

Abstract
In the genetic mutant mouse models ob/ob or db/db, leptin deficiency or resistance, respectively, results in severe obesity and the development of a syndrome resembling NIDDM. One of the earliest manifestations in these mutant mice is hyperinsulinemia, suggesting that leptin may normally directly suppress the secretion of insulin. Here, we show that pancreatic islets express a long (signal-transducing) form of leptin-receptor mRNA and that β-cells bind a fluorescent derivative of leptin (Cy3-leptin). The expression of leptin receptors on insulin-secreting β-cells was also visualized utilizing antisera generated against an extracellular epitope of the receptor. A functional role for the β-cell leptin receptor is indicated by our observation that leptin (100 ng/ml) suppressed the secretion of insulin from islets isolated from ob/ob mice. Furthermore, leptin produced a marked lowering of [Ca2+]i in ob/ob β-cells, which was accompanied by cellular hyperpolarization and increased membrane conductance. Cell-attached patch measurements of ob/ob β-cells demonstrated that leptin activated ATP-sensitive potassium channels (KATP) by increasing the open channel probability, while exerting no effect on mean open time. These effects were reversed by the sulfonylurea tolbutamide, a specific inhibitor of KATP. Taken together, these observations indicate an important physiological role for leptin as an inhibitor of insulin secretion and lead us to propose that the failure of leptin to inhibit insulin secretion from the β-cells of ob/ob and db/db mice may explain, in part, the development of hyperinsulinemia, insulin resistance, and the progression to NIDDM.
The obesity hormone leptin is produced by adipose tissue and acts at the hypothalamus to reduce food intake and to increase energy expenditure (1-7). In leptin-deficient ob/ob mice treated with leptin, reductions of hyperinsulinemia cannot be entirely accounted for by changes in food intake or body weight (1,2,8), suggesting that leptin directly regulates insulin secretion. Previously, we reported that leptin receptors (Ob-Rs) are expressed in rat islets of Langerhans and suggested that leptin may directly regulate the secretion of insulin as part of an adipoinsular endocrine axis (9). This concept was recently supported by the finding that leptin inhibits the secretion of insulin in perfused pancreases of ob/ob mice (10). To date, it is not known which hormone-secreting cells of the islets of Langerhans express receptors for leptin, and the cellular mechanisms by which leptin regulates islet cell function are unknown.
In the present report, we examined the expression of Ob-R mRNA in islets and tumor-derived cell lines representative of distinct islet cell types. We further examined the distribution of Ob-R expression on immunologically identified islet cells, utilizing a fluorescent derivative of leptin and an epitope-specific antiserum to the Ob-R. Finally, we determined the effect of leptin on islet hormone release and used electrophysiological techniques to determine the mechanism of leptin action on ATP-sensitive potassium channels (KATP).
RESEARCH DESIGN AND METHODS
Expression of Ob-R mRNA
Total RNA was isolated from freshly excised rat hypothalamus, rat islets, and cultured INS-1, RIN-B2 and INR1G-9 cells, using RNA STAT-60 (Tel-Test “B,” Friendswood, TX). The extracted RNA preparations were reverse transcribed, and the resultant cDNA was amplified by polymerase chain reaction (PCR) with the following primers: 5′-ATGAAGTGGCTTAGAATCCCTTCG-3′ and 5′-ATATCACTGATTCTGCATGCT-3′ for Ob-Rb; 5′-AACCTTGCCACCAGAGACTTCATC-3′ and 5′-CAGGGCGGTAACTTCAAAACGAG-3′ for glucagon. The PCR conditions were 95°C for 5 min, followed by 36 cycles at 95°C for 30 s, 50°C for 30 s, and 72°C for 1.5 min. A total of 5 μl of each PCR product was analyzed on a 1.5% agarose gel stained with Sybr Green I dye (Molecular Probes, Eugene, OR) and scanned with a FluorImager 575 (Molecular Dynamics, Sunnyvale, CA). The PCR products were transferred to Magna NT nylon membranes (Micron Separations, Westboro, MA) and probed with a 32P-labeled oligonucleotide (5′-ACACTGTTAATTTCACACCAGAG-3′), using Rapid-hyb buffer (Amersham Life Science, Arlington Heights, IL), following the manufacturer’s instructions.
