

Abstract
Background and purpose:
Purinoceptors containing the P2X3 subunit (P2X3 homotrimeric and P2X2/3 heterotrimeric) are members of the P2X family of ion channels gated by ATP and may participate in primary afferent sensitization in a variety of pain-related diseases. The current work describes the in vitro pharmacological characteristics of AF-353, a novel, orally bioavailable, highly potent and selective P2X3/P2X2/3 receptor antagonist.
Experimental approach:
The antagonistic potencies (pIC50) of AF-353 for rat and human P2X3 and human P2X2/3 receptors were determined using methods of radioligand binding, intracellular calcium flux and whole cell voltage-clamp electrophysiology.
Key results:
The pIC50 estimates for these receptors ranged from 7.3 to 8.5, while concentrations 300-fold higher had little or no effect on other P2X channels or on an assortment of receptors, enzymes and transporter proteins. In contrast to A-317491 and TNP-ATP, competition binding and intracellular calcium flux experiments suggested that AF-353 inhibits activation by ATP in a non-competitive fashion. Favourable pharmacokinetic parameters were observed in rat, with good oral bioavailability (%F = 32.9), reasonable half-life (t1/2 = 1.63 h) and plasma-free fraction (98.2% protein bound).
Conclusions and implications:
The combination of a favourable pharmacokinetic profile with the antagonist potency and selectivity for P2X3 and P2X2/3 receptors suggests that AF-353 is an excellent in vivo tool compound for study of these channels in animal models and demonstrates the feasibility of identifying and optimizing molecules into potential clinical candidates, and, ultimately, into a novel class of therapeutics for the treatment of pain-related disorders.
Keywords: P2X, purinergic, ATP, ion channel, antagonist, urinary bladder
Introduction
ATP, a ubiquitous energy donor and receptor ligand in living cells, gates a family of ion channels known as P2X receptors, which, not surprisingly, are themselves localized widely in cell types of nearly every origin, including neuronal, muscular, epithelial and immune (Burnstock and Knight, 2004). The functional P2X channel is assembled as hetero- or homo-trimers from protein subunits encoded by one or more of seven genetically distinct subtypes, P2X1-7 (North and Surprenant, 2000; Gever et al., 2006; nomenclature follows Alexander et al., 2008). The distribution of one protein subunit, P2X3, is somewhat more limited than the other subtypes, with mRNA and/or protein found largely in small-diameter, unmyelinated C-fiber sensory neurons, and, possibly, in some epithelial cells and enteric neurons (Chen et al., 1995; Lewis et al., 1995; Vulchanova et al., 1998; Jin et al., 2004; Wang et al., 2005; Burnstock, 2008). Since C-fiber sensory neurons have been shown to be important for the detection of noxious stimuli in damaged or sensitized tissues, P2X3-containing channels have emerged as targets of significant interest for the treatment of certain types of pain and disorders of visceral sensory function, such as overactive bladder (Burnstock, 2007).
Accordingly, P2X3 receptor gene deletion results in a markedly attenuated nocifensive phenotype in mice, including altered sensitivity to thermal stimuli and decreased pain-related behaviours after intraplantar injection of carageenan or formalin (Cockayne et al., 2000). Reduction of P2X3 receptor expression through intrathecal administration of P2X3-selective antisense or siRNA also causes a significant decrease in behavioural signs of chronic inflammatory and neuropathic pain in mice (Barclay et al., 2002; Honore et al., 2002a; Dorn et al., 2004). The role of the P2X3 protein in the function of visceral organs, such as the urinary bladder and small intestine, is suggested by the sensory deficits observed in P2X3 receptor-KO mice, that included urinary and intestinal hyporeflexia (Cockayne et al., 2000; Bian et al., 2003; Ren et al., 2003). Thus, it appears that P2X3 protein subunits are involved in sensitization of many types of somatic and visceral afferent circuits.
Until recently, pharmacological studies linking P2X3 with nociception relied on the use of molecules with poor potency, selectivity and permeability, such as pyridoxal phosphate-6-azo (benzene-2,4-disulphonic acid) (PPADS), suramin, reactive blue 2, and/or low metabolic stability, such as 2′,3′-O-(2,4,6-trinitrophenyl) ATP (TNP-ATP), making them less than ideal for critical in vivo studies (Jarvis et al., 2001; Honore et al., 2002b; Ueno et al., 2003). A more selective and potent, low molecular weight, dual hP2X3/hP2X2/3 antagonist, A-317491 (Ki = 9–22 nM; >1000-fold selective over other P2X channels), was described by Jarvis et al. (2002). However, undesirable characteristics, including very high protein binding (>99.9%), low oral bioavailability and poor CNS penetration, also limit its use as an in vivo tool.
A-317491 was reported to behave as a competitive antagonist on the basis of functional curve shift experiments using the slowly desensitizing P2X2/3 receptor. Several research groups have created chimeric P2X protein subunits by combining the N-terminus and first transmembrane domain of the slowly desensitizing P2X2 channel with the remaining extracellular portion, second transmembrane domain and intracellular C-terminus of either P2X1 or P2X3 in order to confer slow desensitization kinetics on the rapidly desensitizing P2X channels while retaining the pharmacological selectivity of the rapidly desensitizing P2X subtype (Werner et al., 1996; Neelands et al., 2003). In this manner, the competitive antagonist TNP-ATP, when tested in curve shift experiments at the chimeric P2X2-3 receptor, behaved in a manner consistent with competitive antagonism, as seen previously with the heteromeric P2X2/3 receptor, whereas it had previously appeared to exhibit insurmountable behavior using the wildtype P2X3 channel (Virginio et al., 1998; Neelands et al., 2003).
In an effort to identify novel antagonists, we conducted a series of chemical library screens at recombinant P2X3 and P2X2/3 channels, and identified several novel and medicinally interesting chemical leads with reasonable potency. Chemical optimization has been successful in the subsequent generation of high affinity, selective pharmacological tools from these leads. In the current work, we present data on a compound of unique chemical structure, AF-353 (Figure 1), demonstrating high antagonist potency at and selectivity for P2X3 and P2X2/3 channels, moderate protein binding, oral bioavailability and attractive pharmacokinetic profile suitable for in vivo studies and an allosteric mechanism of antagonism.
Figure 1.
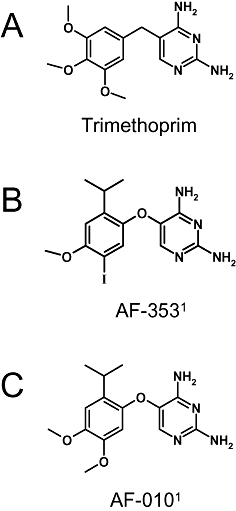
Chemical structures of (A) trimethoprim; MW = 290.32, (B) AF-353; MW = 400.21 and (C) AF-010; MW = 304.35. 1Note that AF-353 and AF-010 were previously known as RO-4 and RO-10, respectively.
