gray) effector of expression of FA-responsive loci. Diamond-shaped nodes indicate kinases, parallelogram-shaped nodes indicate phosphatases, octagonal nodes indicate regulators, circular nodes indicate transcription factors, and rounded rectangular nodes indicate phosphorylation substrates. The network is provided as a Cytoscape file (supplemental FA_Net_Saleem_MCP.cys) to facilitate interactive viewing.
DISCUSSION
Signal transduction via the reversible phosphorylation of proteins is a universal mechanism in virtually all cellular processes (1, 2). Here, quantitative mass spectrometry was used to reveal the changes of the phosphoproteome that accompany the transition of cells from conditions that repress (glucose) or induce (oleate) peroxisome proliferation in yeast. Integration of these data with physical and ontological data from the literature enabled the identification of an extensive phosphoproteome network governing the response. From more than 1,300 peptides mapping to 697 proteins, this study identified 480 proteins in which phosphorylation significantly changed when cells were transferred from glucose to oleic acid. From this study, 54 novel phosphoproteins were identified, and another 214 new phosphopeptides were identified in 179 previously known phosphoproteins. We note that this assay does not measure absolute protein levels but rather quantitatively interrogates the relative changes in the phosphoproteome. Using these data alone, it is not always possible to distinguish between changes in protein abundance and changes in the posttranslational state of a protein. We can, however, rule out dramatic changes in gene expression as underlying the vast majority of the changes in the phosphoproteome. Microarray analysis under the same conditions demonstrated that there is no correlation, on a genome-wide level, between the changes in expression and changes in the phosphoproteome (supplemental Fig. S3).
We previously mapped the signalome portion of the FA-responsive network using a representative protein of the peroxisomal matrix (8); viable deletions of signaling molecules were individually assayed to determine the contribution of each signaling molecule to the FA response. Many of the kinases and phosphatases identified in that study as regulating peroxisome biogenesis were also found here to be differentially phosphorylated. These key components of the FA response include regulators of glucose repression (Hsl1p, Ypk1p, and Apl5p) and regulators required for robust activation of an FA-induced gene encoding a peroxisomal reporter (Inp52p, Ste20p, Tps3p, Ypk2p, Cdc19p, Gin4p, Lcb4p, Tor1p, and Reg1p).
A key goal of this quantitative analysis of the phosphoproteome was the generation of new hypotheses that would have otherwise remained obscured. Importantly, oleate-specific enrichments in the phosphorylation status of two transcription factors, Pip2p and Adr1p, which are known to be essential for activation of oleate-responsive loci, were detected. Mutagenesis of the novel phosphorylated residue on Pip2p revealed its role in modulating the oleate-mediated transcriptional responses of Pip2p targets. Although previously not linked to the oleate response, Cst6p is known to be involved in the utilization of non-optimal carbon sources (46) and was identified as a strongly enriching component of the oleate-induced phosphorylome. This raises the possibility that Cst6p is enriched in response to the detergent used to solubilize the oleic acid rather than oleic acid itself. Although this detergent alone has no observable effect on peroxisome induction, the sensitivity of the proteomics approaches may identify molecular changes not evident at the morphological level. Nonetheless, analysis of the role of Cst6p demonstrates a role in the expression of oleate-responsive loci. Indeed, like Pip2p, the phosphorylation of Cst6p modulates the response of oleate-induced genes; however, it appears that these posttranslational modifications play different time-dependent roles. As the transcriptional regulation of oleate-induced peroxisome proliferation is used as a paradigm for understanding transcriptional network dynamics, it will be important to incorporate these new insights into future models (3, 4, 47–49).
Although signaling pathways are often depicted as discreet, linear pathways, it is increasingly recognized that dense network architectures actually underlie signaling processes (50, 51) and that cells must naturally coordinate activities in response to a myriad of complex molecular stimuli. This appears to be a feature of the network governing the response to FAs. Through analyses of the signalome (8) and the phosphorylome (this study), a number of pathways were observed to be activated in response to exposure to FAs. These include networks responsible for glucose sensing, alterations in cell cycle, and mediators controlling the phosphorylation status of lipids. By phosphoproteomics analysis, not only were key signaling molecules of the FA-responsive network detected, but also signaling molecules previously shown to be involved in the signal transduction of a number of other pathways (Fig. 4), including Hog1p and Ste20p of the MAPK pathways, were discovered. This study demonstrates that the phosphorylation status of these components changes in response to stimulation with glucose or oleic acid, highlighting the integration of these components into what forms the larger FA-responsive network (Fig. 5). However, it should be noted that multiple factors have the potential to influence the network response, and the challenge remains to determine to what extent such a complex network response can be deconvolved into its component responses. Indeed, whether the activation of these pathways is required for the response to oleate-inducing medium or is a dispensable secondary effect due to cross-talk of network components remains an active area of investigation. Microarray analyses of HOG1 and STE20 deletion strains indicate that the effects of these two genes are at least partially integrated; a number of oleate-responsive loci are down-regulated in comparison with wild-type cells when either of these deletions strains is exposed to FAs (data not shown).
