

Abstract
Genetic predisposition to type 1 diabetes (T1D) has been associated with a chromosome 11 locus centered on the proinsulin gene (INS) and with differential steady-state levels of INS RNA from T1D-predisposing and -protective haplotypes. Here, we show that the haplotype-specific expression is determined by INS variants that control the splicing efficiency of intron 1. The adenine allele at IVS1-6 (rs689), which rapidly expanded in modern humans, renders the 3′ splice site of this intron more dependent on the auxiliary factor of U2 small nuclear ribonucleoprotein (U2AF). This interaction required both zinc fingers of the 35-kD U2AF subunit (U2AF35) and was associated with repression of a competing 3′ splice site in INS exon 2. Systematic mutagenesis of reporter constructs showed that intron 1 removal was facilitated by conserved guanosine-rich enhancers and identified additional splicing regulatory motifs in exon 2. Sequencing of intron 1 in primates revealed that relaxation of its 3′ splice site in Hominidae coevolved with the introduction of a short upstream open reading frame, providing a more efficient coupled splicing and translation control. Depletion of SR proteins 9G8 and transformer-2 by RNA interference was associated with exon 2 skipping whereas depletion of SRp20 with increased representation of transcripts containing a cryptic 3′ splice site in the last exon. Together, these findings reveal critical interactions underlying the allele-dependent INS expression and INS-mediated risk of T1D and suggest that the increased requirement for U2AF35 in higher primates may hinder thymic presentation of autoantigens encoded by transcripts with weak 3′ splice sites.
Electronic supplementary material
The online version of this article (doi:10.1007/s00439-010-0860-1) contains supplementary material, which is available to authorized users.
Introduction
The majority of vertebrate genes contain introns that must be removed from precursor messenger RNA (pre-mRNA) by splicing to ensure correct gene expression (Burge et al. 1999). This process requires pre-mRNA signals at the 5′ and 3′ splice sites (5′ss and 3′ss), branch points, polypyrimidine tracts (PPTs) and splicing silencers and enhancers that inhibit or promote exon inclusion in mRNA by altering RNA structure and/or binding of splicing factors, such as serine/arginine-rich (SR) proteins or heterogenous nuclear ribonucleoproteins (hnRNPs) (Burge et al. 1999). Because silencers and enhancers are highly prevalent in both exons and introns, a significant fraction of naturally occurring gene variability that confers a risk of genetic disease may operate at the level of pre-mRNA splicing (Graveley 2008; Kralovicova et al. 2004). The splicing process is, however, tightly coupled to other gene expression pathways, including transcription, polyadenylation, mRNA export and translation (Maniatis and Reed 2002; Moore and Proudfoot 2009). For example, promoter strength, RNA polymerase II elongation rate and transcriptional activators have been shown to alter splicing; conversely, efficient intron removal by the spliceosomal machinery promotes transcription (Brinster et al. 1988; de la Mata et al. 2003) and translation (Nott et al. 2004). However, the extent to which splicing variants contribute to disease susceptibility at each level is poorly understood.
Type 1 diabetes (T1D) results from insulin deficiency caused by the autoimmune destruction of pancreatic β cells. T1D aetiology has a strong genetic component, which is conferred by the major histocompatibility complex and a number of modifying chromosome loci (Davies et al. 1994). The strongest modifier was identified in the proinsulin gene (INS) region on chromosome 11 (termed IDDM2) (Davies et al. 1994) and fine-mapping of IDDM2 suggested that INS is the most likely IDDM2 target (Barratt et al. 2004). The genetic risk to T1D at IDDM2 has been attributed to differential steady-state RNA levels transcribed from predisposing and protective haplotypes, potentially involving a minisatellite sequence upstream of INS, termed VNTR (Barratt et al. 2004; Vafiadis et al. 1997) and references therein). However, the underlying mechanism that would link the VNTR to altered INS expression and T1D susceptibility has been elusive. In addition, reanalysis of IDDM2 genotypes in T1D families did not support the original finding of a difference in association between VNTR lineages that had previously enabled the exclusion of intragenic polymorphisms (Barratt et al. 2004).
Here, we demonstrate that differential mRNA and proinsulin expression from T1D-susceptibility and -protective haplotypes is due to INS variants that control the efficiency of intron 1 removal from the pre-mRNA. We also show that the adenine allele at rs689 (IVS1-6A/T), a critical polymorphism in this process, impairs recognition of the 3′ss of intron 1 by the auxiliary factor of U2 small nuclear RNP (U2AF). This interaction required both zinc finger domains of the 35-kD U2AF subunit, which inhibited a competing 3′ss in INS exon 2. The increased requirement for U2AF35 coevolved with the relaxation of 3′ss in higher primates and is here proposed to restrict thymus presentation of autoantigens encoded by alleles containing weak 3′ss.
Materials and methods
Reporter constructs
Reporter constructs and their splicing products are summarized in Fig. 1a and their haplotypes in Fig. 1b. Cloning of reporters A–C and D–E was described previously (Kralovicova et al. 2006a). D–F constructs were obtained with primer F (5′-cat gcc tgc tat tgt ctt ctc caa gag tcc aga gct act), which contains the BbsI site (underlined). For cloning, we employed previously haplotyped DNA samples as a template for PCR (Stead et al. 2003). The VNTR-containing reporter, which has a minisatellite sequence upstream of the IC haplotype constructs, was obtained by cloning PCR products amplified with primers J (5′-atc caa gct tgt cct aag gca ggg tgg gaa cta; HindIII site is underlined) and C into pCR3.1 and propagated in bacterial cells with genotypes designed to stabilize direct repeats (MAX Efficiency Stbl2, Invitrogen). Stable propagation of highly repetitive elements of class III haplotypes was unsuccessful even in this system. Two-exon minigenes were cloned into HindIII/XbaI sites using primers K (5′-ata aag ctt atc act gtc ctt ctg cca) and R (5′-ata tct aga atg ggc agt tgg ctc acc c) (Fig. 1a).
Fig. 1.
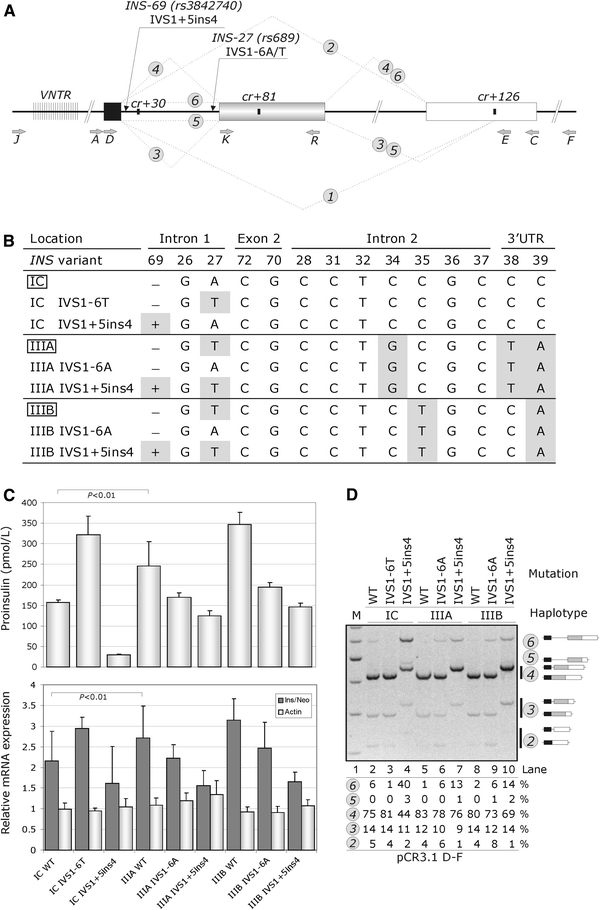
Haplotype-dependent proinsulin expression is determined by INS variants in intron 1. a INS splicing and reporter constructs. Exons 1, 2 and 3 are shown as black, grey and white boxes, respectively. Thick lines denote introns, dotted lines represent mRNA isoforms (numbered in circles). Primers and the most important gene variants are shown by grey and black arrows, respectively. The VNTR is denoted by closely spaced vertical lines. Cryptic splice sites are shown as black rectangles. b The INS haplotype structure and corresponding wild-type (boxed) and mutated reporter constructs. Variant nucleotides are highlighted. Designation of INS variants and haplotypes is as described (Stead et al. 2003); IVS intervening sequence or intron. c Haplotype-dependent proinsulin expression (upper panel) and corresponding relative mRNA levels (lower panel). The mean ratios of INS mRNA to RNA transcribed from the vector neomycin gene (neo) are shown as dark grey bars; the relative expression of endogenous β-actin is shown as light grey bars. Error bars represent s.d. d Splicing pattern of the INS reporter constructs. M 100-nt size marker. RNA isoforms are shown schematically to the right and are also numbered as in a . Percentage of splicing of each isoform is shown at the bottom, except for isoform 1 (expressed at <1%). Amplification was with primers PL3 (Kralovicova et al. 2006a) and E
Intron 1 and exon 2 deletion constructs were prepared using overlap-extension PCR as described (Kralovicova and Vorechovsky 2007). Serial deletions of intron 1 were made with the D–E reporter that lacked intron 2. For adenovirus preparation, the D–F constructs carrying haplotypes IC and IIIA were cut with BbsI, filled with the Klenow enzyme and subcloned into HindIII and blunted BglII sites of the shuttle plasmid pVQ-CMV-K-NpA (Anderson et al. 2000). Viruses were produced by recombination as described (Anderson et al. 2000) and supplied by ViraQuest Inc. (North Liberty, IA, USA). Adenovirus was purified over two rounds of CsCl gradients and was dialyzed against a storage buffer (Anderson et al. 2000).
The U2AF35 reporters were cloned into BamHI/EcoRI sites of pcDNA3.1/His (Invitrogen) using primers 5′-att gga tcc tgg gaa atg gcg gag tat ctg and 5′-ata gaa ttc ata agg taa aaa tgg cat ggc. Plasmids containing the 65-kD subunit of U2AF were subcloned into BamHI/XhoI sites of the same vector. ZRSR2 was prepared as a BamHI and XhoI amplicon with primers 5′-ata gga tcc gcg ccc gag aag atg acg tt and 5′-atc ctc gag tta ttt gga ttt ggg act ctg. Murine U2AF26 was cloned into BamHI/EcoRI sites of pcDNA3.1/His using primers 5′-ata gga tcc cgg gta aaa atg gct gaa ta and 5′-atc gaa ttc ccg tct cag aag cga cca tgc. Mammalian expression constructs encoding splicing factors were as described (Kralovicova et al. 2004).
Cell culture and RNA/cDNA preparations
Cells were cultured as previously described (Kralovicova et al. 2004, 2006a). Transfections of reporter constructs were carried out in 6- or 12-well plates using siPORT XP-1 (Ambion, USA). Total RNA was extracted using TRI reagent (Ambion) 48 h post-transfection, treated with DNase I (DNA-free™; Ambion, USA) and transcribed using the Moloney murine leukaemia virus reverse transcriptase (Promega, USA) and oligo-d(T)15 according to the manufacturers’ recommendations.