Methods
Cell culture
Chinese hamster ovary (CHO-K1) cells stably expressing recombinant human P2X4, rat P2X5 or human P2X7 were cultured in Ham's F12 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, 250 µg·mL−1 G418 (Invitrogen) and 100 µg·mL−1 hygromycin B. Expression of recombinant human P2X1 and rat P2X3 in CHOK-K1 cells was regulated through the use of a tetracycline-controlled transactivator (tTA) gene expression system, such that cells grown in the presence of 0.1 µg·mL−1 tetracycline did not express P2X1 or P2X3 protein subunits, but upon removal of tetracycline from the growth medium, abundant expression of P2X1 or P2X3 was achieved within 7 days (Lachnit et al., 2000). 1321N1 Human astrocytoma cells expressing hP2X2, hP2X3 and hP2X2/3 were cultured in a base medium of Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and either 275 µg·mL−1 G418 (hP2X2), 0.25 µg·mL−1 puromycin (hP2X3), or 300 µg·mL−1 G418 and 0.25 µg·mL−1 puromycin (hP2X2/3). 1321N1 astrocytoma cells expressing chimeric human P2X2-3 were prepared as described previously (Neelands et al., 2003).
Intracellular calcium flux
Receptor-evoked changes in intracellular calcium were measured using calcium-selective fluorescent dyes quantitated with a fluorometric imaging plate reader (FLIPR; Molecular Devices, Sunnyvale, CA, USA). CHO-K1 cells (transfected with recombinant human P2X1, rat P2X3, human P2X4, human P2X5 or human P2X7 receptor subunits) and 1321N1 astrocytoma cells (transfected with cloned human P2X2, human P2X3 or human P2X2/3) were passaged in flasks in commercial media containing and lacking phenol red (CHOK1 in Ham's F-12 and 1321N1 in DMEM; Invitrogen). Eighteen to 24 h before the FLIPR experiment, cells were released from their flasks, centrifuged and resuspended in nutrient medium at 2.5 × 105 cells·mL−1. The cells were aliquoted into black-walled 96-well plates at a density of 5 × 104 cells per well and incubated overnight in 5% CO2 at 37°C. On the day of the experiment, cells were washed in FLIPR buffer (FLIPR buffer: calcium- and magnesium-free Hank's balanced salt solution, 10 mM HEPES, 2 mM CaCl, 2.5 mM probenecid; FB) and loaded with fluorescent dye Fluo-3 AM (2 µM final conc.). After a 1 h dye loading incubation at 37°C, the cells were washed to remove extracellular dye, and test compounds (dissolved with DMSO at 10 mM and serially diluted with FB) or vehicle were added to each well and allowed to equilibrate for 20 or 60 min at room temperature. The plates were then placed in the FLIPR, and a baseline fluorescence measurement (excitation @ 488 nm and emission @ 510–570 nm) was obtained for 10 s before agonist or vehicle addition. The agonist α,β-methyleneATP (α,β-meATP) was added to produce a final concentration ranging from 3 nM to 10 µM. Fluorescence was measured for an additional 2 min at 1 s intervals after agonist addition. A final addition of ionomycin (5 µM, final concentration) was made to each well of the FLIPR test plate to establish cell viability and maximum fluorescence of dye-bound cytosolic calcium. Peak fluorescence in response to the addition of α,β-meATP (in the absence and presence of test compounds) was measured and inhibition curves generated using nonlinear regression (Prism v.4, GraphPad Software, San Diego, CA, USA) employing a four parameter logistic equation (Y = Bottom + (Top − Bottom)/(1 + 10^(LogEC50 − X)*HillSlope). PPADS, a ‘standard’ P2X antagonist, was used as a positive control.
Pharmacological selectivity
The selectivity of AF-353 for P2X3 and P2X2/3 channels over other homomeric P2X channels was established by measuring the potency of antagonism by AF-353 of agonist-evoked intracellular calcium flux in cell lines expressing recombinant P2X channels (see above). Additionally, AF-353 was examined in two broad commercially available panels of selectivity, one covering 75 receptors, channels, enzymes and transporters (Cerep, Poitiers, France), and a second one covering more than 100 kinases (Ambit).
Radioligand binding
Radioligand binding experiments were conducted in membranes derived from Chinese hamster ovary cells (CHO) expressing the rat P2X3 (CHO-rP2X3) ion channel using a tetracycline-off expression vector (Lachnit et al., 2000) or 1321N1 human astrocytoma cells expressing hP2X3 or hP2X2/3. Cells were harvested in 1X Versene (Invitrogen) and homogenized by a Polytron (Kinematica, Lucerne, Switzerland) in ice-cold 50 mM Tris pH 7.4 with 1X Complete™ protease inhibitor cocktail (Roche Molecular Systems, Pleasanton, CA, USA). Plasma membranes were isolated by a two-step centrifugation. Homogenized membranes were centrifuged at 1000×g for 15 min at 4°C. The 1000×g pellet was discarded, and the supernatant was centrifuged at 43 000×g for 30 min at 4°C. The 43 000×g supernatant was discarded, and the pellet was stored at −70°C until assayed. Tritium-labelled AF-353 (81.2 Ci·mmol−1) was synthesized by the Radiochemistry Department at Roche (Palo Alto, CA, USA); purity was confirmed by HPLC to be >97%. The ligand affinities at P2X3 and P2X2/3 membranes were determined under equilibrium-binding conditions in 50 mM Tris pH 7.4. [3H]-AF-353 (1.7–140 nM for homomeric and 1.3–660 nM for heteromeric ion channels) was incubated with membranes (200–350 µg·mL−1) in the absence or presence of 10 µM of an unlabelled AF-353 analogue, AF-010 (to define non-specific binding) for 2–5 h at 22°C to determine its equilibrium dissociation affinity constant (Kd), as well as the receptor expression level (Bmax) of the membranes. For unlabelled molecules, dissociation affinity constants (KB) were determined by co-incubating unlabelled molecules (serially diluted over a million-fold concentration range) with [3H] AF-353 (1–5 nM) and CHO-rP2X3 membranes. In all cases, incubation was ended by filtration with ice-cold 50 mM Tris (pH 7.4) on GF/B filters. Filters were soaked in MicroScint-20 scintillation cocktail (PerkinElmer Life Sciences, Boston, MA) for at least 3 h prior to quantification of filter-trapped radioactivity using a Perkin Elmer TopCount plate reader. Competition binding data were analyzed by non-linear regression using a four-parameter hyperbolic function to estimate curve maxima, curve minima, Hill slope and IC50; KB estimates were calculated from observed IC50 values using the Cheng–Prusoff equation (Cheng and Prusoff, 1973). Affinities are presented as the mean and standard deviations determined over two to four repeat experiments.
Assessment of competition in the binding modes between [3H] AF-353 and other unlabelled test ligands was based on the expectations of the Cheng–Prusoff relationship that describes binding of two ligands in a mutually exclusive manner (Cheng and Prusoff, 1973). This relationship is described by the equation  , where [A*] represents the radioligand concentration, Kd the equilibrium dissociation constant of A*, KB the equilibrium dissociation constant of the unlabelled test compound, and IC50 is the test compound concentration that displaces 50% of the binding of A*. Radioligand binding studies were conducted using the conditions described above by incubating CHO-rP2X3 membranes with [3H]-AF-353 in the absence or presence of competing agents. For this analysis, IC50 values were determined for each unlabelled test compound over a range of 5–8 different radioligand concentrations ([3H]-AF-353 0.1–60 nM). The observed IC50 values were plotted as a ratio of IC50/KB versus [A*]/Kd for all concentrations of radioligand (A*) tested. From the Cheng–Prusoff equation, a competitive interaction between unlabelled ligand and [3H] AF-353 will plot as a line with a slope of unity, and a Y-axis intercept of 1.