Finally, beyond the hypotheses generated from the interactions within the network, the architecture of this nutrient-responsive network itself reveals additional biological insight. Within the FA-responsive network, highly connected nodes correspond to genes that when deleted show the most dramatic phenotypes with respect to peroxisome biogenesis and oleate-responsive gene expression. In other data sets, highly connected nodes correlate with essentiality (52). This quality of a few, highly influential nodes, representing network hubs, with numerous less influential nodes is a trait of scale-free networks and is a common network organization in biological systems (53). This FA-responsive network extends the concept of highly connected nodes being essential for cell viability, showing the same topology at the nutrient utilization level. Thus, although the network presented herein is dense and contains pathway components that encompass all the requisite programs for S. cerevisiae to respond to FAs as a nutrient source, scale-free properties are conserved in the signaling network governing responses to FAs (Fig. 6C).
This FA-responsive network represents a potential, static map of the components of the network and provides an extensive framework from which to map FA signaling in a dynamic manner, including temporal organization and information flow via network perturbations. The comprehensive and quantitative nature of this phosphoproteomics analysis suggests that identification of large numbers of phosphorylated peptides in the various states is not sufficient to define the cellular response. Analogous to an expression microarray, it is the relative abundance of the phosphorylated peptides in different conditions that defines the cellular state, and deciphering this code is a major collective challenge. Understanding cellular behaviors at a systems-wide level will require the elucidation of these networks and ultimately derivation of the information flow within the network. Global phosphoproteomics analyses of these cell states provide an opportunity to generate the data necessary for such a systems-level view of these networks.
Supplementary Material
Acknowledgments
We thank Abraham Armstrong and Christina Arens for technical assistance. We also thank the Luxembourg Centre for Systems Biomedicine and the University of Luxembourg for support.
* This work was supported, in whole or in part, by National Institutes of Health Grants GM075152, RR022220, and GM076547. This work was also supported by Canadian Institutes of Health Research Grant 53326.
 This article contains supplemental Figs. S1–S3, Tables S1–S6, spectra, and Cytoscape and MATLAB files.
This article contains supplemental Figs. S1–S3, Tables S1–S6, spectra, and Cytoscape and MATLAB files.
2 D. Shteynberg, E. W. Deutsch, H. Lam, J. K. Eng, Z. Sun, N. Tasman, L. Mendoza, R. L. Moritz, R. Aebersold, and A. Nesvizhskii, manuscript in preparation.
1 The abbreviations used are:
- FA
- fatty acid
- MSA
- multistage activation
- FDR
- false discovery rate
- HILIC
- hydrophilic interaction liquid chromatography
- ASAP
- automated statistical analysis of peptide
- Ascore
- ambiguity score
- QTIPS
- quantification by total identified peptides for SILAC
- RGT
- restores glucose transport
- TOR
- target of rapamycin
- SPS
- ssy1-ptr3-ssy5
- GCN
- general control system for amino acid biosynthesis.