Detection of spliced products and real-time PCR
RNA products of transfected plasmids were visualized using RT-PCR with vector primers described previously (Kralovicova et al. 2004, 2006a). A combination of cDNA and vector primers was used as indicated to validate the ratios of RNA products. RT-PCR amplifications were for 28 cycles to maintain approximately linear relationship between the RNA input and signal. PCR products were separated on 6% polyacrylamide gels and stained with ethidium bromide. Signal intensity from alternatively spliced products was quantified as described (Kralovicova et al. 2004). To confirm the identity of each product, DNA fragments were excised from the gels and sequenced.
For real-time PCR, we used the FAM-labelled TaqMan probe (5′-ccc tcc agg aca ggc tgc atc ag), a forward primer (5′-cca agc tgg cta gcg ttt aaa) and reverse primers directed to exon 1 [5′-(a/g/c/t)(a/g/c)c tgc ttg atg gcc tctt] or exons 1 and 2 (5′-aga agg aca gtg atc tgc ttg). The former reverse primer amplifies all transcripts, whereas the latter detects correctly spliced RNAs lacking intron 1. Transcripts from the neomycin gene in pCR3.1 were measured by real-time PCR with primers neo-R (5′-gcc gga tca agc gta tgc) and neo-F (5′-ctc ctg ccg aga aag tat cca) and the TaqMan probe 5′-cgc cgc att gca tca gcc at. The endogenous expression of β-actin was quantified as described (Kralovicova and Vorechovsky 2005). For all real-time PCR experiments, we employed the 7700 sequencer and the SDS analysis software (ABI). Standard curves were constructed using serial dilutions of the corresponding plasmid DNA as described previously (Kralovicova and Vorechovsky 2005).
Alternatively spliced U2AF35 isoforms were visualized by a HinfI digest using PCR primers reported previously (Pacheco et al. 2004). Dual-specificity splice sites in DIABLO and UBE2C were amplified as described (Zhang et al. 2007).
Expression and purification of recombinant proteins
The pET28a-PTB-His construct was generously provided by D. Black (UCLA). The pET28a-PUF60-His was prepared by subcloning a BamHI-EcoRI fragment of the pcDNA3.1-PUF60 vector (cloning primers 5′-caa gat ggc gac ggc gac c and 5′-gag agg gac cac tgt cac g). Following transformation, BL21 Star cells (Invitrogen) were grown to optical density of 0.8 at 37°C. Protein expression was induced with 0.25 mM IPTG for 3 h. Pellets were resuspended in a lysis buffer (50 mM Tris, pH 8.0, 300 mM NaCl, 1 mM DTT) and sonicated. Proteins were purified from supernatant using the Ni–NTA agarose (Qiagen). Bound proteins were eluted using 250 mM imidazole, 50 mM Tris and 300 mM NaCl and dialysed against buffer D (25 mM Tris, pH 7.5, 1 mM DTT, 300 mM NaCl, 15% glycerol).
Recombinant U2AF65 were produced as GST fusion proteins with pGEX6P-U2AF65 generously provided by Berglund (Berglund et al. 1998). U2AF65 was purified from the BL21 cell lysate using Glutathione Sepharose 4B, cleaved from the GST tag with the PreScission protease (GE Healthcare) and dialysed. Protein concentrations were measured using the Bradford assay (Biorad) and by comparing protein preparations to serial dilutions of bovine serum albumin on SDS-PAGE gels stained with Coomassie Brilliant Blue G (Sigma).
Gel shift assays
Oligoribonucleotides (MWG Biotech) were 5′ end-labeled with [γ-32P]ATP (3000 Ci/mmol; PerkinElmer) and T4 polynucleotide kinase (Promega), gel purified and incubated with the indicated protein concentrations at room temperature for 15 min. The reaction mix (0.5 nM of labeled oligoribonucleotide, 50 mM Tris, pH 7.5, 70 mM NaCl, 1 mM DTT, 3.5% glycerol, 5 mM MgCl2, 0.25 mg/mL heparin, 0.3 mg/mL BSA, and 2U RNasin) was loaded onto native polyacrylamide gels (5%, 37.5:1 mono:bis acrylamide, 0.5xTBE) and run at 100 V at 4°C for 2 h. Gels were dried and exposed to phosphor screens. The signal was measured using the ImageQuant TL software (Amersham). For competition experiments, the indicated concentrations of unlabeled oligoribonucleotides were added to the protein-RNA complex after 15 min incubation for additional 10 min before gel loading.
Western blot analysis
Cells were washed twice with PBS and lysed in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0). Equivalent amount of protein lysates were loaded on 10% SDS-PAGE gels and transferred to PVDF (Amersham) membranes. The membranes were incubated with mouse PTB (kindly provided by Smith), mouse Puf60 (generously provided by Krainer), mouse U2AF65 (Sigma), rabbit U2AF35 (Protein Tech Group), mouse tubulin (ABcam) and rabbit actin (ABcam) antibodies and appropriate horse radish peroxidase-conjugated secondary antibodies (ABcam). Protein bands were detected using the Immun-Star WesternC Kit (Biorad) according to the manufacturer’s instructions.
RNA interference
Mammalian cells were plated at 8 × 104 cells per well in 12-well plates to achieve ~40% confluence. The next day, HiPerFect (Qiagen) was combined with Opti-MEM medium (Invitrogen) and siRNAs (MWG Biotech). Before adding to cells, the mixture was left at room temperature for 20 min. Cells were transfected with reporter constructs 48 h later and harvested after additional 24 h.
For rescue experiments, the cells were split into 6-well plates following the initial hit with either U2AF35-UTR or U2AF35ab siRNAs. Next day, the cells received the second hit. After 8 h, they were co-transfected with the reporter and rescue plasmids using the FuGENE HD transfection reagent (Roche) and 48 h later, they were harvested for protein/RNA isolation. For depletion experiments with U2AF35ab, we introduced silent mutations in the targeted region of rescue plasmids using a primer 5′-gaa aag gct gta atc gat tta aat aac cgt tgg tt. Final PCR products were inserted into NheI/NotI sites of the pCI vector (Promega).
Proinsulin secretion
Total proinsulin levels were measured in coded samples of culture supernatants by dissociation-enhanced lanthanide fluoroimmunoassay using monoclonal antibodies 3B1 and CPT3F11 as described (Kralovicova et al. 2006a).
Statistical analysis
Multiple comparisons of exogenous INS/neo mRNA expression across experiments were carried out with the SigmaStat (v. 3.5; Systat Software Inc., USA) and XLStat 2007 using the Fisher LSD and Holm-Sidak methods. Spearman correlation coefficients shown throughout the text were computed with the SigmaStat.
Results
Haplotype-dependent proinsulin expression is determined by differential splicing efficiency of INS intron 1
To identify gene variants that contribute to the allele-specific INS expression, we prepared reporter constructs carrying T1D-predisposing (class IC) and -protective (class IIIA and IIIB) haplotypes (Fig. 1a, b). These haplotypes are common in population and differ at only 5 of 14 known INS polymorphisms (Stead et al. 2003). We transiently expressed the wild-type reporters in 293T and HeLa cells, examined their splicing pattern, measured their steady-state mRNA levels and determined the proinsulin concentration in culture supernatants. Cells expressing the class IC haplotype secreted less proinsulin than those with class III haplotypes (Fig. 1c, upper panel), which was mirrored by their steady-state mRNA levels, as measured by real-time PCR with TaqMan probes in the first INS exon and in the neomycin gene of the vector (lower panel). Visualization of alternatively spliced RNA products using RT-PCR confirmed that the reporter constructs with adenine (A) at position IVS1-6 (also known as rs689, INS-27 or HphI+/−) showed an increased intron retention (IR) and exon skipping (ES) as compared to those with the T allele (cf. isoforms 6 and 2 in lanes 2, 5 and 8; Fig. 1d).
Systematic mutagenesis of each variant revealed that the A > T mutation at IVS1-6 introduced in the IC construct increased mRNAs/proinsulin to levels observed for the class III haplotypes (Fig. 1c). Conversely, the T > A mutation in both class III reporters reduced proinsulin to levels typical of the IC construct. In addition to IVS1-6, the 4-bp insertion at IVS1+5ins4 (also known as INS-69ins4), which activates a cryptic 5′ss of intron 1 (Kralovicova et al. 2006a), increased IR and lowered proinsulin secretion, particularly on the class IC haplotype background (cf. lanes 4, 7 and 10; Fig. 1d). The remaining 12 polymorphisms altered the relative expression of RNA products only to a minor extent (INS-26), or not at all, despite creating/eliminating predicted silencers/enhancers (Supplemental Fig. 1a, b). The splicing pattern of constructs carrying African haplotypes Ja, Ma and Ta (IVS1+5ins4) and Na, Va and Ya (IVS1+5del4), which all have the T allele at IVS1-6 and a wide range of class I, II and III VNTRs (Stead et al. 2003), was also similar. The addition of ~0.5 kb sequence upstream of INS containing the class I VNTR did not influence the INS splicing either (Supplemental Fig. 1c, d). Finally, the allele-specific splicing pattern was confirmed upon transfection/transduction of plasmid/adenovirus reporter constructs into mouse thymic epithelial cell lines, a macrophage cell line and control cells (Supplemental Fig. 2a).
Together, these results demonstrate that IVS1-6T/A and IVS1+5ins/del4 are key variants underlying the haplotype-dependent proinsulin expression through altered efficiency of intron 1 splicing. These findings support a model, in which T1D predisposition at IDDM2 results from a failure to adequately present proinsulin peptides of the low-expressing and T1D-predisposing IVS1-6A allele in the developing thymus (Supplemental Fig. 2b).
Identification of splicing regulatory elements in INS intron 1
Although the haplotype-specific INS expression is largely determined by intron 1 variants, this intron may contain additional signals that may improve its splicing efficiency and can be exploited in the future to correct the defect. To identify these motifs, we systematically examined splicing and proinsulin secretion of reporter constructs with a series of overlapping deletions (Fig. 2a). IR levels were altered in 9/14 deletion mutants and correlated positively with the G content of deleted segments (r = 0.62, P < 0.01, F test) and negatively with their C (r = −0.47, P < 0.05) and A (−0.43, P = 0.05) contents (Fig. 2b). Each deletion that increased IR also removed one or more Gn (where n > 2) repeats, except for del14, which encompassed a predicted branchpoint and showed an increased ES. In contrast, IR was diminished for deletions that recreated these motifs (del5, del9), except for del6, which lacked a G repeat altogether. The concentration of secreted proinsulin correlated negatively with IR (r = −0.66, P < 0.01) and positively with mRNA levels (r = 0.89, P < 0.0001). In cells depleted of hnRNP F and H, which were shown to recognize G-rich motifs (Caputi and Zahler 2001), IR was increased whereas transcripts spliced to cryptic 3′ss in exon 3 (cr3′ss+126) were diminished. In contrast, transient overexpression of hnRNPs H/F in 293T cells revealed the opposite pattern (Supplemental Fig. 3).
Fig. 2.