, where [A*] represents the radioligand concentration, Kd the equilibrium dissociation constant of A*, KB the equilibrium dissociation constant of the unlabelled test compound, and IC50 is the test compound concentration that displaces 50% of the binding of A*. Radioligand binding studies were conducted using the conditions described above by incubating CHO-rP2X3 membranes with [3H]-AF-353 in the absence or presence of competing agents. For this analysis, IC50 values were determined for each unlabelled test compound over a range of 5–8 different radioligand concentrations ([3H]-AF-353 0.1–60 nM). The observed IC50 values were plotted as a ratio of IC50/KB versus [A*]/Kd for all concentrations of radioligand (A*) tested. From the Cheng–Prusoff equation, a competitive interaction between unlabelled ligand and [3H] AF-353 will plot as a line with a slope of unity, and a Y-axis intercept of 1.
Whole cell voltage clamp electrophysiology
Standard giga-seal patch clamp technique was employed to study the P2X2/3 channel for these experiments. The patch clamp rig consisted of the following components: Anti-vibration table (TMC, Peabody, MA, USA), microscope (Axiovert 100, Zeiss, Thornwood, NY, USA), micromanipulator (MP285, Sutter Instruments, Novato, CA, USA), patch-clamp amplifier (Axopatch 200B, Molecular Devices, Sunnyvale, CA, USA), digitizer (Digidata 1200, Molecular Devices), drug perfusion system (Dynaflow ProII Perfusion system, Cellectricon, Rockville, MD, USA), and acquisition software (Molecular Devices PClamp9).
For recordings, the bath solution consisted of (in mM) 155 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 d-glucose, 10 HEPES, pH 7.4 with NaOH, 310 mOsM, and the pipette intracellular solution consisted of (in mM) 130 CsF, 10 NaCl, 10 EGTA, 1 MgCl2, 10 HEPES, pH 7.2 with CsOH, 290mOsM. Standard wall borosilicate glass electrodes (OD 1.50 mm, ID 0.87 mm, with filament) were pulled with a Sutter Instruments P-87 pipette puller. The average resistance of the electrodes used was 3.5 MOhm. To activate the P2X2/3 heteromeric channel, 10 µM α,β-meATP solution (pH adjusted with NaOH) was used. This value is approximately the EC50 for the channel under the conditions of the experiment. The channel was activated for 2 s, at regular intervals of approximately 30 s. Test compound was added when the current from the channel was consistent for at least three agonist applications (about 90 s). The block due to the compound was monitored until equilibrium was achieved, and then compound was washed out to determine the off-rate kinetics. Data were analyzed using Molecular Devices ClampFit, as well as OriginLab (Northampton, MA, USA).
Pharmacokinetics
Animals
All animal care and experimental protocols for the pharmacokinetic studies were approved by the Institutional Animal Care and Use Committee at Roche, LLC. Male Hanover Wistar rats (240–300 g) with a single catheter inserted into the jugular vein were obtained from Charles River Laboratories (Hollister, CA, USA). Intravenous dosing was via the tail vein and oral administration was with disposable Teflon gavage needles.
To prepare brain samples, rats were killed with isoflurane, the skin was removed from the skull and the skull opened with sharp scissors. The skull flaps were removed, the brain was carefully removed with a spatula and gently blotted dry with Kim Wipes before weighing in tared scintillation vials and stored frozen at −80°C until used.
Blood collection
Blood was collected at pre-determined time points, using lithium heparin as anticoagulant from the jugular vein. After centrifugation at 3000×g for 5 min, plasma was obtained and stored at −80°C until analysis.
Plasma protein binding
Heparinized rat plasma was obtained from Pel-Freez® Biologicals (Rogers, AR, USA) and stored at −80°C until use. Centrifree Micropartition Devices (Millipore, Bedford, MA, USA) were used to separate unbound from protein-bound material. AF-353 was added to heparinized ultrafiltered plasma (n = 3) to yield a final concentration between 200 and 5000 ng·mL−1. One millilitre of the plasma solutions and 0.3 mL of the ultrafiltrate solution were added to the filtration device and centrifuged (fixed angle) for 20 min at 2000×g. Protein binding was calculated according to:
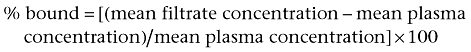 |
Determination of brain to plasma ratio
Three parts (weight) of saline were added to one part of brain tissue. The brain was minced and subsequently homogenized (2 × 10 s) on ice using a sonicator (Ultrasonic Processor XL, Heat Systems, Inc., Farmingdale, NY, USA). Sample preparation was the same as for plasma samples. Brain to plasma ratios were calculated according to:
 |
Bioanalytical method for AF-353
Plasma concentrations of AF-353 were determined using a LC-MS/MS method. Samples were analysed using a LC-MS/MS system consisting of an Agilent 1100 HPLC pump (Agilent, Santa Clara, CA, USA), a LEAP HTC PAL autosampler (LEAP Technologies, Carrboro, NC, USA), interfaced with a Waters Quattro Ultima mass spectrometer (Waters, Milford, MA, USA), which was equipped with an ion electrospray ionization (ESI) interface acting in positive mode. Multiple reaction monitoring (MRM) scanning was used for ion monitoring. Instrument control and data acquisition was performed using the MassLynx software version 4.0 (Waters Corporation, Milford, MA, USA). Sample preparation was as follows. Briefly, 10–100 µL of sample were transferred to a 96-well plate. Volume was brought to a final volume of 100 µL with blank rat plasma, and 50 µL of a methanol/water (1:1) mixture was added. Internal standard (Roche proprietory compound) was added at a final concentration of 2.5 ng·mL−1. Acetonitrile (150 µL) was added to precipitate the protein. After briefly vortexing, sample plates were centrifuged for 5 min. One hundred microlitres of the supernatant were transferred to a deep-well plate containing 100 µL of 0.1% formic acid in acetonitrile and vortexed. Chromatographic separation was achieved using a 50 × 2 mm, 5 µm ACE C8 column coupled to a 10 × 2 mm, 5 µm Keystone BDS Hypersil C8 guard column at ambient temperature. Mobile phase A (MA) and B (MB) consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile : methanol (50:50, v:v) respectively. The gradient run from 25% MB (T = 0 min) to 80% MB (1 min) held at 80% MB (3 min) and 25% MB (4 min). The flow was 0.25 mL·min−1, and the injection volume was 5 µL. The method was linear between 1 and 1000 ng·mL−1. Concentrations were estimated utilizing standard curves fitted by linear regression and 1/x weighing.
Pharmacokinetic analysis
The maximum drug concentration in the plasma (Cmax) and time (Tmax) thereof was determined from the observed values. The area under the plasma concentration-time curve (AUC) was calculated using the trapezoidal rule and extrapolation to infinity using the elimination rate constant. The bioavailability (%F) was calculated as %F = (AUC oral dose)/(AUC iv dose) × 100.
Materials
AF-353 (5-(5-iodo-2-isopropyl-4-methoxy-phenoxy)-pyrimidine-2,4-diamine; Figure 1B) and AF-010 (5-(2-isopropyl-4,5-dimethoxy-phenoxy)-pyrimidine-2,4-diamine; Figure 1C) were synthesized and characterized in the Department of Medicinal Chemistry at Roche Palo Alto. The anhydrous mono-hydrochloride salt of AF-353 is a white crystalline solid with a melting point of 260°C; in the solid state, it is stable at 40°C with 75% relative humidity for at least 2 months. The full chemical synthesis of AF-353 and related diaminopyrimidine analogues has been published (Carter et al., 2009; Jahangir et al., 2009). Note that AF 353 and AF 010 were previously known as RO-4 and RO-10 respectively. Solutions (2 mg·mL−1) and suspensions of AF-353 in acidic media are physically and chemically stable for at least 4 weeks at room temperature.