Distribution of Ob-R in islets
Murine leptin (PeproTech, Rocky Hill, NJ) was coupled with iodocarbocyanine (Cy3) monofunctional dye (Biological Detection Systems, Pittsburgh, PA) in 0.1 mol/l carbonate buffer, pH 9.2, and purified on an Econo-Pac 10DG column (Bio-Rad Laboratories, Richmond, CA) in phosphate-buffered saline (PBS). The purified Cy3-leptin fraction contained no free Cy3 as assessed by gel electrophoresis visualized with a FluorImager 575 (Molecular Dynamics). Islets were isolated from rat pancreases by collagenase digestion and then dispersed into single cells, using trypsin-EDTA, and seeded onto poly-l-lysine–coated glass microscope slides in RPMI1640 medium supplemented with 10% fetal bovine serum. After overnight culture, slides were moved to 4°C, washed with ice-cold PBS, and incubated at 4°C for 1 h with ~40 nmol/l Cy3-leptin or boiled Cy3-leptin (controls) in Hanks’ balanced salt solution containing 5 mmol/l glucose and 0.5% bovine serum albumin. After several rinses with ice-cold PBS, the cells were then fixed for 10 min with ice-cold 4% paraformaldehyde, rinsed again, and coverslipped.
For immunocytochemistry, following Cy3-leptin binding and fixation, slides were rinsed and permeabilized with methanol for 10 min at −20°C and blocked with 3% normal donkey serum for 30 min at room temperature. Slides were then incubated overnight at 4°C with primary antisera against insulin, glucagon, or somatostatin. Slides were then rinsed with PBS and blocked for another 10 min. The hormones were localized with appropriate secondary antisera coupled with dichlorotriazinylamino fluorescein (DTAF) for 30 min at room temperature. After final rinses, slides were coverslipped and viewed first under a DTAF filter, allowing identification of hormone-positive cells (green). The same fields were then viewed under a Cy3 filter, allowing identification of Cy3-leptin–positive cells (orange).
To determine the distribution of Ob-R immunoreactivity, a synthetic peptide corresponding to a specific epitope in the extracellular region of the mouse Ob-R (GPPNTTDDSFLC) was coupled with maleimide-activated keyhole limpet hemocyanin and was used to raise antisera in rabbits. Slides with dispersed islets were prepared as described above and were incubated overnight at 4°C with the Ob-R antisera in addition to antisera against insulin, glucagon, or somatostatin. Slides were then rinsed and blocked, the Ob-R immunoreactivity was localized with an anti-rabbit antiserum coupled with Cy3, and hormones were localized with appropriate secondary antisera coupled with DTAF. Following coverslipping, slides were viewed with the appropriate filters, allowing for the detection of Ob-R immunoreactivity (orange) or hormone immunoreactivity (green). All images were obtained with a Nikon Optiphot-2 microscope equipped with an Optronics TEC-470 CCD camera (Optronics Engineering, Goleta, CA) interfaced with a Power Macintosh 7100 installed with a RasterOps frame grabber and IPLab Spectrum analysis software (Signal Analytics, Vienna, VA).
Effects of leptin on islet hormone release
Islets were isolated by collagenase digestion of pancreases from 10-week-old female ob/ob mice, and 10 islets per well were transferred into 12-well culture plates, each well in triplicate, containing 1 ml RPMI medium supplemented with 1% bovine serum albumin and 1% aprotinin, at either 5.5 mmol/l or 11.0 mmol/l glucose in the presence or absence of 100 ng/ml murine leptin (Pepro Tech, Rocky Hill, NJ) and/or 10 nmol/l glucagon-like peptide (GLP)-I(7-36) amide (Peninsula Laboratories, Belmont, CA). Islets were incubated for 1 h at 37°C, following which the medium was removed and assayed for both insulin and somatostatin by radioimmunoassay.