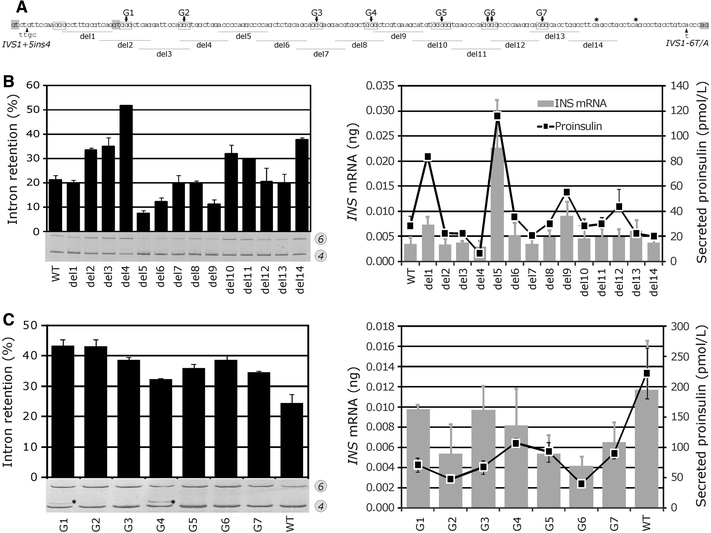
Identification of splicing enhancers and silencers in INS intron 1. a Intron 1 deletion constructs. Deletions 1–14 (horizontal lines) were made in reporter D–F on the IC haplotype background. G-runs (boxed) were numbered G1–G7; G > C substitutions in their central residues are denoted by arrows. 5′ss GT and 3′ss AG dinucleotides are highlighted in grey; stars denote predicted branchpoints. Variant alleles are indicated by arrowheads. b Intron retention, steady-state mRNA levels and proinsulin secretion of wild-type and deletion constructs. Error bars indicate s.d. determined from transfection experiments in duplicate. Secreted proinsulin was measured in culture supernatants 48 h post-transfection with 750 ng of the indicated reporter plasmid DNA. c Cytosine substitutions of central guanosines in Gn runs increase intron 1 retention and lower proinsulin secretion. Stars indicate activation of the cryptic 5′ss at position +30 of intron 1
To determine the importance of each G run, we introduced G > C substitutions in their central residues. Remarkably, each substitution significantly increased IR, but only to a minor extent. In addition, two of these substitutions activated a cryptic 5′ss at position +30 of intron 1 (cr5′ss+30), but only one of them (G4 in Fig. 2c) mediated a long-range interaction. Deletions containing CCC triplets (del5, del11 and del12, Fig. 2b) that may act as splicing regulatory elements (Kennedy and Berget 1997; Murray et al. 2008) did not show consistent IR changes.
Together, intron 1 splicing was facilitated by G-rich enhancers that appeared to act additively to increase INS expression, possibly through interactions with hnRNPs F/H and/or structural RNA motif(s) required for the cr5′ss+30 inhibition. These results also revealed a G > C>C > T hierarchy in regulatory signals that facilitate intron 1 removal and confirmed the importance of efficient splicing for mRNA/protein expression.
Primate evolution of coupled splicing and translation regulatory elements in the 5′ untranslated region
If G runs are functionally important for efficient intron 1 splicing, they should be conserved in evolution. To test this, we sequenced intron 1 in a number of primate species (Supplemental Fig. 4). Multiple sequence alignments revealed that all G triplets were absolutely conserved in Great Apes and Old World Monkeys, while most of them were present in New World Monkeys and Strepsirrhini (Supplemental Table 1).
Of two large intron 1 deletions that took place during primate evolution, only the Hominini-specific event influenced splicing by enhancing IR, most likely by eliminating a G5-containing enhancer, whereas the largest but splicing-neutral deletion was observed in colobines (Supplemental Figs. 4, 5). A G > A transition 24 nucleotides upstream of the 3′ss, which was found only in orangutans, was predicted to alter branchpoint position in this species (Supplemental Fig. 6). However, the most important ancestral event at intron 1 splice sites was a Hominidae-specific substitution at first position of exon 2 (E2+1G > A), which reduced the predicted strength of this 3′ss (Supplemental Table 2).
In addition to splicing signals, Homininae acquired a short upstream open reading frame, which encodes a 3-aa peptide (uORF2 in Fig. 3a). Because intron 1-containing transcripts are efficiently exported from the nucleus (Wang et al. 1997) and are overrepresented in expressed sequenced tags with the class IC haplotype as compared to the class III haplotypes (Kralovicova et al. 2006a), this uORF may curtail translation and further contribute to the lower proinsulin output of unspliced transcripts. To test this, we examined RNA and proinsulin levels for reporters with serial, 12- to 14-nt deletions of intron 1. As expected, constructs lacking the short uORF exhibited high levels of proinsulin secretion. In contrast, proinsulin concentration was diminished for uORF-containing reporters, except for deletion constructs capable of intron 1 splicing, in which the increase in proinsulin levels was roughly proportional to the spliced fraction (Fig. 3b).
Fig. 3.
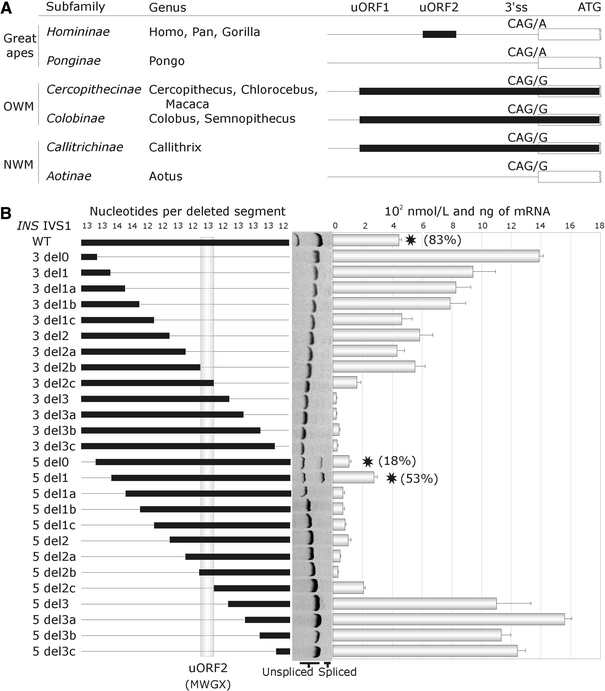
Evolution of coupled splicing and translational regulation of the proinsulin gene. a Primate phylogeny of upstream open reading frames and 3′ss of intron 1. uORFs are shown as black rectangles. Their position in the 5′ untranslated region is shown at the top. OWM/NWM, Old/New World Monkeys. b Upstream open reading frame in INS intron 1 is a dominant regulatory motif inhibiting translation. Proinsulin secretion in culture supernatants of 26 constructs with serial 12- to 14-nt deletions transiently transfected into 293T cells. Deletion constructs (left panel) were derived from a reporter lacking intron 2 because this intron is spliced much more efficiently than intron 1 and has a strong translation-enhancing effect (data not shown). Thin horizontal lines represent deleted segments of intron 1, thick lines indicate the remaining sequence. Homininae-specific upstream open reading frame is denoted by a grey vertical column; encoded amino acids are shown at the bottom. Schematic representation of deletions is followed by their splicing pattern (middle panel). The right panel shows proinsulin concentrations in cultures; error bars represent s.d. Stars denote reporter constructs that were spliced; the percentage of splicing is shown in parentheses
In contrast to Homininae, all Old and New World Monkeys had a distinct uORF further upstream that was in-frame with the canonical start codon (uORF1 in Fig. 3a). The GTG > ATG mutation creating uORF1 in the D-F construct increased IR/ES and lowered proinsulin secretion (Supplemental Fig. 7). In contrast, the ATG > GTG mutation in uORF2 had a negligible effect on intron 1 splicing, but raised proinsulin secretion. Interestingly, orangutans lacked intron 1 uORFs altogether (Fig. 3a, Supplemental Fig. 4). As predicted by calculating the intrinsic strength of 3′ss, reporter constructs carrying G at the first position of exon 2, which is specific for lower primates, consistently generated less IR/ES and more proinsulin than the A-containing constructs.
Taken together, splicing of intron 1 was weakened in higher primates by the 3′ss mutation E2+1G > A in Hominidae and the 16-nt deletion in Homininae, which was compensated by coupled splicing (G-rich intronic enhancers) and translational (loss of uORF1, gain of uORF2) regulatory motifs. Evolution of splicing regulatory motifs in intron 1 culminated in further relaxation of the 3′ss upon fixation of the IVS1-6A allele in modern humans, providing a means of rapidly fine-tuning INS expression, but reducing the intron-mediated translational yield. We propose that this reduction could result in insufficient pool of antigenic peptides for presentation in the foetal thymus and T1D susceptibility (Supplemental Figs. 2, 9).
Interactions of poly(Y)-binding proteins with IVS1-6
Experiments described so far indicated that the allele-specific INS expression is determined by the efficiency of intron 1 removal through two natural variants IVS1+5ins/del4 and IVS1-6A/T located at its 5′ss and 3′ss, respectively, and is influenced by a number of regulatory motifs, some of which apparently compensate for the 3′ss relaxation in higher primates. Because genetic susceptibility to T1D at IDDM2 in the white population cannot be attributed to the African-specific insertion IVS1+5ins4, we set out to characterize RNA interactions affected by IVS1-6A/T. Since the IVS1-6A allele weakens the PPT and may reduce binding of U2 snRNP auxiliary factor (U2AF) and/or other poly(Y)-binding proteins, including PTB (Sauliere et al. 2006; Singh et al. 1995) and PUF60 (Hastings et al. 2007; Page-McCaw et al. 1999), we first determined their binding affinity to the 3′ss sequences in vitro. Synthetic, end-labeled oligoribonucleotides representing IVS1-6A, IVS1-6T and a strong PPT as a control were incubated with recombinant U2AF65, PUF60 and PTB (Fig. 4a). Binding of each protein to the A allele was weaker as compared to the T allele and a T-rich control (Fig. 4b–d), which was more pronounced for PTB and PUF60 than for U2AF65 and which was further supported by competition experiments with unlabelled RNA probes (Fig. 4e).
Fig. 4.