Other chemicals, ATP, α,β-meATP, TNP-ATP, PPADS, were obtained from Sigma-Aldrich, St. Louis, MO, USA. Isoflurane was purchased from VWR Scientific Products (Batavia, IL, USA).
Results
AF-353 (Figure 1B) was synthesized following the optimization of an active, but weaker, screening hit derived from the bacterial dihydrofolate reductase inhibitor trimethoprim (Figure 1A) discovered by high-throughput screening of the Roche compound collection. A close structural analogue, AF-010 (Figure 1C), was also used in the current work for some mechanistic experiments.
AF-353 is a highly potent inhibitor of α,β-meATP-evoked intracellular calcium flux in cell lines expressing recombinant rat and human P2X3 and human P2X2/3 channels (Figure 2A, Table 1). It is an equally potent inhibitor of human and rat P2X3 (pIC50 = 8.0), and can also block human P2X2/3 channel function with marginally reduced potency (pIC50 = 7.3). The functional potency of AF-353 was confirmed by whole-cell voltage clamp recordings using the same recombinant cell lines employed for the calcium flux experiments (Figure 2B, Table 1). Additionally, electrophysiological recordings were obtained from acutely dissociated rat dorsal root and nodose ganglion neurons, which are known to endogenously express significant populations of P2X3 and P2X2/3 channels, respectively (Rae et al., 1998; Virginio et al., 1998). Inhibition by AF-353 of α,β-MeATP-evoked inward currents was of similar potency in cells expressing either recombinant or native channels with potency estimates (pIC50) of 8.4 and 8.5 for recombinant and native rP2X3, respectively, and 7.7 and 7.6 for recombinant human and native rat P2X2/3 respectively.
Figure 2.

In vitro potency estimates of AF-353 antagonism at native and recombinant P2X3 and P2X2/3 receptors. (A) Inhibition by AF-353 of α,β-meATP-evoked intracellular calcium flux through recombinantly expressed rat P2X3 (CHOK1 cells) or human P2X3 and P2X2/3 (1321N1 cells). Potency estimates (pIC50) were 8.06, 8.05 and 7.41 for human P2X3, rat P2X3 and human P2X2/3 respectively. Inhibition curves were constructed in the presence of approximately EC80 concentrations of α,β-meATP: 1 µM (P2X3) and 5 µM (P2X2/3). (B) Inhibition by AF-353 of α,β-meATP-evoked inward currents through recombinantly expressed rat P2X3 (CHOK1 cells) or human P2X2/3 (1321N1 cells) and through isolated rat dorsal root (P2X3) or nodose (P2X2/3) ganglion neurons. Potency estimates (pIC50) were 8.42 and 7.73 for recombinantly expressed rat P2X3 and human P2X2/3, respectively, and 8.51 and 7.56 for acutely dissociated rat dorsal root (P2X3) and nodose (P2X2/3) ganglion neurons respectively.
Table 1.
Potency and affinity estimates of AF-353
| Assay |
hP2X3 |
rP2X3 |
hP2X2/3 |
|||
|---|---|---|---|---|---|---|
| n | pIC50 ± SEM | na | pIC50 ± SEM | na | pIC50 ± SEM | |
| Intracellular calcium flux (FLIPR)b | 3 | 8.06 ± 0.15 | 9 | 8.05 ± 0.03 | 9 | 7.41 ± 0.08 |
| Voltage clamp electrophysiology (recombinant cell lines)b | n.d. | 3–5 | 8.42 ± 0.05 | 3–4 | 7.73 ± 0.08 | |
| Voltage clamp electrophysiology (rat sensory neurons)c | n.d.c | 3–8 | 8.51 ± 0.10 | 3–8 | 7.56 ± 0.06d | |
| n | KD ± SEM | n | KD ± SEM | n | KD ± SEM | |
| Radioligand bindinge | 3 | 15 ± 1 nM | 3 | 30 ± 1 nM | 3 | 14 ± 2 nM |
Replicates for FLIPR and radioligand binding studies were entirely separate experiments conducted on separate days; inhibition curves constructed from voltage clamp electrophysiology experiments used recordings from 3 to 8 different cells per antagonist concentration.
Recombinant cell lines used for FLIPR and voltage clamp electrophysiology were: 1321N1-hP2X3, CHOK1-rP2X3 and 1321N1-hP2X2/3.
Not determined.
Voltage clamp electrophysiological recordings of rat sensory neurons utilizing acutely dissociated rat dorsal root ganglion and rat nodose ganglion neurons for the columns headed by P2X3 and P2X2/3 respectively.
Radioligand binding experiments were done using membrane homogenates of the same recombinant cell lines used for FLIPR and voltage clamp electrophysiology experiments.
The kinetics of the interaction between AF-353 and the hP2X2/3 channel was determined using manual patch clamp electrophysiology coupled with the Cellectricon Dynaflow high-speed perfusion system. Inhibition of α,β-meATP-evoked inward currents was concentration dependent and complete, and reversible upon washout of the antagonist (Figure 3A,B). Each recording consisted of a Kobs measurement at some particular concentration of AF-353, as well as an off-rate (Koff) measurement. The values of these parameters were determined by fitting a simple exponential function to the data (Figure 3A,B). Kobs was found to be linearly dependent on the concentration of AF-353, whereas Koff was found to be independent of antagonist concentration, consistent with a simple, second-order kinetic model (Figure 3D). We determined the value of Kon by fitting the following linear equation:
Figure 3.

Antagonism by AF-353 of α,β-meATP-evoked inward currents in 1321N1 astrocytoma cells expressing hP2X2/3. Agonist pulses consist of 10 µM α,β-meATP at 30 s intervals, 2 s of agonist exposure per pulse. Application of (A) 300 nM AF-353 or (B) 100 nM AF-353 produced τobs (association kinetics) of 48.5 and 112.6 s, respectively, and τoff (dissociation kinetics) of 157.6 and 188.8 s respectively. (C) Enlargement of α,β-meATP-evoked inward currents (10 µM) in the presence (red) or absence (black) of 30 nM AF-353. The traces on the left are unscaled, and the traces on the right are scaled to peak in order to overlay the traces. (D) Plotting the relationship of AF-353 concentration and Kobs shows there is a linear dependence, consistent with a simple, second-order kinetic model.
Based on this fit, we found Kon = 7.49 × 10−5 ± 0.41 × 10−5·s−1·nM−1 and Koff = 3.5 × 10−3 ± 0.29 × 10−3·s−1. Using these rate constants, we can estimate the KD of AF-353 at the heteromeric P2X2/3 receptor as 47 nM.