REFERENCES
- 1.Cohen P. (2000) The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem. Sci. 25, 596–601 [DOI] [PubMed] [Google Scholar]
- 2.Delom F., Chevet E. (2006) Phosphoprotein analysis: from proteins to proteomes. Proteome Sci. 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ratushny A. V., Ramsey S. A., Roda O., Wan Y., Smith J. J., Aitchison J. D. (2008) Control of transcriptional variability by overlapping feed-forward regulatory motifs. Biophys. J. 95, 3715–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith J. J., Ramsey S. A., Marelli M., Marzolf B., Hwang D., Saleem R. A., Rachubinski R. A., Aitchison J. D. (2007) Transcriptional responses to fatty acid are coordinated by combinatorial control. Mol. Syst. Biol. 3, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith J. J., Aitchison J. D. (2009) Regulation of peroxisome dynamics. Curr. Opin. Cell Biol. 21, 119–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marelli M., Smith J. J., Jung S., Yi E., Nesvizhskii A. I., Christmas R. H., Saleem R. A., Tam Y. Y., Fagarasanu A., Goodlett D. R., Aebersold R., Rachubinski R. A., Aitchison J. D. (2004) Quantitative mass spectrometry reveals a role for the GTPase Rho1p in actin organization on the peroxisome membrane. J. Cell Biol. 167, 1099–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saleem R. A., Smith J. J., Aitchison J. D. (2006) Proteomics of the peroxisome. Biochim. Biophys. Acta 1763, 1541–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saleem R. A., Knoblach B., Mast F. D., Smith J. J., Boyle J., Dobson C. M., Long-O'Donnell R., Rachubinski R. A., Aitchison J. D. (2008) Genome-wide analysis of signaling networks regulating fatty acid-induced gene expression and organelle biogenesis. J. Cell Biol. 181, 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gruhler A., Olsen J. V., Mohammed S., Mortensen P., Faergeman N. J., Mann M., Jensen O. N. (2005) Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell. Proteomics 4, 310–327 [DOI] [PubMed] [Google Scholar]
- 10.Wilson-Grady J. T., Villén J., Gygi S. P. (2008) Phosphoproteome analysis of fission yeast. J. Proteome Res. 7, 1088–1097 [DOI] [PubMed] [Google Scholar]
- 11.Albuquerque C. P., Smolka M. B., Payne S. H., Bafna V., Eng J., Zhou H. (2008) A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell. Proteomics 7, 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McNulty D. E., Annan R. S. (2008) Hydrophilic interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol. Cell. Proteomics 7, 971–980 [DOI] [PubMed] [Google Scholar]
- 13.Villén J., Gygi S. P. (2008) The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 3, 1630–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ficarro S. B., McCleland M. L., Stukenberg P. T., Burke D. J., Ross M. M., Shabanowitz J., Hunt D. F., White F. M. (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20, 301–305 [DOI] [PubMed] [Google Scholar]
- 15.Larsen M. R., Thingholm T. E., Jensen O. N., Roepstorff P., Jørgensen T. J. (2005) Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 4, 873–886 [DOI] [PubMed] [Google Scholar]
- 16.Pinkse M. W., Uitto P. M., Hilhorst M. J., Ooms B., Heck A. J. (2004) Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal. Chem. 76, 3935–3943 [DOI] [PubMed] [Google Scholar]
- 17.Tao W. A., Wollscheid B., O'Brien R., Eng J. K., Li X. J., Bodenmiller B., Watts J. D., Hood L., Aebersold R. (2005) Quantitative phosphoproteome analysis using a dendrimer conjugation chemistry and tandem mass spectrometry. Nat. Methods 2, 591–598 [DOI] [PubMed] [Google Scholar]
- 18.Zhou H., Watts J. D., Aebersold R. (2001) A systematic approach to the analysis of protein phosphorylation. Nat. Biotechnol. 19, 375–378 [DOI] [PubMed] [Google Scholar]
- 19.de la Fuente van Bentem S., Mentzen W. I., de la Fuente A., Hirt H. (2008) Towards functional phosphoproteomics by mapping differential phosphorylation events in signaling networks. Proteomics 8, 4453–4465 [DOI] [PubMed] [Google Scholar]
- 20.Blagoev B., Ong S. E., Kratchmarova I., Mann M. (2004) Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat. Biotechnol. 22, 1139–1145 [DOI] [PubMed] [Google Scholar]
- 21.Dengjel J., Akimov V., Olsen J. V., Bunkenborg J., Mann M., Blagoev B., Andersen J. S. (2007) Quantitative proteomic assessment of very early cellular signaling events. Nat. Biotechnol. 25, 566–568 [DOI] [PubMed] [Google Scholar]
- 22.Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 23.Dilworth D. J., Saleem R. A., Rogers R. S., Mirzaei H., Boyle J., Aitchison J. D. (2010) QTIPS: a novel method of unsupervised determination of isotopic amino acid distribution in SILAC experiments. J. Am. Soc. Mass Spectrom. 21, 1417–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 25.MacLean B., Eng J. K., Beavis R. C., McIntosh M. (2006) General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics 22, 2830–2832 [DOI] [PubMed] [Google Scholar]
- 26.Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 27.Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., Aebersold R. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller A., Eng J., Zhang N., Li X. J., Aebersold R. (2005) A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 30.Li X. J., Zhang H., Ranish J. A., Aebersold R. (2003) Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal. Chem. 75, 6648–6657 [DOI] [PubMed] [Google Scholar]