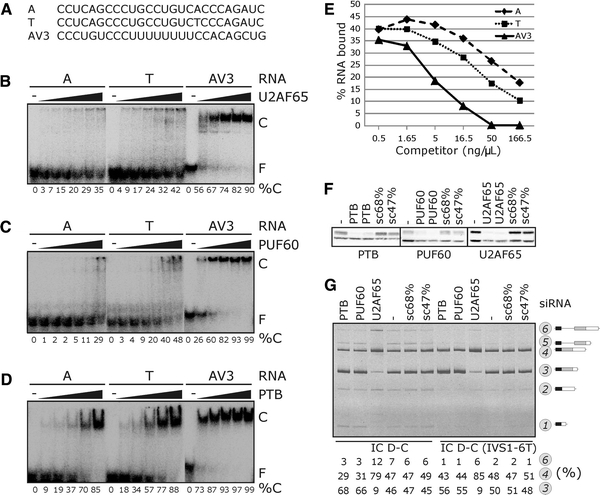
Interaction of poly(Y)-binding proteins with allele-specific INS transcripts. a Oligoribonucleotides representing IVS1-6A and IVS1-6T alleles and a control (AV3) used in gel shift experiments shown in b–d . b Concentration of recombinant U2AF65 was 0.14, 0.28, 0.56, 1.12, 2.24 and 4.48 μM. C, protein-RNA complex; F, free probe; %C, % bound. c Concentration of recombinant PUF60 was 0.34, 0.68, 1.36, 2.72, 5.44 and 10.88 μM. d Concentration of recombinant PTB was 0.32, 0.64, 1.28, 2.56 and 5.12 μM. e Example of a competition experiment with PUF60, 32P-labeled T probe and increasing concentrations of unlabeled competitors. f Western blot analysis of 293T cells transfected with siRNAs shown at the top (in duplicate); antibodies are shown at the bottom. The lower band corresponds to β-tubulin in each panel. sc, scrambled control siRNAs with the indicated GC content. g Opposite effects of U2AF65 and PUF60/PTB on intron 1 retention and activation of cryptic 3′ss +126. siRNAs (100 nM each) are at the top, reporters are at the bottom, spliced products to the right
Interestingly, RNA interference-mediated depletion of each poly(Y)-binding protein (Fig. 4f) revealed that IR was increased and utilization of cr3′ss+126 was decreased in U2AF65-depleted cells, but both PTB- and PUF60-depleted cells exhibited the opposite pattern. The same outcome of U2AF65/PTB/PUF60 depletion was observed also for the IVS1-6T allele and D–F constructs (Fig. 4g and data not shown) and for other 3′ss, such as LIPC intron 1, pointing to a more general role of these proteins in 3′ss control (J.K., I.V, manuscript in preparation). Thus, although binding to the IVS1-6A allele was reduced in vitro, U2AF65 acted antagonistically to PTB and PUF60 in vivo, implicating additional interactions.
T1D-protective allele at IVS1-6 relieves the requirement of intron 1 splicing for U2AF35
Recruitment of the U2 snRNP to the branch site is facilitated by the U2AF heterodimer, which consists of the PPT-interacting U2AF65 (Zamore and Green 1989) and the 3′AG-binding U2AF35 (Merendino et al. 1999; Wu et al. 1999; Zorio and Blumenthal 1999). Because interaction between U2AF35 and 3′AG can stabilize U2AF65 binding to weak PPT (Merendino et al. 1999; Wu et al. 1999), we tested if the A allele at IVS1-6 renders the 3′ss of intron 1 more dependent on U2AF35. Interestingly, RNA interference-mediated U2AF35 depletion in Hela cells (Fig. 5a; Supplemental Fig. 9) increased IR and ES, but also activated a cryptic 3′ss 81 nucleotides downstream of the authentic 3′ss (cr3′ss+81) and inhibited cr3′ss+126 of intron 2 (Fig. 5b). Importantly, the 3′ss requirement for U2AF35 was dramatically relieved for the IVS1-6T allele and also for the E2+1G mutation, which is characteristic of lower primates (Fig. 3a). The U2AF35-specific splicing pattern was observed with additional three small interfering RNAs (siRNAs) targeting this protein, but never for scrambled controls or siRNAs against >30 other splicing factors (see below). The same change in utilization of both cryptic 3′ss was found in U2AF35-deficient cells transfected with reporters containing only exons 1–2 and exons 2–3 (Fig. 5c), indicating that they are used independently of each other. Depletion of U2AF65, which also reduced U2AF35 levels (Pacheco et al. 2006a) (Supplemental Fig. 9), did not activate cr3′ss+81, whereas codepletion of both U2AF subunits reduced its use as compared to the U2AF35 depletion (Fig. 5d). Separate knockdown of alternatively spliced isoforms U2AF35a and U2AF35b (Pacheco et al. 2004) did not induce cr3′ss+81, but their combination was effective (Fig. 5e, f). Overexpression of wild-type U2AF plasmids in cells symmetrically depleted of both subunits resulted in more dramatic alterations of relative U2AF35/U2AF65 levels. A higher U2AF35/U2AF65 ratio was associated with lower IR. In contrast, cr3′ss+126 was repressed when the ratio was altered in either direction, whereas cr3′ss+81 was preferred only at low U2AF35/U2AF65 ratios (Fig. 5g).
Fig. 5.

IVS1-6T relieves the requirement of intron 1 splicing for U2AF35. a Western blot analysis of HeLa cells transfected with the U2AF35ab siRNA (final concentration of 3, 9, 27 and 81 nM) sc scrambled control (68% GC). Antibodies are shown to the right. b RT-PCR with total RNA extracted from HeLa cells that were transfected with INS reporters shown at the bottom. The wild-type reporter D–F contained haplotype IC. RNA products are to the right. Sequence chromatogram shows activation of cr3′ss+81. c Splicing pattern of two-exon minigenes in U2AF35-depleted cells. Final concentration of U2AF35ab was 30 nM. Reporters are shown at the bottom; location of cloning primers is shown in Fig. 1a. d Inhibition of 3′ss+81 upon cotransfection of U2AF35ab (30 nM) and U2AF65 K (45 nM) siRNAs. e INS splicing pattern upon separate knockdown of U2AF35a and U2AF35b isoforms. The final concentration of siRNAs in each lane was 150 nM (100 nM of U2AF35a and 50 nM of U2AF35b in lane 3). f The relative expression of U2AF35a and b isoforms in HeLa cells transfected with siRNAs shown at the top. RT-PCR products were digested with HinfI, which cuts only U2AF35b (Pacheco et al. 2004). The last two lanes are HinfI-digested PCR products amplified using template plasmids carrying U2AF35a- and -b isoforms as controls. g upper panel. INS splicing pattern in cells with varying U2AF35/U2AF65 ratios. The amount of wild-type plasmid DNAs (shown at the top) was 0.3, 1.0 and 3.0 μg. siRNAs (shown at the bottom) were added at final concentrations of 90 nM (U2AF65 K) and 10 nM (U2AF35ab). Lower panel shows Western blot analysis with U2AF65 and U2AF35 antibodies. Ex, exogenously expressed subunit. h upper panel. Multiple alignment of C3H ZF proteins. Histidines and cysteines (numbered in U2AF35) coordinated to zinc are highlighted in black, residues involved in base stacking interactions in TIS11d in light grey and other TIS11d residues interacting with RNA in dark grey. Lower panel, rescue of INS splicing with wild-type and mutated U2AF35 isoforms. The extent of rescue is shown on a semiquantitave scale, reflecting decrease of isoform 6 and cr3′ss+81 upon cotransfection of U2AF35-depleted cells with the IC D–F reporters. Rescue plasmids are shown at the top
Although binding of U2AF35 to 3′AG is well-established (Merendino et al. 1999; Wu et al. 1999; Zorio and Blumenthal 1999), it has been unclear which residues contact RNA and how these interactions control 3′ss selection. U2AF35 contains two C3H-type zinc fingers (ZF1, ZF2), highly prevalent eukaryotic domains increasingly implicated in RNA binding (Lai et al. 2002; Liang et al. 2008). To test the importance of ZF1/ZF2, we made a series of substitutions of U2AF35 amino acids that corresponded to RNA-interacting residues in tristetraprolin/TIS11d, the best studied C3H ZF proteins (Hudson et al. 2004; Lai et al. 2002) (Fig. 5h, upper panel). Unlike the wild-type plasmids expressing U2AF35a and U2AF35b isoforms, substitutions C33R in ZF1 and C169R in ZF2 failed to rescue the INS splicing pattern in U2AF35 deficient cells (Fig. 5h, lower panel). Interestingly, a partial rescue phenotype was observed for a conservative substitution F171Y in ZF2, but not for F171L, in agreement with similar RNA binding affinities of tristetraprolin mutants containing identical substitutions at F164 (Lai et al. 2002). Corresponding positions in TIS11d and MBNL1 were essential for high-affinity RNA binding by forming stacking interactions with RNA bases (Hudson et al. 2004; Teplova and Patel 2008). U2AF35 carrying a substitution of T45, a residue previously proposed to contact RNA (Kielkopf et al. 2001), had no obvious effect on U2AF35 activity as well as a C-terminal deletion removing amino acids 195–240 that constitute the RS domain. Finally, INS splicing was also partially rescued by U2AF35-related proteins, namely by murine U2AF26 and human ZRSR2, but not by ZRSR2 lacking ZF2 or by empty plasmids (Fig. 5h and data not shown).
U2AF35 has a preference for 3′AGs that are followed by G at the first exon position (Wu et al. 1999). To test whether this preference is maintained for the U2AF35-sensitive cr3′ss+81, we mutated the first exon positions systematically and examined splicing of the mutated constructs in U2AF35-depleted cells. We found that the G>A>C>T hierarchy in splicing efficiency was identical for both competing 3′ss (Fig. 6a, b) and corresponded to nucleotide frequencies at position +1 of human exons and disease-causing pseudoexons (Fig. 6c–g). These frequencies were very similar to those previously obtained by in vitro U2AF35 selection (Wu et al. 1999). We did not see activation of cr3′ss+81 in cells treated with siRNAs targeting other U2AF35- and -65 related proteins, including ZRSR2/URP (Tronchere et al. 1997), U2AF26 (Shepard et al. 2002) and CAPERα and/or CAPERβ (data not shown). U2AF35 was significantly depleted with U2AF35ab siRNA even at the very low concentration (<1 nM), however, a faint U2AF35 signal was still detectable on Western blots at much higher concentrations of this duplex (Supplemental Fig. 10).
Fig. 6.
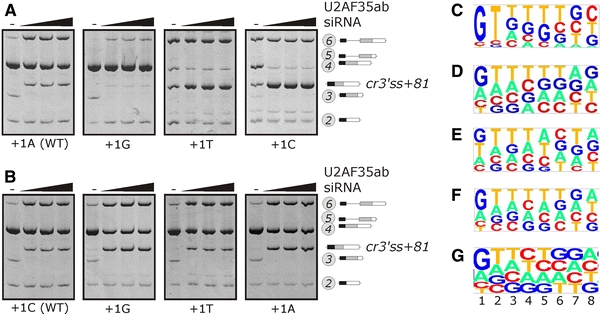
Splicing of INS reporters mutated at first exon position of authentic and cryptic 3′ss of intron 1 upon U2AF35 depletion. a Authentic 3′ss. b Cr3′ss+81. The final concentration of the U2AF35ab duplex was 0, 3, 9 and 27 nM. RNA products are shown to the right. The 3′ss consensus of the wild-type reporters was CAG/A (a) and CAG/C (b); their predicted intrinsic strength is shown in Supplemental Table 2. c–g Consensus sequences for the first 8 positions of human exons. c 30 clones obtained after 6 rounds of selection with U2AF35 (Wu et al. 1999). d 444 non-coding human exons (Zhang and Chasin 2004). e 78 disease-causing human pseudoexons that were generated by intronic mutations, most of them in transposable elements (Vorechovsky 2010). f 25,000 randomly selected human–mouse conserved exons (Carmel et al. 2004). g 39,862 human exons from the Alternative splicing Database (Stamm et al. 2006). The relative nucleotide frequencies at each position were plotted with a pictogram utility available at http://genes.mit.edu/pictogram.html. The height of each letter is proportional to the frequency of the corresponding base at the given position
Taken together, the T1D-susceptibility allele IVS1-6A and the ancestral E2+1A mutation increased the requirement of intron 1 splicing for U2AF35. This interaction required both ZFs of U2AF35, which may employ similar contacts with RNA as other C3H ZFs. Finally, rather than by U2AF35- or U2AF65-related proteins, selection of competing 3′ss appeared to be controlled by the balance between the small and large subunit of U2AF, with cr3′ss+81 preferred at low U2AF35 concentrations.