The selectivity of AF-353 for P2X3 and P2X2/3 over other P2X channels was established by testing the ability of AF-353 to block agonist-evoked intracellular calcium flux in cell lines expressing recombinant human P2X1, P2X2, P2X4, P2X5 or P2X7 receptors (Table 2). In all cases, AF-353 produced no inhibition up to a concentration of 10 µM. Additionally, two general screens of selectivity, one comprising 75 receptors, channels, enzymes and transporters (Cerep), and a second one covering more than 100 kinases (Ambit) established AF-353 to be a highly selective molecule (data not shown). Finally, because of the structural similarity of AF-353 to well-known bacterial dihydrofolate reductase inhibitors, AF-353 was also tested for the ability to inhibit the human isoform of this enzyme and found to be weakly active (pIC50 = 6.0 compared with the positive control, pyrimethamine, pIC50 = 7.1; data not shown).
Table 2.
Pharmacological selectivity of AF-353
| Receptor | n | pIC50 ± SEMa |
|---|---|---|
| hP2X3 | 3 | 8.06 ± 0.15 |
| rP2X3 | 9 | 8.05 ± 0.03 |
| hP2X2/3 | 9 | 7.41 ± 0.08 |
| hP2X1 | 3 | <5 |
| hP2X2 | 3 | <5 |
| hP2X4 | 3 | <5 |
| rP2X5 | 3 | <5 |
| hP2X7 | 3 | <5 |
Potency estimates for all of the P2X channels were obtained from FLIPR experiments using recombinant cell lines.
In order to measure the affinity for the P2X3 or P2X2/3 channels directly using radioligand binding displacement studies, AF-353 was tritium labelled on the methoxy group at the four position of the phenyl ring. Saturation-binding experiments were conducted using [3H]-AF-353, and cell membrane homogenates prepared from the same cell lines employed for intracellular calcium flux experiments; the specific binding window was significant for all three cell lines and was best fitted with a one-site binding model (Figure 4A–C). The affinity of AF-353 was equal at human and rat P2X3 (KD = 15 and 14 nM, respectively), and was marginally lower at the heteromeric human P2X2/3 receptor as well (KD = 30 nM, see Table 1). In competition-binding experiments, [3H]-AF-353 binding was completely eliminated by concurrent incubation with either AF-010 or α,β-meATP, but the inhibition potency (IC50) of AF-010 alone was sensitive to the concentration of [3H]-AF-353 (data not shown). A graph relating the ratio of IC50/Kb of AF-010 to the ratio of [AF-353]/KD of AF-353 resulted in a straight line following closely that expected of a purely competitive agent, as would be expected for such a close structural analogue of AF-353 (Figure 5). However, the IC50/Kb ratio for α,β-meATP was insensitive to radioligand concentration, suggesting AF-353 and α,β-meATP do not behave in a purely competitive manner.
Figure 4.
Specific binding of [3H]-AF-353 to CHOK1 membranes expressing (A) hP2X3 or (B) rP2X3 receptors or (C) 1321N1 astrocytoma cells expressing hP2X2/3 receptors. Specific binding was fit simultaneously using total binding and non-specific binding (determined using unlabelled AF-010).
Figure 5.

Competition binding experiments between [3H]-AF-353 (0.65, 1.24, 13, 33.9, 74.5, 158, 296; in nM) and unlabelled AF-010 or α,β-meATP employing membrane homogenates from CHOK1 cells expressing recombinant rat P2X3. The relationship of inhibition potencies and concentration of radio-labelled AF-353 is normalized to affinity estimates for each compound.
To better understand the mechanism of antagonism, a series of functional curve shift experiments, based on the original work of Arunlakshana and Schild (Arunlakshana and Schild, 1959), were conducted using intracellular calcium flux as the functional readout. As previously stated, a slowly desensitizing, chimeric P2X2–3 channel was used in place of the wild-type homomeric P2X3 channel. Figure 6A,B show the rightward, parallel, fully surmountable shifts of the agonist concentration-effect curves to α,β-meATP by TNP-ATP using cell lines expressing either heteromeric P2X2/3 or chimeric P2X2–3 receptors, confirming previously published conclusions that TNP-ATP is a competitive antagonist of these channels (Neelands et al., 2003). Relating the dose-ratios for the half-maximal calcium influxes (EC50) to the concentration of antagonist required for these dose ratios (i.e. Schild plot) produced a linear relationship with a slope of 1.2 (in both cases) and pA2 affinity estimates for TNP-ATP of 7.4 and 8.3 for P2X2/3 and P2X2–3, respectively (Figure 6A,B). However, in the same cell lines, AF-353 produced insurmountable shifts of the agonist concentration-effect curve consistent with the possibility (though not proving) that AF-353 does not act as a purely competitive antagonist (Figure 6C,D).
Figure 6.

Shifts of α,β-meATP-evoked intracellular calcium flux by varying concentrations of TNP-ATP (left panels) or AF-353 (right panels) in hP2X2/3 (A,C) and hP2X2-3 (B,D). Concentrations of antagonists used are shown in the key to the figure. Insets in panels A and B represent the relationship of the logarithm (dose ratio – 1) to the antagonist concentration (i.e. ‘Schild plot’). The dose ratio is defined as EC50(antagonist)/EC50(vehicle). The slopes of these lines were 1.2 in both cases and the X axis intercepts were 7.4 and 8.3 for hP2X2/3 and hP2X2-3 respectively.
To further explore if TNP-ATP and AF-353 blocked channel function through independent or common mechanisms, α,β-meATP concentration-effect curves were constructed after incubation with both antagonists in combination. Figure 7A shows that both 32 nM AF-353 and 10 nM TNP-ATP produced approximately threefold shifts in the α,β-meATP concentration-effect curves in cells expressing hP2X2–3. When the same cells expressing hP2X2–3 were equilibrated with a combination of 32 nM AF-353 and 10 nM TNP-ATP, the α,β-meATP pEC50 was shifted 10-fold, a multiplicative effect. AF-010, a close structural analogue of AF-353 that would be expected to act at precisely the same binding site (as shown in displacement studies), produces rightward shifts that are only marginally greater in combination with AF-353 than alone in either the chimeric P2X2–3 or the heteromeric P2X2/3 receptor (Figure 7B,C). From these experiments, it would be reasonable to conclude that molecules from the diaminopyrimidine chemical series represented by AF-353 and AF-010 do not act through the same competitive mechanism as TNP-ATP, and therefore bind allosterically to the ATP binding site.
Figure 7.
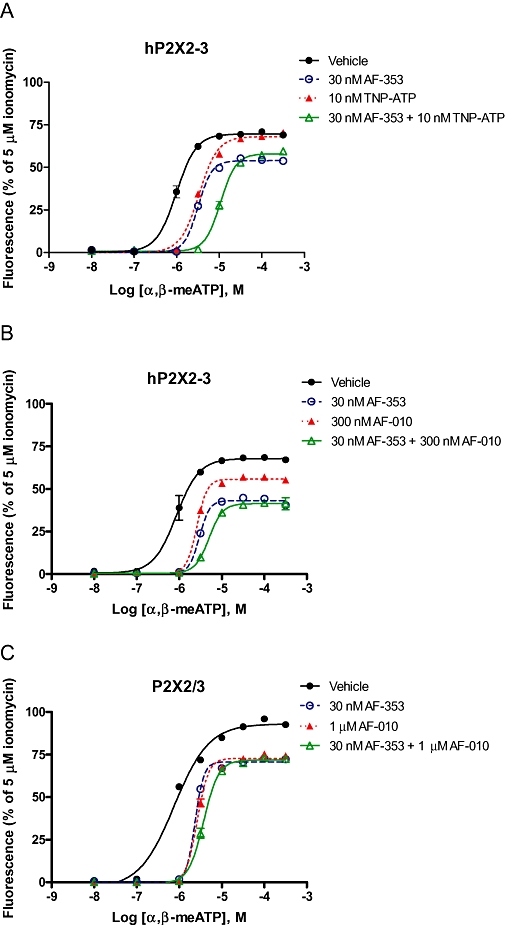
Shifts of α,β-meATP-evoked intracellular calcium flux by varying concentrations of antagonist in hP2X2-3 (A,B) and hP2X2/3 (C). Concentrations of antagonists used are shown in the key to the figure.