Effects of leptin on β-cell [Ca2+]i, membrane potential, membrane conductance, and single KATP channel activity
Isolated dispersed mouse pancreatic islets were prepared as above and cultured overnight on glass coverslips coated with concanavalin-A. Cells were prepared for the measurement of [Ca2+]i by incubation in fura-2 acetoxymethyl ester (fura-2 AM; Molecular Probes, Eugene, OR). Cells were loaded in a standard extracellular solution (SES; 138 mmol/l NaCl, 5.6 mmol/l KC1, 2.6 mmol/l CaCl2, 1.2 mmol/l MgCl2, 10.0 mmol/l HEPES, 4.0 mmol/l d-glucose, and 295 mOsm, pH adjusted to 7.4 with ~4 mmol/l NaOH) supplemented with 2% fetal bovine serum, 0.03% pluronic F-127, and 1 μmol/l fura-2 AM for 15–20 min at room temperature (20–22°C). Experiments were conducted at 32°C, using an IonOptix video imaging system (Milton, MA). [Ca2+]i was estimated from the ratio of 510-nm emission fluorescences from excitation by 350- and 380-nm wavelength light from the equation: [Ca2+]i = Kdβ(R − Rmin)/(Rmax−R)], where Kd is the dissociation constant of fura-2 AM (225 nmol/l), β is the ratio of 380-nm induced fluorescences of free/bound fura-2 AM, R is the measured ratio of the 350- and 380-nm fluorescences, and Rmin and Rmax are the 350 and 380 nm fluorescence ratios in zero [Ca2+] and saturating [Ca2+], respectively (11). Values of β, Rmin and Rmax were determined using fura-2 pentapotassium salt and calibration solutions from Molecular Probes Inc. K+-pipette solution for perforated patch measurements contained 95 mmol/l K2SO4,7 mmol/l MgCl2, 5 mmol/l HEPES (pH adjusted to 7.4 with NaOH; final concentration of 2 mmol/l Na+). Pipettes were then back-filled with the same solution to which nystatin (240 μg/ml) was added. For on-cell single-channel recordings of KATP, the pipette was filled with 140 mmol/l KCl, 2.6 mmol/l CaCl2, 1.2 mmol/l MgCl2 10 mmol/l HEPES, and ~4 mmol/l NaOH (pH 7.4). Single-channel currents were stored on videotape, low-pass filtered at 2 kHz (eight-pole Bessel filter, −3 decibel attenuation), and digitized at 4 kHz, using an Axon Instruments DigiData interface (Burlingame, CA). Data were analyzed using Axon Instruments pClamp 6.0 software. The patch pipette was connected to an Heka Electronik EPC-9 patch clamp amplifier (Instrutech, Mineola, NY) interfaced with a Macintosh Quadra 840AV computer running Pulse 8.0 software (Instrutech).
RESULTS
Expression of Ob-R mRNA
As a first test of our hypothesis that leptin directly suppresses insulin release, we examined pancreatic islets for the expression of the signal-transducing long form of the Ob-R (Ob-Rb). Using reverse transcriptase (RT)-PCR with primers designed to amplify specifically a region of Ob-Rb, the products of the predicted size (376 bp) were obtained from both rat islets and tumor-derived cell lines representative of pancreatic β- and δ-cells (Fig. 1). PCR products were obtained in cDNAs prepared with reverse transcriptase (RT+) and were undetectable in controls (RT−). No product was observed in cDNA generated from the α-cell line INR1G-9. To ensure the quality of the α-cell cDNA, primers designed to amplify glucagon cDNA gave a product of the predicted size (307 bp) (Fig. 1C, right panel).
FIG. 1.