- 31.Ulintz P. J., Yocum A. K., Bodenmiller B., Aebersold R., Andrews P. C., Nesvizhskii A. I. (2009) Comparison of MS(2)-only, MSA, and MS(2)/MS(3) methodologies for phosphopeptide identification. J. Proteome Res. 8, 887–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beausoleil S. A., Villén J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 33.Bodenmiller B., Campbell D., Gerrits B., Lam H., Jovanovic M., Picotti P., Schlapbach R., Aebersold R. (2008) PhosphoPep—a database of protein phosphorylation sites in model organisms. Nat. Biotechnol. 26, 1339–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amanchy R., Periaswamy B., Mathivanan S., Reddy R., Tattikota S. G., Pandey A. (2007) A curated compendium of phosphorylation motifs. Nat. Biotechnol. 25, 285–286 [DOI] [PubMed] [Google Scholar]
- 35.Wheeler D. L., Church D. M., Lash A. E., Leipe D. D., Madden T. L., Pontius J. U., Schuler G. D., Schriml L. M., Tatusova T. A., Wagner L., Rapp B. A. (2001) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 29, 11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cherry J. M., Adler C., Ball C., Chervitz S. A., Dwight S. S., Hester E. T., Jia Y., Juvik G., Roe T., Schroeder M., Weng S., Botstein D. (1998) SGD: Saccharomyces Genome Database. Nucleic Acids Res. 26, 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gray M., Kupiec M., Honigberg S. M. (2004) Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wan Y., Saleem R. A., Ratushny A. V., Roda O., Smith J. J., Lin C. H., Chiang J. H., Aitchison J. D. (2009) Role of the histone variant H2A.Z/Htz1p in TBP recruitment, chromatin dynamics, and regulated expression of oleate-responsive genes. Mol. Cell. Biol. 29, 2346–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith J. J., Marelli M., Christmas R. H., Vizeacoumar F. J., Dilworth D. J., Ideker T., Galitski T., Dimitrov K., Rachubinski R. A., Aitchison J. D. (2002) Transcriptome profiling to identify genes involved in peroxisome assembly and function. J. Cell Biol. 158, 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz D., Gygi S. P. (2005) An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 41.Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 42.Bare J. C., Shannon P. T., Schmid A. K., Baliga N. S. (2007) The Firegoose: two-way integration of diverse data from different bioinformatics web resources with desktop applications. BMC Bioinformatics 8, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramos H., Shannon P., Aebersold R. (2008) The protein information and property explorer: an easy-to-use, rich-client web application for the management and functional analysis of proteomic data. Bioinformatics 24, 2110–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koerkamp M. G., Rep M., Bussemaker H. J., Hardy G. P., Mul A., Piekarska K., Szigyarto C. A., De Mattos J. M., Tabak H. F. (2002) Dissection of transient oxidative stress response in Saccharomyces cerevisiae by using DNA microarrays. Mol. Biol. Cell 13, 2783–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith J. J., Sydorskyy Y., Marelli M., Hwang D., Bolouri H., Rachubinski R. A., Aitchison J. D. (2006) Expression and functional profiling reveal distinct gene classes involved in fatty acid metabolism. Mol. Syst. Biol. 2, 2006.0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Gimeno M. A., Struhl K. (2000) Aca1 and Aca2, ATF/CREB activators in Saccharomyces cerevisiae, are important for carbon source utilization but not the response to stress. Mol. Cell. Biol. 20, 4340–4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gurvitz A., Rottensteiner H. (2006) The biochemistry of oleate induction: transcriptional upregulation and peroxisome proliferation. Biochim. Biophys. Acta 1763, 1392–1402 [DOI] [PubMed] [Google Scholar]
- 48.Karpichev I. V., Durand-Heredia J. M., Luo Y., Small G. M. (2008) Binding characteristics and regulatory mechanisms of the transcription factors controlling oleate-responsive genes in Saccharomyces cerevisiae. J. Biol. Chem. 283, 10264–10275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Litvak V., Ramsey S. A., Rust A. G., Zak D. E., Kennedy K. A., Lampano A. E., Nykter M., Shmulevich I., Aderem A. (2009) Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat. Immunol. 10, 437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Friedman A., Perrimon N. (2007) Genetic screening for signal transduction in the era of network biology. Cell 128, 225–231 [DOI] [PubMed] [Google Scholar]
- 51.Jordan J. D., Landau E. M., Iyengar R. (2000) Signaling networks: the origins of cellular multitasking. Cell 103, 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jeong H., Mason S. P., Barabási A. L., Oltvai Z. N. (2001) Lethality and centrality in protein networks. Nature 411, 41–42 [DOI] [PubMed] [Google Scholar]
- 53.Barabási A. L., Oltvai Z. N. (2004) Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 5, 101–113 [DOI] [PubMed] [Google Scholar]
- 54.Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.