Identification of U2AF35-dependent 3′ splice sites
Previous studies employing three-exon reporter constructs demonstrated exon skipping as a result of U2AF35 depletion in vivo (Pacheco et al. 2006a), suggesting that this protein may preferentially promote intron-proximal 3′ss. Rather than for splicing of all pre-mRNAs with a weak PPT, U2AF35 appears to be required for a specific subset of 3′ss (Pacheco et al. 2006a). To extend and better define this subset, we examined splicing of tandem 3′ss on endogenous transcripts (Akerman and Mandel-Gutfreund 2007) (Supplemental Table 3), minigenes with competing 3′ss (Kralovicova et al. 2006b; Kralovicova and Vorechovsky 2007), and a CUED1 transcript as a positive control, in which U2AF35 depletion promoted intron-distal 3′ss (Pacheco et al. 2006a). We also analyzed dual-specificity splice sites that can be used as either 5′ss or 3′ss in endogenous DIABLO and UBE2C pre-mRNAs (Zhang et al. 2007). We observed a reproducible shift in 3′ss utilization for 4/14 (~29%) endogenous transcripts and for each minigene and dual-specificity site (Supplemental Table 4). Reporters containing short middle exons, such as DQB1 (Kralovicova et al. 2004) and TH (Kralovicova et al. 2006b), showed increased ES in U2AF35-depleted cells (Supplemental Fig. 11 and 12; data not shown). Altogether, intron-distal 3′ss was promoted in 4 cases and intron-proximal 3′ss in 7 cases. In U2AF65-depleted cells, we consistently observed the same direction in the utilization of competing 3′ss as in U2AF35-depleted cells. Comparison with the predicted strength of 3′ss revealed that U2AF promoted intrinsically weaker 3′ss in 7 cases and stronger 3′ss in 4 cases, with both INS pairs in the latter category. Thus, although we identified a number of new U2AF35-dependent 3′ss, this extended sample failed to show a clear bias toward intron-proximal or intrinsically weaker sites.
A search for cis- and trans-acting regulators of INS exon 2 inclusion
Because U2AF35 may function as a mediator of enhancer-dependent splicing (Zuo and Maniatis 1996), we next carried out a systematic deletion analysis of exon 2 to identify putative enhancers promoting the 3′ss of intron 1. Deletions in the 5′ half of this exon tended to induce IR, whereas 3′ deletions often promoted ES (Supplemental Fig. 13a). Of 20 deletion constructs, two activated cr3′ss+81 (del3 and del9 in Supplemental Fig. 13b). Del9 improved the exonic portion of cr3′ss+81 (CAG/C > CAG/G), but only removal of exon positions 22–34 in del3 revealed a genuine long-range effect. This deletion resulted in a loss of a putative enhancer UGUGGA that has its 3′ portion in a predicted single-stranded conformation (Supplemental Fig. 13c). Point mutations of loop-closing base-pairs and the first loop G increased ES and IR, however, they did not activate cr3′ss+81. The highest ES was observed for a G > C transversion predicted to form the first position of a GNUR-type tetraloop, but the double mutation swapping the loop-closing nucleotides did not rescue the splicing phenotype (Supplemental Fig. 13c, d). Apart from del3, cr3′ss+81 was also activated by distant single-nucleotide substitutions +5C > T, +10C > T and +11T > C, all located within ~12 nt from the 3′ss where U2AF35 may bind (Wu et al., 1999). These changes repressed canonical transcripts and proinsulin secretion (Supplemental Fig. 13E-F) and altered putative binding sites for PTB, but their splicing pattern in PTB-/neuronal PTB-depleted cells was similar to that in untreated cells (data not shown).
Splicing enhancers, including the newly identified element in INS exon 2, may interact with trans-acting factors that promote exon 2 inclusion, such as SR proteins (Burge et al. 1999). Individual depletion of human SR proteins revealed that exogenous INS exon 2 was skipped in HeLa cells treated with siRNAs targeting 9G8 (Fig. 7a, left panel). In addition, depletion of SRp20 was associated with an increased representation of transcripts spliced to cr3′ss+126. Moreover, codepletion of the human homologs of D. melanogaster transformer-2 Tra2α and Tra2β was associated with increased ES and IR. We observed similar effects with the reporter carrying the IVS1-6T allele (right panel) and their specificity was also confirmed by independent siRNAs and rescue experiments with plasmids expressing wild-type SR proteins (Fig. 7b). Finally, coexpression of a panel of SR proteins with INS reporters in 293T cells was associated with increased exon 2 inclusion. In contrast, coexpression of INS with a subset of hnRNPs appeared to promote exon skipping and/or cr3′ss+126 (hnRNP C1 and F; Supplementary Fig. 14).
Fig. 7.
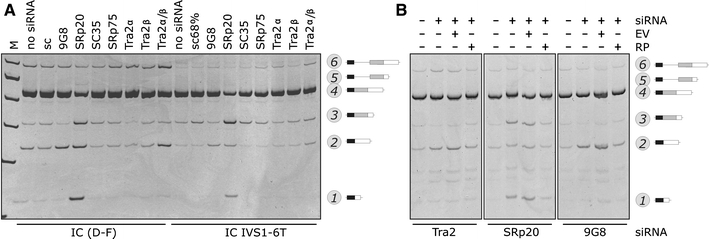
SR proteins in INS splicing a INS splicing pattern in cells depleted of SR proteins. HeLa cells were transfected with a battery of optimized siRNAs individually targeting human SR or SR-related proteins (shown at the top). Haplotype-specific construct and mutation are shown at the bottom, RNA products to the right. b Specificity of SR-mediated INS splicing for three SR proteins. EV empty vector (pCR3.1), RP rescue plasmid expressing wild-type SR proteins shown at the bottom
Discussion
Variable intron retention in the 5′ untranslated region and coupled splicing and translation control
Our study is the first to experimentally determine the effects of each disease gene variant/haplotype on splicing, mRNA levels and protein expression. This was facilitated by the small size of INS, the established allelic structure in several human populations (Stead et al. 2003) and by the haplotype-specific and highly transfectable reporter system capable of secreting exogenous proinsulin (Fig. 1). The overall proportion of variants that influenced splicing was ~20% (3/15), which is lower than estimates obtained for RNA-based mutation screens of large disease genes with many introns (Teraoka et al. 1999), but higher than that initially predicted for monogenic disease (Krawczak et al. 1992).
The higher proinsulin output from T1D-protective haplotypes involved at least two levels of tightly coupled gene expression pathways. A major determinant was the increase of mRNA upon splicing (Fig. 1), in agreement with earlier observations of diminished mRNA levels upon intron removal from genomic constructs derived from several species (Brinster et al. 1988; Rose 2004), including the rat proinsulin (Lu and Cullen 2003). This has been attributed to the enhancement of RNA polymerase II initiation or processivity (Furger et al. 2002), improved 3′ end processing (Lu and Cullen 2003) or modulation of nucleosome positions (Liu et al. 1995). The overall increase of gene expression by intron splicing was usually moderate, with the rat proinsulin close to the average (Lu and Cullen 2003; McKenzie and Brennan 1996).
Our work also shows that splice variants in the 5′ untranslated region can alter gene expression through gain of uORFs in a weakly spliced intron and extends the concept of translational pathophysiology (Cazzola and Skoda 2000) to complex traits (Supplemental Fig. 8). Coupled splicing and translation control is likely to reflect the relative depletion of ATGs in the 5′ untranslated region from yeasts to mammals and their higher level of conservation than any other trinucleotides (Churbanov et al. 2005). Upstream ORFs starting from conserved ATGs are, on average, shorter than those starting from non-conserved ATGs (uORF2 in Fig. 3). Thus, purifying selection against deleterious ATGs has been accompanied by translation attenuation through conserved upstream ATGs (Churbanov et al. 2005). The lack of uORFs in orangutans (Fig. 3a) and their distinct predicted branchpoint (Supplemental Fig. 6) suggests that the translational/splicing control may be less effective, speculatively linking these features to the propensity to diabetes proposed for this species (Gresl et al. 2000).
Splicing of INS intron 1 appears to be regulated during early development in chicken and mouse (Mansilla et al. 2005). Embryonic proinsulin levels decreased from gastrulation to neurulation, but mRNAs increased, which was attributed to the increasing amount of intron 1-containing transcripts (Mansilla et al. 2005). Additional level of developmental regulation was shown in chicken embryos, in which the transcriptional start site is extended 32 nt upstream, introducing an extra upstream ATG (Hernandez-Sanchez et al. 2003). Extending the 5′ untranslated region in our A–C reporters to accommodate the upstream ATG creates an out-of-frame uORF with the authentic start codon in correctly spliced RNAs but in-frame in intron 1-containing transcripts. As the out-of-frame uORF dramatically reduced proinsulin translation (Kralovicova et al. 2006a), future studies should establish if alternative transcriptional initiation sites that are present in developing vertebrates (Hernandez-Sanchez et al. 2003) also exist in humans.
Balancing act of U2AF subunits in 3′ss selection
Our study has identified the first U2AF35-sensitive cryptic 3′ss (Fig. 5) and extended the number of U2AF35-dependent authentic 3′ss (Supplemental Table 4), but did not find a clear bias toward intron-proximal or intrinsically weaker sites. The latter finding is in agreement with a lack of correlation between the requirement for either subunit and the PPT length/sequence in S. pombe (Webb et al. 2005). In addition, S. pombe introns that were most highly dependent on the large subunit also exhibited a marked dependence on the small subunit (Webb et al. 2005). Similarly, the direction of 3′ss choice upon depletion of human subunits was identical in each case (Supplemental Table 4). These results are consistent with a competition rather than directional scanning mechanism of 3′ss selection, as supported by earlier studies (Crotti and Horowitz 2009; Luukkonen and Seraphin 1997; Patterson and Guthrie 1991).