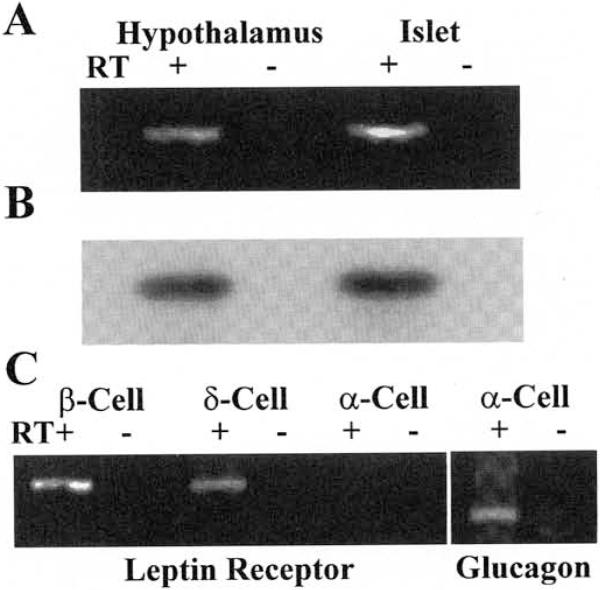
A: RT-PCR of rat hypothalamic and islet cDNA using a specific primer pair to amplify a 376-bp region of the Ob-Rb. The preparation of the cDNA was either in the presence (RT+) or absence (RT−) of reverse transcriptase. B: autoradiogram of a Southern blot of the above gel probed with a 32P-labeled oligo with a complementary sequence internal to the PCR primers. C: RT-PCR for Ob-Rb in tumor-derived cell lines representative of islet β-cells (INS-1), δ-cells (RIN-B2), and α-cells (INR1G-9); RT-PCR of α-cell (INR1G-9) cDNA using a specific primer pair to amplify a 307-bp region of glucagon.
Distribution of Ob-R in islets
To determine which islet hormone-producing cell types bind Cy3-leptin, cells were immunostained for glucagon, somatostatin, or insulin, following incubation with Cy3-leptin. No fluorescence was detectable on dispersed islet cells incubated with purified Cy3-leptin, which was heat-inactivated as a control (Fig. 2A, left panel). However, distinct membrane fluorescence could be found on a population of cells incubated with Cy3-leptin that had not been inactivated (Fig. 2A, right panel). The majority of cells immunoreactive for glucagon (α-cells) did not bind Cy3-leptin (Fig. 2B, hollow arrows). However, cells immunoreactive for insulin β-cells) or somatostatin (δ-cells) were found with distinct membrane fluorescence indicating the presence of Ob-Rs on these cell types (Fig. 2B, solid arrows). The distribution of the Ob-Rs on specific islet endocrine cells was also determined using an antiserum raised against a specific epitope of the Ob-R. Ob-R immunoreactivity was found on β-cells and δ-cells (Fig. 2C, solid arrows) but not α-cells (Fig. 2C, hollow arrows). No Ob-R immunofluorescence was detected on cells incubated with preimmune serum (not shown). Collectively, these data indicate that Ob-Rs are expressed on β- and δ-cells, but are absent from the majority of glucagon-producing α-cells.
FIG. 2.

A: binding of purified boiled (left panel) or nonboiled (right panel) Cy3-leptin to dispersed rat islet cells. B: combined Cy3-leptin binding and immunofluorescence of dispersed rat islet cells. Both columns show the same fields of view, either under a DTAF filter to allow hormone detection (green, left column) or under a Cy3 filter to allow detection of the Cy3-leptin (orange, right column). Hormone immunoreactive cells are indicated as either positive (solid arrows) or negative (hollow arrows) for Cy3-leptin binding. C: dual immunofluorescence of dispersed rat islet cells for hormone immunoreactivity (green, left column) and Ob-R immunoreactivity (orange, right column).