To assess the utility of AF-353 as a tool to investigate antagonism of P2X3 and P2X2/3 receptors in vivo, rats were dosed with 2 mg·kg−1 of AF-353 i.v. and orally as a suspension. The relevant pharmacokinetic parameters that were determined are shown in Table 3. AF-353 is orally bioavailable (F = 32.9%) with a Tmax of ∼30 min and plasma half-life of 1.63 h. CNS penetration was determined by measuring the brain to plasma ratio (B/P); AF-353 is highly CNS penetrant, with a B/P ratio of 6 (total brain extracted concentration/total plasma concentration). In addition; the in vitro protein binding was determined to be 98.2% in rat plasma.
Table 3.
Pharmacokinetic parameters of AF-353 in rat
| Parameter |
Result |
|
|---|---|---|
| AF-353 | A-317491a | |
| Bioavailability (F) | 32.9% | 0% |
| Time to peak plasma concentration (Tmax) | 0.5 h | NA |
| Plasma half-life (T1/2) | 1.63 h | NA |
| CNS penetration (brain/plasma ratio) | 6 | 0 |
| Plasma protein binding (bound) | 98.2% | 99.9% |
Estimates of bioavailability, CNS penetration and plasma protein binding were conducted at Roche Palo Alto using the same protocols as those used for AF-353 and employing powders synthesized in the research site's Department of Medicinal Chemistry.
Discussion
Here we present a detailed pharmacological characterization of AF-353, a selective, dual P2X3/P2X2/3 receptor antagonist with pharmacokinetic and pharmacodynamic properties that allow its use in vivo. At the initiation of our medicinal chemistry efforts, the majority of the known P2X3 ligands were either nucleotides or high molecular weight, polyacidic dyes, all of which offered poor selectivity of action. In an attempt to discover novel chemotypes with the physicochemical and pharmacokinetic properties required for a viable drug candidate, a high-throughput screen of the Roche chemical library was performed looking for inhibitors of α,β-meATP-evoked intracellular calcium flux employing cell lines expressing recombinant forms of P2X3 or P2X2/3 receptors. One of several initial hits was structurally related to trimethoprim, a bacterial dihydrofolate reductase inhibitor used as an antibiotic; this was subjected to extensive chemical optimization, yielding several chemical analogues. The current work describes the characterization of a potent representative molecule from this series, AF-353.
AF-353 is a dual P2X3/P2X2/3 receptor antagonist capable of inhibiting agonist-evoked intracellular calcium flux and inward currents at concentrations in the nanomolar range in cell lines recombinantly expressing rat or human P2X3 and human P2X2/3 receptors, and also natively expressed P2X3 and P2X2/3 receptors naturally present in the dorsal root and nodose ganglia of rat. Having established both potency at, and selectivity for, P2X3 and P2X2/3 channels, we conducted a series of experiments to better understand the mechanism of inhibition of AF-353. A classical experiment, which provides evidence (though not proof) for whether an agent is acting orthosterically or allosterically, is the curve shift experiment pioneered by Arunlakshana and Schild (1959), in which the concentration-effect relationship of an agonist acting on a receptor or tissue is constructed in the presence and absence of an antagonist. Modifications of this experiment were carried out using both radioligand binding and intracellular calcium flux as the readouts for this effect.
First, using a concentration of [3H]-AF-353 ranging from 0.04 to 20 times KD binding to membrane homogenates from cell lines recombinantly expressing rP2X3 channels, inhibition curves were constructed using α,β-meATP and a close structural analogue, AF-010. When the IC50 values of AF-010 are related to radioligand concentration (normalized to their own affinities for rP2X3), a linear relationship very close to the theoretical relationship of a purely competitive agent was observed; this was expected given the chemical similarity of these two compounds. In contrast, although binding of α,β-meATP was mutually exclusive with that of AF-353, the resulting IC50 values did not follow the relationship expected of a purely competitive agent and in fact was relatively insensitive to radioligand concentration. These data suggest AF-353 does not behave as a competitive antagonist with the nucleotide agonists, but rather is able to bind in a manner that modulates nucleotide interaction allosterically.
We wanted to confirm this result with functional evidence, but one of the ideal requirements for these types of mechanistic experiments is the establishment of an agonist–equilibrium, made difficult when agonist-induced function is prone to desensitization. Consequently, the slowly desensitizing heteromeric P2X2/3 receptor was used, as well as a chimeric P2X2–3 receptor modified to desensitize slowly, much like the P2X2/3 receptor (Werner et al., 1996; Neelands et al., 2003). Confirming previously published results, TNP-ATP, a nucleotide analog of ATP, inhibited α,β-meATP-evoked intracellular calcium flux in cell lines expressing either the chimeric hP2X2–3 or the heteromeric P2X2/3 channels, and shifted the agonist concentration-effect relationship to the right in a fully surmountable manner with no change in the Hill slopes. Relating the agonist ‘concentration-ratios’ (CR) resulting in half-maximal increases in intracellular calcium flux (log(CR − 1)) with the corresponding antagonist concentration produced a linear plot with slopes close to unity (n = 1.2 for both channels) and pA2 estimates, represented by the x-intercept, of 8.3 and 7.4 for hP2X2–3 and P2X2/3 respectively. However, for both the chimeric hP2X2–3 (used as a surrogate of wild-type P2X3 pharmacology) and the heteromeric P2X2/3 channels, AF-353 shifted the agonist-effect curves in a non-parallel, insurmountable fashion, suggesting that AF-353 does not behave in a simple, reversible competitive manner. It should be noted that even though peak α,β-meATP-evoked intracellular calcium flux was measured after 5 min of equilibration between agonist and antagonist, it is possible that complete equilibrium was not established due to the slow off-rate of AF-353 and may have caused, in part, the observed insurmountability of the response.
To address this further, the concentration-effect relationship of α,β-meATP was observed in the presence of AF-353 and TNP-ATP, an antagonist already established to be acting at the orthosteric binding site. If two antagonists are acting through entirely independent mechanisms, the shift of the agonist concentration-effect curve in the presence of both antagonists might be expected to be multiplicative relative to their individual shifts, whereas two antagonists acting at the same binding site will result in an additive shift (Paton and Rang, 1965). Concentrations of AF-353 and TNP-ATP individually resulting in a half-log increase of the pEC50 of α,β-meATP at the chimeric hP2X2–3 channel produced a full-log increase when used in combination, a multiplicative rather than additive increase, suggesting that AF-353 does not block P2X2–3-mediated intracellular calcium increases syntopically. Since TNP-ATP acts at the ATP binding site, these data provide further evidence that AF-353 acts allosterically. Supporting this notion, the combination of AF-353 and AF-010 resulted in a less than multiplicative (essentially additive) effect on agonist pEC50 shifts, consistent with the presumption that these structurally similar chemical analogues bind at precisely the same site. Thus, when the radioligand binding and functional curve shift experiments are viewed in their entirety, it is reasonable to conclude that AF-353 binds allosterically to the ATP binding site, and consequently behaves as a non-competitive antagonist.