Contrary to preferential inhibition of a subset of weak 3′ss in U2AF35-depleted cells (Pacheco et al. 2006a), intrinsically weaker 3′ss were preferred at low U2AF35 concentration in both INS introns (Fig. 5; Supplemental Table 2), supporting the importance of other factors in 3′ss selection. Activation of cr3′ss+81 upon depletion of U2AF35, but not U2AF65, is likely to reflect the specific involvement of U2AF35 in the second step of splicing of ‘AG-dependent’ 3′ss (Guth et al. 1999; Reed 1989), and is consistent with an additional function of this molecule besides stabilizing U2AF65 binding to the PPT (Guth et al. 2001). Our results suggest that the balance of U2AF subunits and their availability in nuclear speckles, probably in complexes with splicing factor 1 (Rino et al. 2008), is important for the accurate 3′ss choice. However, U2AF35 depletion alters expression levels of additional mRNAs that may themselves influence INS splicing or harbor U2AF35-dependent 3′ss; on the other hand, very few known splicing factors were affected (Pacheco et al. 2006b). Alternatively, activation of weak cr3′ss+81 in U2AF35-depleted cells could be promoted by an upstream G-repeat that is likely to act as an intronic splicing enhancer (Fig. 2a). In addition, exon 2 segment upstream of cr3′ss+81 lacks adenosines in an optimal branchpoint consensus (Supplementary Fig. 13), therefore this cryptic 3′ss may use the same branchpoint/PPT unit as its authentic counterpart. Moreover, cr3′ss+81 has a consensus CAG/CC that deviates from the optimal U2AF35 binding site CAG/GT (Wu et al. 1999) and may thus bind a U2AF35-related protein with a higher affinity. HeLa cells transfected with siRNAs targeting ZRSR2 (Tronchere et al. 1997) and U2AF26 (Shepard et al. 2002) did not activate cr3′ss+81, however, antibodies against these proteins were not available to us and we could not exclude inefficient knockdown. Similarly, treatment of HeLa cells with two siRNAs targeting SRrp53 (RSRC1), which interacts with U2AF35 and CAPERα and activates a weak adenovirus 3′ss with cytosines at exon positions +1+2 (Cazalla et al. 2005), did not induce cr3′ss+81. Finally, hSlu7 depletion also failed to activate this 3′ss (J.K., I.V., unpublished data), which may be attributed to a larger distance from the predicted branch point of exon 2 beyond which aberrant 3′ss were not observed in hSlu7-depleted extracts (Chua and Reed 1999).
Apart from U2AF35, U2AF65 depletion repressed a stronger 3′ss of intron 2 and promoted authentic but weaker competitor (Fig. 4g). A BLAST search of expressed sequence tags with a sequence string bridging the stronger cr3′ss+126 identified at least 13 transcripts in cDNA libraries from insulinoma (n = 11) and isolated pancreatic islets (n = 2) (J.K. and I.V., unpublished data). This suggests that cr3′ss+126 is selected with an appreciable frequency in vivo, which was recapitulated in our reporters (Fig. 1d). As U2AF65 binds to nascent transcripts immediately after transcription and assists RNA polymerase II in returning to transcriptional competence when it pauses downstream of a PPT, a lack of U2AF65 would be predicted to increase polymerase II pausing (Ujvári and Luse 2004). Promotion of the authentic, intron-proximal 3′ss may thus be explained by longer dwell times at a pause site and a slower elongation rate between the two competing 3′ss of intron 2.
A putative model for RNA binding of U2AF35 ZF
ZFs are one of the most abundant protein domains in eukaryotes, with over 50 C3H-type ZF proteins encoded in the human genome, many involved in RNA metabolism, immune responses and macrophage activation (Liang et al. 2008). Each cysteine and histidine of U2AF35 ZFs is invariant from fly to man, but the number of identical amino acids in ZF orthologs is higher in ZF1 than in ZF2, suggesting that ZF1 interacts with a more phylogenetically conserved signal. Although both ZFs are likely to contribute to RNA binding, comparable mutations of the last three ZF residues of the S. pombe ortholog were lethal for ZF1 but not for ZF2 (Webb et al. 2005). Because ZF proteins often bind overlapping 4-nucleotide motifs (Hudson et al. 2004) (and references therein), it is tempting to speculate that U2AF35 ZF1/ZF2 could bind the extended CAG/ R UUY consensus of the 3′ss. In addition, since repeated C3H motifs have been associated with RNase activity and hairpin cleavage (Bai and Tolias 1996), it is conceivable that U2AF35 ZFs may be directly involved in shaping the 3′ss structure. Apart from U2AF35, INS intron 1 splicing can also be regulated by other RNA-binding ZF proteins. For example, tandem ZF3 and ZF4 of the alternative splicing regulator MBNL1 were shown to preferentially recognize sequences upstream of 3′ss (Teplova and Patel 2008). Finally, because ZF proteins have a pivotal role in the development of eukaryotes and are promising targets for drug design, future studies should explore the potential of ZF-based approaches to increase efficiency of intron 1 splicing, ultimately reducing IDDM2-mediated risk of T1D.
U2AF35 as a possible mediator of environmental risk of T1D
The ZF domain structure is stabilized by zinc, but affinities of ZF domains/proteins vary over many orders of magnitude (Hanas et al. 2005). Zinc concentration in the cell is low and zinc metalloproteins do not acquire this metal from a pool of free ions (Finney and O’Halloran 2003); instead, zinc uptake is maintained by metal trafficking proteins (Jacob et al. 1998). As a result, zinc deficiency may impair function of ZF proteins and diminish accurate recognition of U2AF35-dependent 3′ss. Interestingly, a long-term exposure to high groundwater concentration of zinc was associated with a decrease in T1D risk, particularly in rural areas in which drinking water was taken from local wells (Haglund et al. 1996), and this association was supported independently (Zhao et al. 2001). Pretreatment with zinc partially prevented diabetes induced by streptozocin (Yang and Cherian 1994). In contrast, xenobiotic metals capable of releasing zinc from ZF coordination spheres, such as cadmium or mercury, would be predicted to increase the T1D incidence. However, it should be emphasized that zinc is important for a large number of biological reactions, including packaging in secretory granules of β cells.
In addition to metals, 3′ss requirement for U2AF35 may be altered in virus-infected cells, as was demonstrated for adenovirus (Lutzelberger et al. 2005). The small subunit was also proposed to bind a Shigella effector IpaH9.8, which may modulate cytokine expression and immune responses (Okuda et al. 2005). Thus, a lack of zinc or microbial infection in early pregnancy might interfere with accurate expression of U2AF35-dependent transcripts.
U2AF35 as a mediator of the IDDM2-related risk of T1D
The increased dependency of the 3′ss of intron 1 on U2AF35 was due to its relaxation in Hominidae and out-of-Africa Homo sapiens, which coevolved with the introduction of uORF2 (Fig. 3a) and splicing regulatory motifs (Fig. 2, Supplemental Table 1). The requirement for U2AF35 was limiting for splicing of the low-expressing and T1D-predisposing allele IVS1-6A (Fig. 5b) and is likely to be restrictive in vivo for transcripts containing U2AF35-dependent 3′ss, including those encoding other autoantigens. In addition, location of specific T-cell epitopes in each proinsulin chain without a clear hot spot argues for the importance of quantitative differences of INS expression in the thymus because of the intron-mediated enhancement (Supplementary Fig. 2b), and not by antigen presentation of de novo peptides resulting from alternative gene splicing.
Rather than being an all-or-none phenomenon, however, U2AF35-dependency is likely to manifest as a continuous spectrum of interactions that promote 3′ss recognition, including cross-exon contacts. U2AF35-related genes such as ZRSR2, which is highly expressed in immune competent cells (Su et al. 2004), and putative partners of U2AF35, such as SR proteins (Wu and Maniatis 1993), may compensate this restriction. This concept is in agreement with the observed rescue of U2AF35-deficient phenotype by ZRSR2. Even if dependence on U2AF35 does not always correlate with the presence of purine-rich enhancers (Guth et al. 2001), future studies should attempt to identify U2AF35 binding partners that contact INS exon 2. This exon is longer than the average and less likely to form a continuous cross-exon protein network (Reed 1996; Zuo and Maniatis 1996), thus providing more space for the activation of cr3′ss+81. Our initial screening identified a subset of SR proteins associated with increased inclusion of this exon and also exon 2 residues required for activation of the U2AF35-sensitive cr3′ss+81 that were engaged in long-range RNA interactions. Future studies should show that these factors contact the INS pre-mRNA and identify their binding sites. Notably, SRp20 RNA binding consensus motifs (Cavaloc et al. 1999) can be found between cr3′ss+126 and its authentic counterpart (data not shown). In addition, this protein was found to be abundantly expressed in the thymus (Ayane et al. 1991).
Does INS VNTR confer genetic predisposition to T1D?
The role of VNTR in T1D was originally supported by cross-haplotype studies, but reanalysis of more genotypes in T1D families revealed that the previous exclusion of intragenic variants was not warranted (Barratt et al. 2004). VNTR-free reporter constructs carrying the T1D-protective haplotype produced consistently higher levels of total proinsulin than plasmids with the T1D-predisposing haplotype (Fig. 1c). The VNTR upstream of the class IC insert did not influence INS splicing (Supplemental Fig. 1d). A previous claim that haplotype-dependent mRNA levels cannot be attributed to intragenic variants and must therefore reflect the VNTR variability (Marchand and Polychronakos 2007; Pugliese et al. 1997; Vafiadis et al. 1997) is convincingly refuted by this and many previous studies (Furger et al. 2002; Kwek et al. 2002; Lu and Cullen 2003) (and references therein). A lack of intron 1-containing transcripts in the foetal thymus where INS is expressed at very low levels does not warrant the conclusion (Marchand and Polychronakos 2007) that polymorphic splicing is irrelevant to T1D predisposition, because even small decrease in splicing kinetics in the developing thymus could reach a threshold required for efficient expression and presentation of self-peptides in carriers of the susceptibility alleles. Although putative long-range transcriptional effects of VNTR on INS expression cannot be excluded (Ladd et al. 2007), our study offers a plausible alternative explanation for the haplotype-specific expression that had been attributed solely to the VNTR (Lucassen et al. 1995; Pugliese et al. 1997). Importantly, the increased IR on the IC haplotypes was observed in many cell types, including rat β-cells, macrophages and thymic epithelial cells (Kralovicova et al. 2006a; Marchand and Polychronakos 2007) (Supplemental Fig. 2a) and also in INS pre-mRNAs transcribed from vectors carrying weaker promoters (pGL3, J.K., unpublished data), consistent with differential recognition of 3′ss signals by key components of the splicing machinery that are ubiquitously expressed and highly conserved in evolution.