Effects of leptin on islet hormone release
To determine whether the insulin-lowering effects of leptin observed in vivo following administration of leptin by either daily injections (1,2) or gene therapy (12,13) involve direct effects of leptin on pancreatic islets, we isolated islets from ob/ob mice and measured the effects of leptin on insulin release. The application of 100 ng/ml (~6 nmol/l) leptin, a concentration within the range of plasma leptin levels in obese animals (14), significantly reduced basal insulin release at 5.5 mmol/l glucose by ~28% (Table 1). This response is similar in magnitude to that recently reported (10). We also observed a significant but smaller reduction of insulin release (13%) in response to leptin in islets incubated at a higher glucose concentration (11.0 mmol/l), and no effect of leptin in the presence of 10 nmol/l GLP-I. Ob-Rs were also detected on δ-cells, raising the possibility that leptin might regulate somatostatin secretion. Therefore, we examined the effects of leptin on the release of somatostatin in islets prepared from ob/ob mice. In contrast to the observed inhibition of insulin release, we observed no significant effect of leptin on somatostatin release from ob/ob islets (Table 1), suggesting that the reduction of insulin secretion was not mediated by somatostatin.
TABLE 1.
Effects of leptin on 1-h insulin and somatostatin release from islets isolated from 10-week-old female ob/ob mice
| Treatment | Insulin (ng/ml) | Somatostatin (pg/ml) |
|---|---|---|
| 5.5 mmol/1 glucose | 7.58 ± 0.74 | 40.42 ± 2.52 |
| 5.5 mmol/1 glucose 100.0 ng/ml leptin |
5.47 ± 0.68* | 40.91 ± 5.77 |
| 11.0 mmol/1 glucose | 8.63 ± 0.48 | 45.31 + 4.50 |
| 11.0 mmol/1 glucose 100.0 ng/ml leptin |
7.53 ± 0.31† | 39.65 ± 3.37 |
| 11.0 mmol/1 glucose 10.0 nmol/1 GLP-I |
13.87 ± 1.01 | 59.19 ± 6.11 |
| 11.0 mmol/1 glucose 10.0 nmol/1 GLP-I 100.0 ng/ml leptin |
14.06 ± 1.17 | 59.98 ± 1.88 |
Data are means ± SE; n = 7; GLP-I, n = 4.
P < 0.0007
P < 0.022 compared with controls in the absence of leptin using Student’s paired t test.
Effects of leptin on β-cell [Ca2+]i, membrane potential, membrane conductance, and single KATP channel activity
It is known that the activity and/or number of KATP regulates the resting potential of pancreatic β-cells (15). Inactivation (closure) of these channels in response to glucose or other insulin secretagogues generates cellular depolarization, activation of voltage-dependent Ca2+ channels, a rise of cytosolic [Ca2+]i, and insulin secretion (15). Thus, to determine whether the suppressive effect of leptin on insulin secretion is mediated by actions on KATP, we performed patch clamp electrophysiological analyses on β-cells isolated from mouse islets. The application of leptin (100 ng/ml) to mouse β-cells produced membrane hyperpolarization and a decrease in the frequency of action potentials (Fig. 3A, upper panel). This reduced frequency of action potentials was accompanied by a fall of [Ca2+]i (Fig. 3A, lower panel). The fall of [Ca2+]i in a ob/ob mouse β-cell resulting from the bath application of 100 ng/ml leptin was reversed by the introduction of 20 mmol/l glucose and 10 nmol/l GLP-I, which resulted in a rapid increase of [Ca2+]i, followed by a return to baseline (Fig. 3B). That the leptin-induced decrease of [Ca2+]i was reversed by the application of glucose plus GLP-I is in agreement with our observation that glucose plus GLP-I can overcome leptin suppression of insulin release (Table 1). The effect of leptin on membrane conductance was determined using the perforated patch technique in ob/ob mouse β-cells. The application of leptin (100 ng/ml) increased membrane conductance, consistent with the activation of KATP (Fig. 3C). Furthermore, this increase in conductance was rapidly reversed by the application of the sulfonylurea tolbutamide (Fig. 3C).
FIG. 3.