A P2X3/P2X2/3 antagonist that is relatively insensitive to the concentration of the endogenous agonist ATP may be a desirable characteristic for a drug targeting these channels. ATP is present in the cytoplasm of most cells at millimolar concentrations, and stored in synaptic vesicles at even higher concentrations, perhaps as high as 200 mM (Pankratov et al., 2006; Burnstock, 2007). Since it has been hypothesized that the peak ATP concentration in the synaptic cleft could approach 500 µM (Pankratov et al., 2006), effective inhibition of P2X channel function through a mechanism relatively insensitive to ATP concentrations might be a desirable attribute.
The only other novel, small molecule P2X3 antagonist for which published data are available, A-317491, has been reported to be fully surmountable in curve shift experiments(Burgard et al., 2000; Neelands et al., 2003), and consequently may prove to be less effective when ATP concentrations are very high. Furthermore, A-317491 displayed very high protein binding (essentially 100%, resulting in a negligible free plasma fraction), negligible oral bioavailability and no CNS penetration; all features limiting its utility as a tool in vivo. For example, in the chronic constriction injury and L5-L6 nerve ligation models of neuropathic pain, A-317491 is effective only if given intrathecally (McGaraughty et al., 2003; Sharp et al., 2006). In models where the peripheral role of P2X channels is presumed to be important, such as the rat Freund's complete adjuvant model of inflammatory pain or in the rat cyclophosphamide-induced bladder cystitis model, A-317491 was effective, but only if administered intravenously (Wu et al., 2004; Ito et al., 2008). In contrast, AF-353 appears to be a significantly improved in vivo tool, demonstrating high oral bioavailability and CNS penetration in addition to high P2X3/P2X2/3 antagonist potency.
When the P2X3 protein was first cloned and characterized, both as a homomeric channel as well as a heteromeric channel formed with P2X2 subunits (Chen et al., 1995; Lewis et al., 1995), it generated much interest as a therapeutic target because it appeared to be located almost exclusively on small diameter, nociceptive neurons. Over the ensuing years, the evidence linking P2X3 and/or P2X2/3 channels with different types of pain, particularly neuropathic and chronic inflammatory pain, as well as afferent sensitization of hollow viscera, has grown dramatically. For example, when P2X3 expression in mice is reduced by intrathecal administration of antisense oligonucleotides (Honore et al., 2002a), allodynic responses in models of neuropathic pain are significantly reversed. Furthermore, local application of A-317491 directly to the spinal cord reduced neuronal excitability in the dorsal horn of rats after chronic constriction injury of the sciatic nerve, a well-established model of neuropathic pain (Bennett and Xie, 1988), whereas intravenous administration (presumed to be acting peripherally due to the very poor CNS penetrance of this compound) had no effect (Sharp et al., 2006). It is estimated that as much as 8% of the population in the developed world suffer from neuropathies (produced by nerve injury, diabetes, herpetic infections, for example), which are treated very poorly with the current medicines (Dworkin et al., 2007). Additionally, a compound targeting P2X3 and P2X2/3 receptors may prove beneficial for patients suffering from other types of pain, as well as disorders of the lower urinary and gastrointestinal tracts (Bian et al., 2003; Ford et al., 2006; Brederson and Jarvis, 2008). A potent antagonist of P2X3 and P2X2/3 receptors may ultimately prove to be a useful tool in the physician's arsenal of medicines, and the identification of antagonists such as AF-219 with excellent pharmacokinetic and pharmacodynamic properties may portend the identification of clinically useful compounds in the near future.
Acknowledgments
This work was funded by Roche Pharmaceuticals.
Glossary
Abbreviations:
- α
β-meATP
- α
β-methyleneATP
- AM
acetoxymethyl ester
- AUC
area under plasma concentration-time curve
- CHO-K1
Chinese hamster ovary
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- FLIPR
fluorometric imaging plate reader
- HPLC
high performance liquid chromatography
- IBS
irritable bowel syndrome
- LC-MS/MS
liquid chromatography-mass spectrometry
- P2X3-KO
P2X3 receptor knockout
- PPADS
pyridoxal phosphate-6-azo (benzene-2,4-disulfonic acid) tetrasodium salt
- TNP-ATP
2′,3′-O-(2,4,6-trinitrophenyl) ATP
- tTA
tetracycline-controlled transactivator
Conflicts of interest
APF is an employee of Afferent Pharmaceuticals Inc., which owns an exclusive license for P2X3 antagonists developed at Roche Pharmaceuticals. None of the other authors have any conflicts of interest.
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):51–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay J, Patel S, Dorn G, Wotherspoon G, Moffatt S, Eunson L, et al. Functional downregulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. J Neurosci. 2002;22:8139–8147. doi: 10.1523/JNEUROSCI.22-18-08139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33(1):87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bian X, Ren J, DeVries M, Schnegelsberg B, Cockayne DA, Ford APDW, et al. Peristalsis is impaired in the small intestine of mice lacking the P2X3 subunit. J Physiol. 2003;551:309–322. doi: 10.1113/jphysiol.2003.044172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brederson JD, Jarvis MF. Homomeric and heteromeric P2X3 receptors in peripheral sensory neurons. Curr Opin Investig Drugs. 2008;9(7):716–725. [PubMed] [Google Scholar]
- Burgard EC, Niforatos W, Van Biesen T, Lynch KJ, Kage KL, Touma E, et al. Competitive antagonism of recombinant P2X2/3 receptors by 2′, 3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP) Mol Pharmacol. 2000;58:1502–1510. doi: 10.1124/mol.58.6.1502. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev. 2007;87:659–797. doi: 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]
- Burnstock G. The journey to establish purinergic signalling in the gut. Neurogastroenterol Motil. 2008;20(Suppl 1):8–19. doi: 10.1111/j.1365-2982.2008.01107.x. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol. 2004;240:31–304. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- Carter DS, Alam M, Cai H, Dillon MP, Ford AP, Gever JR, et al. Identification and SAR of novel diaminopyrimidines. Part 1: the discovery of RO-4, a dual P2X3/P2X2/3 antagonist for the treatment of pain. Bioorg Med Chem Lett. 2009;19:1628–1631. doi: 10.1016/j.bmcl.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Chen CC, Akopian AN, Sivilotti L, Colquhoun D, Burnstock G, Wood JN. A P2X purinoceptor expressed by a subset of sensory neurons. Nature. 1995;377:428–431. doi: 10.1038/377428a0. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cockayne DA, Hamilton SG, Zhu QM, Dunn PM, Zhong Y, Novakovic S, et al. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature. 2000;407:1011–1015. doi: 10.1038/35039519. [DOI] [PubMed] [Google Scholar]