In conclusion, we have identified critical INS variants responsible for the haplotype-specific expression and their important interactions. U2AF35 contacts with 3′ss of INS intron 1 represent a new target for drug design to explore the prophylactic potential of ZF proteins and reduce genetic risk of T1D conferred by the strongest non-MHC locus. Finally, these results challenge the VNTR hypothesis of disease susceptibility and provide new insights into environmental aspects of T1D aetiology.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank D. Black, UCLA, G. Dreyfuss, HHMI, C. Smith, University of Cambridge, G. Ast, Tel Aviv University, A. Krainer, Cold Spring Harbor Lab, M. Hastings, University of Chicago, G. Screaton, University of Oxford, G. Anderson, University of Birmingham, K. Henscheid and A. Berglund, University of Oregon, J. Cáceres, University of Edinburgh, and I. Eperon, University of Leicester, for providing antibodies/plasmids and for helpful discussions. We also thank C. Glenn, Southampton Hospital NHS Trust, for proinsulin measurements, and G. Multani, E. Matrai, P. Duriez and P. Skipp, University of Southampton, for technical advice or help. We are grateful to J. Stead, Carlton University, and Sir A. Jeffreys, University of Leicester, for providing template DNA from previously typed individuals and S. Kanthaswamy, M. Krug, K. Petrzelkova, J. Gromoll and C. Roos for sharing primate DNA/samples. This study was supported by grants from the Juvenile Diabetes Research Foundation (1-2006-263 and 1-2008-047) and the Diabetes UK (09/3962) to I.V.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- INS
Gene for human proinsulin
- T1D
Type 1 diabetes
- IDDM2
T1D susceptibility locus
- VNTR
Variable number of tandem repeats
- U2AF
Auxiliary factor of the U2 small nuclear ribonucleoprotein
- U2AF65
65-kD subunit of U2AF
- U2AF35
35-kD subunit of U2AF
- hnRNPs
Heterogenous nuclear ribonucleoproteins
- SR proteins
Serine/arginine-rich proteins
- PPT
Polypyrimidine tract
- PTB
Polypyrimidine tract-binding protein
- ES
Exon skipping
- IR
Intron retention
- ZF
Zinc finger
- 3′ and 5′ss
3′ and 5′ splice sites
References
- Akerman M, Mandel-Gutfreund Y. Does distance matter? Variations in alternative 3′ splicing regulation. Nucleic Acids Res. 2007;35:5487–5498. doi: 10.1093/nar/gkm603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RD, Haskell RE, Xia H, Roessler BJ, Davidson BL. A simple method for the rapid generation of recombinant adenovirus vectors. Gene Ther. 2000;7:1034–1038. doi: 10.1038/sj.gt.3301197. [DOI] [PubMed] [Google Scholar]
- Ayane M, Preuss U, Kohler G, Nielsen PJ. A differentially expressed murine RNA encoding a protein with similarities to two types of nucleic acid binding motifs. Nucleic Acids Res. 1991;19:1273–1278. doi: 10.1093/nar/19.6.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C, Tolias PP. Cleavage of RNA hairpins mediated by a developmentally regulated CCCH zinc finger protein. Mol Cell Biol. 1996;16:6661–6667. doi: 10.1128/mcb.16.12.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt BJ, Payne F, Lowe CE, Hermann R, Healy BC, Harold D, Concannon P, Gharani N, McCarthy MI, Olavesen MG, et al. Remapping the insulin gene/IDDM2 locus in type 1 diabetes. Diabetes. 2004;53:1884–1889. doi: 10.2337/diabetes.53.7.1884. [DOI] [PubMed] [Google Scholar]
- Berglund JA, Abovich N, Rosbash M. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev. 1998;12:858–867. doi: 10.1101/gad.12.6.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Allen JM, Behringer RR, Gelinas RE, Palmiter RD. Introns increase transcriptional efficiency in transgenic mice. Proc Natl Acad Sci USA. 1988;85:836–840. doi: 10.1073/pnas.85.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge CB, Tuschl T, Sharp PA. Splicing of precursors to mRNAs by the spliceosome. In: Gesteland RF, Cech TR FAJ, editors. The RNA World. New York: Cold Spring Harbor Laboratory Press; 1999. pp. 525–560. [Google Scholar]
- Caputi M, Zahler AM. Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H’/F/2H9 family. J Biol Chem. 2001;276:43850–43859. doi: 10.1074/jbc.M102861200. [DOI] [PubMed] [Google Scholar]
- Carmel I, Tal S, Vig I, Ast G. Comparative analysis detects dependencies among the 5′ splice-site positions. RNA. 2004;10:828–840. doi: 10.1261/rna.5196404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaloc Y, Bourgeois CF, Kister L, Stévenin J. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA. 1999;5:468–483. doi: 10.1017/S1355838299981967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Newton K, Cáceres JF. A novel SR-related protein is required for the second step of Pre-mRNA splicing. Mol Cell Biol. 2005;25:2969–2980. doi: 10.1128/MCB.25.8.2969-2980.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M, Skoda RC. Translational pathophysiology: a novel molecular mechanism of human disease. Blood. 2000;95:3280–3288. [PubMed] [Google Scholar]
- Chua K, Reed R. The RNA splicing factor hSlu7 is required for correct 3′ splice-site choice. Nature. 1999;402:207–210. doi: 10.1038/46086. [DOI] [PubMed] [Google Scholar]
- Churbanov A, Rogozin IB, Babenko VN, Ali H, Koonin EV. Evolutionary conservation suggests a regulatory function of AUG triplets in 5′-UTRs of eukaryotic genes. Nucleic Acids Res. 2005;33:5512–5520. doi: 10.1093/nar/gki847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti LB, Horowitz DS (2009) Exon sequences at the splice junctions affect splicing fidelity and alternative splicing. Proc Natl Acad Sci USA [DOI] [PMC free article] [PubMed]
- Davies JL, Kawaguchi Y, Bennett ST, Copeman JB, Cordell HJ, Pritchard LE, Reed PW, Gough SC, Jenkins SC, Palmer SM, et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature. 1994;371:130–136. doi: 10.1038/371130a0. [DOI] [PubMed] [Google Scholar]
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell. 2003;12:525–532. doi: 10.1016/j.molcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Finney LA, O’Halloran TV. Transition metal speciation in the cell: insights from the chemistry of metal ion receptors. Science. 2003;300:931–936. doi: 10.1126/science.1085049. [DOI] [PubMed] [Google Scholar]
- Furger A, O’Sullivan JM, Binnie A, Lee BA, Proudfoot NJ. Promoter proximal splice sites enhance transcription. Genes Dev. 2002;16:2792–2799. doi: 10.1101/gad.983602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR. The haplo-spliceo-transcriptome: common variations in alternative splicing in the human population. Trends Genet. 2008;24:5–7. doi: 10.1016/j.tig.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresl TA, Baum ST, Kemnitz JW. Glucose regulation in captive Pongo pygmaeus abeli P.p. pygmaeus, and P.p. abel x P.p. pygmaeus orangutans. Zoo Biology. 2000;19:193–208. doi: 10.1002/1098-2361(2000)19:3<193::AID-ZOO3>3.0.CO;2-M. [DOI] [Google Scholar]
- Guth S, Martinez C, Gaur RK, Valcárcel J. Evidence for substrate-specific requirement of the splicing factor U2AF(35) and for its function after polypyrimidine tract recognition by U2AF(65) Mol Cell Biol. 1999;19:8263–8271. doi: 10.1128/mcb.19.12.8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guth S, Tange TO, Kellenberger E, Valcárcel J. Dual function for U2AF(35) in AG-dependent pre-mRNA splicing. Mol Cell Biol. 2001;21:7673–7681. doi: 10.1128/MCB.21.22.7673-7681.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund B, Ryckenberg K, Selinus O, Dahlquist G. Evidence of a relationship between childhood-onset type I diabetes and low groundwater concentration of zinc. Diabetes Care. 1996;19:873–875. doi: 10.2337/diacare.19.8.873. [DOI] [PubMed] [Google Scholar]
- Hanas JS, Larabee JL, Hocker JR. Zinc finger interactions with metals and other small molecules. In: Iuchi S, Kuldell N, editors. Zinc finger proteins: from atomic contact to cellular function. New York: Kluwer Academic Publishers; 2005. pp. 39–46. [Google Scholar]
- Hastings ML, Allemand E, Duelli DM, Myers MP, Krainer AR. Control of pre-mRNA splicing by the general splicing factors PUF60 and U2AF. PLoS ONE. 2007;2:e538. doi: 10.1371/journal.pone.0000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Sánchez C, Mansilla A, de la Rosa EJ, Pollerberg GE, Martinez-Salas E, de Pablo F. Upstream AUGs in embryonic proinsulin mRNA control its low translation level. EMBO J. 2003;22:5582–5592. doi: 10.1093/emboj/cdg515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson BP, Martinez-Yamout MA, Dyson HJ, Wright PE. Recognition of the mRNA AU-rich element by the zinc finger domain of TIS11d. Nat Struct Mol Biol. 2004;11:257–264. doi: 10.1038/nsmb738. [DOI] [PubMed] [Google Scholar]
- Jacob C, Maret W, Vallee BL. Control of zinc transfer between thionein, metallothionein, and zinc proteins. Proc Natl Acad Sci USA. 1998;95:3489–3494. doi: 10.1073/pnas.95.7.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy CF, Berget SM. Pyrimidine tracts between the 5′ splice site and branch point facilitate splicing and recognition of a small Drosophila intron. Mol Cell Biol. 1997;17:2774–2780. doi: 10.1128/mcb.17.5.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielkopf CL, Rodionova NA, Green MR, Burley SK. A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell. 2001;106:595–605. doi: 10.1016/S0092-8674(01)00480-9. [DOI] [PubMed] [Google Scholar]
- Kralovicova J, Vorechovsky I. Intergenic transcripts in genes with phase I introns. Genomics. 2005;85:431–440. doi: 10.1016/j.ygeno.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Kralovicova J, Vorechovsky I. Global control of aberrant splice site activation by auxiliary splicing sequences: evidence for a gradient in exon and intron definition. Nucleic Acids Res. 2007;35:6399–6413. doi: 10.1093/nar/gkm680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovicova J, Houngninou-Molango S, Krämer A, Vorechovsky I. Branch sites haplotypes that control alternative splicing. Hum Mol Genet. 2004;13:3189–3202. doi: 10.1093/hmg/ddh334. [DOI] [PubMed] [Google Scholar]
- Kralovicova J, Gaunt TR, Rodriguez S, Wood PJ, Day INM, Vorechovsky I. Variants in the human insulin gene that affect pre-mRNA splicing: is -23HphI a functional single nucleotide polymorphism at IDDM2? Diabetes. 2006;55:260–264. doi: 10.2337/diabetes.55.01.06.db05-0773. [DOI] [PubMed] [Google Scholar]
- Kralovicova J, Haixin L, Vorechovsky I. Phenotypic consequences of branchpoint substitutions. Hum Mutat. 2006;27:803–813. doi: 10.1002/humu.20362. [DOI] [PubMed] [Google Scholar]
- Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- Kwek KY, Murphy S, Furger A, Thomas B, O’Gorman W, Kimura H, Proudfoot NJ, Akoulitchev A. U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat Struct Biol. 2002;9:800–805. doi: 10.1038/nsb862. [DOI] [PubMed] [Google Scholar]