A: simultaneous perforated patch recordings of membrane potential (top trace) and fura-2 measurement of [Ca2+]i (lower trace) from a normal mouse β-cell bathed in SES containing 5.5 mmol/l glucose. A 3-min application of 100 ng/ml leptin is indicated by the bar. B: measurement of [Ca2+]i from an ob/ob mouse β-cell bathed in SES containing 5.5 mmol/l glucose. The introduction of 100 ng/ml leptin is indicated by the lower bar. The contents of the bath were switched to a solution containing 100 ng/ml leptin, 20 mmol/l glucose, and 10 nmol/l GLP-I, as indicated by the step of the upper bar. C: recordings from an ob/ob mouse β-cell bathed in SES containing 5.5 mmol/l glucose and voltage clamped at −70 mV using the perforated patch technique. The pipette potential was shifted by ±10 mV (1 s duration) to monitor the membrane conductance. Leptin (100 ng/ml) and tolbutamide (100 μmol/l) application (indicated by bars) were from puffer pipettes placed close to the cell. D: single channel records in an on-cell patch from an ob/ob mouse β-cell bathed in SES containing 5.5 mmol/l glucose. The currents shown in each panel are continuous stretches of data. Control currents are shown in the left panel, and currents beginning 4 min after the start of a 3-min pulse of leptin (100 ng/ml) are shown in the right panel. Channel openings are shown as downward current deflections indicated by the opening of 1, 2, or 3 channels from a closed state (0). E: between 10 and 13 min after the start of the leptin pulse, the patch pipette was stepped to different potentials to obtain the single-channel current-voltage relationship to measure the conductance of the channel. Single-channel current amplitudes at the different potentials were obtained from Gaussian fits to all-points histograms of digitized current records in D. F: the mean value for open probability of KATP plotted against time (in minutes) from six on-cell patches from ob/ob mouse β-cells at a pipette potential of 0 mV.
To investigate the effects of leptin on KATP in greater detail, we also obtained measurements of single-channel activity in on-cell patches of ob/ob mouse β-cells. Leptin increased the KATP open channel probability with no significant effect on open times. During the control period (Fig. 3D, left panel), the channel open probability was ~0.005, and the control mean channel open times were fitted with two exponential components with time constants of 0.63 and 2.28 ms. Four minutes following the start of a 3-min application of 100 ng/ml leptin (Fig. 3D, right panel), the open probability of the channels increased to 0.051 with no effect on the mean open times (0.57 and 2.16 ms). The peak value for the open probability was 0.09 at 18 min after the start of the leptin application in this patch. The single-channel conductance was measured as 80 picosiemens (Fig. 3E), consistent with these channels being KATP. The extrapolated reversal potential of the currents was 46 mV, relative to the resting potential of the cell. The mean value for open probability of KATP versus time appeared to show a biphasic time course (Fig. 3F), similar to that seen in Fig. 3C, for the whole cell conductance measurements.
DISCUSSION
In these studies, we demonstrated that the long form of the leptin receptor (Ob-Rb) is expressed in pancreatic β- and δ-cells, although it appears that the shorter transcripts predominate (10,16). It is generally believed that Ob-Rb is the signal-transducing receptor; no clear functions are as yet attributable to the shorter receptor isoforms (17). However, downstream signaling effects of leptin on human hepatic cells have been reported in which only a short variant of the leptin receptor was detected, suggesting the involvement of an accessory receptor subunit or inherent signaling capabilities of the short form (18). Indeed, a short variant receptor has been reported to induce gene expression in transfected Chinese hamster ovary cells (19). We found an inhibitory effect of leptin (100 ng/ml) on insulin secretion, which was reduced by increasing the glucose concentration and ameliorated by GLP-I. In support of our finding, Emilsson et al. (10) also demonstrated significant effects of leptin on the inhibition of insulin secretion at leptin concentrations ≥10 nmol/l but no effect at 1 nmol/l (~16 ng/ml). Leclercq-Meyer et al. (16), using rat perfused pancreases, argued against a role for leptin in regulating insulin secretion. However, in their studies, they used only a single dose of 1 nmol/l of recombinant human leptin at a background glucose concentration of 8.3 mmol/l.