- Dorn G, Patel S, Wotherspoon G, Hemmings-Mieszczak M, Barclay J, Natt FJC, et al. siRNA relieves chronic neuropathic pain. Nucleic Acids Res. 2004;32:e49. doi: 10.1093/nar/gnh044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin RH, O'Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132:237–251. doi: 10.1016/j.pain.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Ford APDW, Gever JR, Nunn PA, Zhong Y, Cefalu JS, Dillon MP, et al. Purinoceptors as therapeutic targets for lower urinary tract dysfunction. Br J Pharmacol. 2006;147:S132–S143. doi: 10.1038/sj.bjp.0706637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gever JR, Cockayne DA, Dillon MP, Burnstock G, Ford APDW. Pharmacology of P2X channels. Pflugers Arch. 2006;452:513–537. doi: 10.1007/s00424-006-0070-9. [DOI] [PubMed] [Google Scholar]
- Honore P, Kage K, Mikusa J, Watt AT, Johnston JF, Wyatt JR, et al. Analgesic profile of intrathecal P2X3 antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain. 2002a;99:11–19. doi: 10.1016/s0304-3959(02)00032-5. [DOI] [PubMed] [Google Scholar]
- Honore P, Mikusa J, Bianchi B, McDonald H, Cartmell J, Faltynek C, et al. TNP-ATP, a potent P2X3 receptor antagonist, blocks acetic acid-induced abdominal constriction in mice: comparison with reference analgesics. Pain. 2002b;96:99–105. doi: 10.1016/s0304-3959(01)00434-1. [DOI] [PubMed] [Google Scholar]
- Ito K, Iwami A, Katsura H, Ikeda M. Therapeutic effects of the putative P2X3/P2X2/3 antagonist A-317491 on cyclophosphamide-induced cystitis in rats. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:483–490. doi: 10.1007/s00210-007-0197-z. [DOI] [PubMed] [Google Scholar]
- Jahangir A, Alam M, Carter DS, Dillon MP, Bois DJ, Ford AP, et al. Identification and SAR of novel diaminopyrimidines. Part 2: the discovery of RO-51, a potent and selective, dual P2X3/P2X2/3 antagonist for the treatment of pain. Bioorg Med Chem Lett. 2009;19:1632–1635. doi: 10.1016/j.bmcl.2009.01.097. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Wismer CT, Schweitzer E, Yu H, van Biesen T, Lynch KJ, et al. Modulation of BzATP and formalin induced nociception: attenuation by the P2X receptor antagonist, TNP-ATP and enhancement by the P2X3 allosteric modulator, cibacron blue. Br J Pharmacol. 2001;132:259–269. doi: 10.1038/sj.bjp.0703793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MF, Burgard EC, McGaraughty S, Honore P, Lynch K, Brennan TJ, et al. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc Natl Acad Sci USA. 2002;99:17179–17184. doi: 10.1073/pnas.252537299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y-H, Bailey TW, Li B, Schild JH, Andresen MC. Purinergic and vanilloid receptor activation releases glutamate from separate cranial afferent terminals in nucleus tractus solitarius. J Neurosci. 2004;24:4709–4717. doi: 10.1523/JNEUROSCI.0753-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachnit WG, Oglesby IB, Gever JR, Gever M, Huang CC, Li XC, et al. Regulated expression of the rat recombinant P2X3 receptor in stably transfected CHO-K1 tTA cells. J Auton Nerv Syst. 2000;81:75–81. doi: 10.1016/s0165-1838(00)00120-x. [DOI] [PubMed] [Google Scholar]
- Lewis C, Neidhart S, Holy C, North RA, Buell G, Surprenant A. Coexpression of P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory neurons. Nature. 1995;377:432–435. doi: 10.1038/377432a0. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Wismer CT, Zhu CZ, Mikusa J, Honore P, Chu KL, et al. Effects of A-317491, a novel and selective P2X3/P2X2/3 receptor antagonist, on neuropathic, inflammatory and chemogenic nociception following intrathecal and intraplantar administration. Br J Pharmacol. 2003;140:1381–1388. doi: 10.1038/sj.bjp.0705574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelands TR, Burgard EC, Uchic ME, McDonald HA, Niforatos W, Faltynek CR, et al. 2′, 3′-O-(2,4,6,trinitrophenyl)-ATP and A-317491 are competitive antagonists at a slowly desensitizing chimeric human P2X3 receptor. Br J Pharmacol. 2003;140:202–210. doi: 10.1038/sj.bjp.0705411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Surprenant A. Pharmacology of cloned P2X receptors. Annu Rev Pharmacol Toxicol. 2000;40:563–580. doi: 10.1146/annurev.pharmtox.40.1.563. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Lalo U, Verkhratsky A, North R. Vesicular release of ATP at central synapses. Pflugers Arch. 2006;452:589–597. doi: 10.1007/s00424-006-0061-x. [DOI] [PubMed] [Google Scholar]
- Paton WDM, Rang HP. The uptake of atropine and related drugs by intestinal smooth muscle of the guinea-pig in relation to acetylcholine receptors. Proc R Soc Lond B. 1965;163:1–44. doi: 10.1098/rspb.1965.0058. [DOI] [PubMed] [Google Scholar]
- Rae MG, Rowan EG, Kennedy C. Pharmacological properties of P2X3-receptors present in neurones of the rat dorsal root ganglia. Br J Pharmacol. 1998;124:176–180. doi: 10.1038/sj.bjp.0701803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Bian X, DeVries M, Schnegelsberg B, Cockayne DA, Ford APDW, et al. P2X2 subunits contribute to fast synaptic excitation in myenteric neurons of the mouse small intestine. J Physiol. 2003;552:809–821. doi: 10.1113/jphysiol.2003.047944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp CJ, Reeve AJ, Collins SD, Martindale JC, Summerfield SG, Sargent BS, et al. Investigation into the role of P2X3/P2X2/3 receptors in neuropathic pain following chronic constriction injury in the rat: an electrophysiological study. Br J Pharmacol. 2006;148:845–852. doi: 10.1038/sj.bjp.0706790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Moriyama T, Honda K, Kamiya H, Sakurada T, Katsuragi T. Involvement of P2X2 and P2X3 receptors in neuropathic pain in a mouse model of chronic constriction injury. Drug Dev Res. 2003;59:104–111. [Google Scholar]
- Virginio C, Robertson G, Surprenant A, North RA. Trinitrophenyl-substituted nucleotides are potent antagonists selective for P2X1, P2X3, and heteromeric P2X2/3 receptors. Mol Pharmacol. 1998;53:969–973. [PubMed] [Google Scholar]
- Vulchanova L, Riedl MS, Shuster SJ, Stone LS, Hargreaves KM, Buell G, et al. P2X3 is expressed by DRG neurons that terminate in inner lamina II. Eur J Neurosci. 1998;10:3470–3478. doi: 10.1046/j.1460-9568.1998.00355.x. [DOI] [PubMed] [Google Scholar]
- Wang ECY, Lee JM, Ruiz WG, Balestreire EM, von Bodungen M, Barrick S, et al. ATP and purinergic receptor-dependent membrane traffic in bladder umbrella cells. J Clin Invest. 2005;115:2412–2422. doi: 10.1172/JCI24086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner P, Seward EP, Buell GN, North RA. Domains of P2X receptors involved in desensitization. Proc Natl Acad Sci USA. 1996;93:15485–15490. doi: 10.1073/pnas.93.26.15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Whiteside GT, Lee G, Nolan S, Niosi M, Pearson MS, et al. A-317491, a selective P2X3/P2X2/3 receptor antagonist, reverses inflammatory mechanical hyperalgesia through action at peripheral receptors in rats. Eur J Pharmacol. 2004;504:45–53. doi: 10.1016/j.ejphar.2004.09.056. [DOI] [PubMed] [Google Scholar]