- Ladd PD, Smith LE, Rabaia NA, Moore JM, Georges SA, Hansen RS, Hagerman RJ, Tassone F, Tapscott SJ, Filippova GN. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet. 2007;16:3174–3187. doi: 10.1093/hmg/ddm293. [DOI] [PubMed] [Google Scholar]
- Lai WS, Kennington EA, Blackshear PJ. Interactions of CCCH zinc finger proteins with mRNA: non-binding tristetraprolin mutants exert an inhibitory effect on degradation of AU-rich element-containing mRNAs. J Biol Chem. 2002;277:9606–9613. doi: 10.1074/jbc.M110395200. [DOI] [PubMed] [Google Scholar]
- Liang J, Song W, Tromp G, Kolattukudy PE, Fu M. Genome-wide survey and expression profiling of CCCH-zinc finger family reveals a functional module in macrophage activation. PLoS One. 2008;3:e2880. doi: 10.1371/journal.pone.0002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Sandgren EP, Palmiter RD, Stein A. Rat growth hormone gene introns stimulate nucleosome alignment in vitro and in transgenic mice. Proc Natl Acad Sci USA. 1995;92:7724–7728. doi: 10.1073/pnas.92.17.7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Cullen BR. Analysis of the stimulatory effect of splicing on mRNA production and utilization in mammalian cells. RNA. 2003;9:618–630. doi: 10.1261/rna.5260303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucassen AM, Screaton GR, Julier C, Elliott TJ, Lathrop M, Bell JI. Regulation of insulin gene expression by the IDDM associated, insulin locus haplotype. Hum Mol Genet. 1995;4:501–506. doi: 10.1093/hmg/4.4.501. [DOI] [PubMed] [Google Scholar]
- Lutzelberger M, Backstrom E, Akusjarvi G. Substrate-dependent differences in U2AF requirement for splicing in adenovirus-infected cell extracts. J Biol Chem. 2005;280:25478–25484. doi: 10.1074/jbc.M413737200. [DOI] [PubMed] [Google Scholar]
- Luukkonen BG, Séraphin B. The role of branchpoint-3′ splice site spacing and interaction between intron terminal nucleotides in 3′ splice site selection in Saccharomyces cerevisiae. EMBO J. 1997;16:779–792. doi: 10.1093/emboj/16.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416:499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- Mansilla A, Lopez-Sanchez C, de la Rosa EJ, Garcia-Martinez V, Martinez-Salas E, de Pablo F, Hernandez-Sanchez C. Developmental regulation of a proinsulin messenger RNA generated by intron retention. EMBO Rep. 2005;6:1182–1187. doi: 10.1038/sj.embor.7400539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand L, Polychronakos C. Evaluation of polymorphic splicing in the mechanism of the association of the insulin gene with diabetes. Diabetes. 2007;56:709–713. doi: 10.2337/db06-0402. [DOI] [PubMed] [Google Scholar]
- McKenzie RW, Brennan MD. The two small introns of the Drosophila affinidisjuncta Adh gene are required for normal transcription. Nucleic Acids Res. 1996;24:3635–3642. doi: 10.1093/nar/24.18.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merendino L, Guth S, Bilbao D, Martinez C, Valcárcel J. Inhibition of msl-2 splicing by Sex-lethal reveals interaction between U2AF35 and the 3′ splice site AG. Nature. 1999;402:838–841. doi: 10.1038/45602. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Murray JI, Voelker RB, Henscheid KL, Warf MB, Berglund JA. Identification of motifs that function in the splicing of non-canonical introns. Genome Biol. 2008;9:R97. doi: 10.1186/gb-2008-9-6-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Le Hir H, Moore MJ. Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev. 2004;18:210–222. doi: 10.1101/gad.1163204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda J, Toyotome T, Kataoka N, Ohno M, Abe H, Shimura Y, Seyedarabi A, Pickersgill R, Sasakawa C. Shigella effector IpaH9.8 binds to a splicing factor U2AF(35) to modulate host immune responses. Biochem Biophys Res Commun. 2005;333:531–539. doi: 10.1016/j.bbrc.2005.05.145. [DOI] [PubMed] [Google Scholar]
- Pacheco TR, Gomes AQ, Barbosa-Morais NL, Benes V, Ansorge W, Wollerton M, Smith CW, Valcárcel J, Carmo-Fonseca M. Diversity of vertebrate splicing factor U2AF35: identification of alternatively spliced U2AF1 mRNAS. J Biol Chem. 2004;279:27039–27049. doi: 10.1074/jbc.M402136200. [DOI] [PubMed] [Google Scholar]
- Pacheco TR, Coelho MB, Desterro JM, Mollet I, Carmo-Fonseca M. In vivo requirement of the small subunit of U2AF for recognition of a weak 3’ splice site. Mol Cell Biol. 2006;26:8183–8190. doi: 10.1128/MCB.00350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco TR, Moita LF, Gomes AQ, Hacohen N, Carmo-Fonseca M. RNA interference knockdown of hU2AF35 impairs cell cycle progression and modulates alternative splicing of Cdc25 transcripts. Mol Biol Cell. 2006;17:4187–4199. doi: 10.1091/mbc.E06-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page-McCaw PS, Amonlirdviman K, Sharp PA. PUF60: a novel U2AF65-related splicing activity. RNA. 1999;5:1548–1560. doi: 10.1017/S1355838299991938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson B, Guthrie C. A U-rich tract enhances usage of an alternative 3’ splice site in yeast. Cell. 1991;64:181–187. doi: 10.1016/0092-8674(91)90219-O. [DOI] [PubMed] [Google Scholar]
- Pugliese A, Zeller M, Fernandez A, Jr, Zalcberg LJ, Bartlett RJ, Ricordi C, Pietropaolo M, Eisenbarth GS, Bennett ST, Patel DD. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet. 1997;15:293–297. doi: 10.1038/ng0397-293. [DOI] [PubMed] [Google Scholar]
- Reed R. The organization of 3’ splice-site sequences in mammalian introns. Genes Dev. 1989;3:2113–2123. doi: 10.1101/gad.3.12b.2113. [DOI] [PubMed] [Google Scholar]
- Reed R. Initial splice-site recognition and pairing during pre-mRNA splicing. Curr Opin Genet Dev. 1996;6:215–220. doi: 10.1016/S0959-437X(96)80053-0. [DOI] [PubMed] [Google Scholar]
- Rino J, Desterro JM, Pacheco TR, Gadella TW, Jr, Carmo-Fonseca M. Splicing factors SF1 and U2AF associate in extraspliceosomal complexes. Mol Cell Biol. 2008;28:3045–3057. doi: 10.1128/MCB.02015-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AB. The effect of intron location on intron-mediated enhancement of gene expression in Arabidopsis. Plant J. 2004;40:744–751. doi: 10.1111/j.1365-313X.2004.02247.x. [DOI] [PubMed] [Google Scholar]
- Sauliere J, Sureau A, Expert-Bezancon A, Marie J. The polypyrimidine tract binding protein (PTB) represses splicing of exon 6B from the beta-tropomyosin pre-mRNA by directly interfering with the binding of the U2AF65 subunit. Mol Cell Biol. 2006;26:8755–8769. doi: 10.1128/MCB.00893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard J, Reick M, Olson S, Graveley BR. Characterization of U2AF(26), a splicing factor related to U2AF(35) Mol Cell Biol. 2002;22:221–230. doi: 10.1128/MCB.22.1.221-230.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Valcárcel J, Green MR. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- Stamm S, Riethoven JJ, Le Texier V, Gopalakrishnan C, Kumanduri V, Tang Y, Barbosa-Morais NL, Thanaraj TA. ASD: a bioinformatics resource on alternative splicing. Nucleic Acids Res. 2006;34:D46–D55. doi: 10.1093/nar/gkj031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead JD, Hurles ME, Jeffreys AJ. Global haplotype diversity in the human insulin gene region. Genome Res. 2003;13:2101–2111. doi: 10.1101/gr.948003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplova M, Patel DJ. Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat Struct Mol Biol. 2008;15:1343–1351. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengut S, Tolun A, Chessa L, Sanal Ö, Bernatowska E, Gatti RA, et al. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999;64:1617–1631. doi: 10.1086/302418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronchere H, Wang J, Fu XD. A protein related to splicing factor U2AF35 that interacts with U2AF65 and SR proteins in splicing of pre-mRNA. Nature. 1997;388:397–400. doi: 10.1038/41137. [DOI] [PubMed] [Google Scholar]
- Ujvári A, Luse DS. Newly Initiated RNA encounters a factor involved in splicing immediately upon emerging from within RNA polymerase II. J Biol Chem. 2004;279:49773–49779. doi: 10.1074/jbc.M409087200. [DOI] [PubMed] [Google Scholar]
- Vafiadis P, Bennett ST, Todd JA, Nadeau J, Grabs R, Goodyer CG, Wickramasinghe S, Colle E, Polychronakos C. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet. 1997;15:289–292. doi: 10.1038/ng0397-289. [DOI] [PubMed] [Google Scholar]
- Vorechovsky I (2010) Transposable elements in disease-associated cryptic exons. Hum Genet 127:135–154 [DOI] [PubMed]
- Wang J, Shen L, Najafi H, Kolberg J, Matschinsky FM, Urdea M, German M. Regulation of insulin preRNA splicing by glucose. Proc Natl Acad Sci USA. 1997;94:4360–4365. doi: 10.1073/pnas.94.9.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb CJ, Lakhe-Reddy S, Romfo CM, Wise JA. Analysis of mutant phenotypes and splicing defects demonstrates functional collaboration between the large and small subunits of the essential splicing factor U2AF in vivo. Mol Biol Cell. 2005;16:584–596. doi: 10.1091/mbc.E04-09-0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-I. [DOI] [PubMed] [Google Scholar]
- Wu S, Romfo CM, Nilsen TW, Green MR. Functional recognition of the 3’ splice site AG by the splicing factor U2AF35. Nature. 1999;402:832–835. doi: 10.1038/45996. [DOI] [PubMed] [Google Scholar]
- Yang J, Cherian MG. Protective effects of metallothionein on streptozotocin-induced diabetes in rats. Life Sci. 1994;55:43–51. doi: 10.1016/0024-3205(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Green MR. Identification, purification, and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc Natl Acad Sci USA. 1989;86:9243–9247. doi: 10.1073/pnas.86.23.9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Chasin LA. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004;18:1241–1250. doi: 10.1101/gad.1195304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Hastings ML, Krainer AR, Zhang MQ. Dual-specificity splice sites function alternatively as 5′ and 3′ splice sites. Proc Natl Acad Sci USA. 2007;104:15028–15033. doi: 10.1073/pnas.0703773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HX, Mold MD, Stenhouse EA, Bird SC, Wright DE, Demaine AG, Millward BA. Drinking water composition and childhood-onset Type 1 diabetes mellitus in Devon and Cornwall, England. Diabet Med. 2001;18:709–717. doi: 10.1046/j.1464-5491.2001.00554.x. [DOI] [PubMed] [Google Scholar]
- Zorio DA, Blumenthal T. Both subunits of U2AF recognize the 3′ splice site in Caenorhabditis elegans. Nature. 1999;402:835–838. doi: 10.1038/45597. [DOI] [PubMed] [Google Scholar]
- Zuo P, Maniatis T. The splicing factor U2AF35 mediates critical protein–protein interactions in constitutive and enhancer-dependent splicing. Genes Dev. 1996;10:1356–1368. doi: 10.1101/gad.10.11.1356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.