Clues to the cellular mechanisms by which leptin suppresses insulin release come from earlier studies utilizing ob/ob and db/db mouse islets. It was reported more than two decades ago that islets from db/db mice are partially depolarized, even in the absence of glucose (20). This circumstance is associated with elevated basal insulin release and unresponsiveness to further elevation of the glucose concentration (20). The K+ permeability of both ob/ob and db/db islets is relatively insensitive to changes in glucose (21-23), and this insensitivity does not appear to be due to a failure of the cells to express functional KATP (24,25). These earlier observations in conjunction with our present findings indicate that the partially depolarized state characteristic of β-cells from leptin deficient (ob/ob) and leptin resistant (db/db) mice can be ascribed to a failure of leptin to activate KATP. The action of leptin so implied would be to hyperpolarize the β-cell and to inhibit insulin secretion.
Notably, we found that leptin produced a biphasic increase in membrane conductance, as expected if hyperpolarization resulted from the activation of KATP. The activation of KATP was also indicated by our finding that tolbutamide, a sulfonylurea that binds to the sulfonylurea receptor subunit of KATP (SUR1) (26,27), reversed the leptin-induced increase in membrane conductance. The time course of the change in open channel probability in response to leptin that we observed was characterized by both early and late components. The delayed effect of leptin that we observed in ob/ob β-cells is similar to that previously observed for potassium channel activation by janus kinase–associated prolactin receptors in Chinese hamster ovary cells (28).
The autonomic nervous system may also contribute to the leptin-mediated reduction of insulin secretion. For example, leptin administration in vivo inhibits neuropeptide Y (NPY) gene expression (2,29) and secretion (2), which in turn could result in reduced insulin secretion by either inhibiting the parasympathetic or activating the sympathetic nervous system. In the absence of leptin (ob/ob mice), NPY signaling is elevated (2,29,30) and thus could contribute to hyperinsulinemia in these animals. Evidence supporting this notion has recently been provided by the observation that insulin levels are reduced by half in ob/ob mice deficient of NPY (31). However, it is important to note that insulin levels in these animals are still ~30-fold higher, compared with wild-type control mice (31), suggesting that leptin reduces insulin secretion via pathways other than central effector actions of NPY, namely by a direct effect on β-cells.
Although the obesity observed in ob/ob and db/db mice is understood to result from deficient leptin regulation of satiety centers (1-4), the reason for the development of NIDDM in these animal models has not been explained. We provide a mechanism for the early development of hyperinsulinemia (32,33) and the resulting predisposition for NIDDM in ob/ob and db/db mice. Evidently, hyperinsulinemia in these animal models of obesity and NIDDM can be ascribed to persistent β-cell depolarization, an effect we attribute to the diminished Ob-R–mediated activation of KATP. This pathophysiological process may offer important clues as to what normal function leptin may play in the regulation of insulin secretion. We propose that leptin acting on KATP serves more as a β-cell controller to dampen basal insulin release during fasting, rather than as an acute regulator of insulin secretion during feeding. In this conceptual model, leptin acts as a brake to inhibit insulin secretion tonically by maintaining the β-cell in a moderately hyperpolarized state. As such, the tonic inhibition can easily be overcome by nutrient and incretin signals (e.g., GLP-I) that accompany feeding, resulting in the depolarization of the β-cell and prompt insulin secretion (33a). This proposal is consistent with the demonstration that plasma leptin levels derived from adipose stores also do not appear to be acutely regulated by feeding (34-36). Taken together, these observations define the existence of an adipoinsular axis by which the body fat mass informs the β-cells to secrete less insulin. It is known that insulin stimulates leptin secretion (37-39), thus establishing a bi-directional feedback loop between adipose tissue and the β-cells via the hormones leptin and insulin, respectively.
ACKNOWLEDGMENTS
This study was supported in part by grants from the U.S. Public Health Service (G.G.H. and J.F.H.) and the American Diabetes Association (G.G.H.). J.F.H. is an investigator with the Howard Hughes Medical Institute. We thank members of the Laboratory of Molecular Endocrinology for helpful discussions, Karen McManus for expert technical assistance, and Townley Budde for help in the preparation of the manuscript.
Glossary
- Cy3
iodocarbocyanine
- DTAF
dichlorotriazinylamino fluorescein
- fura-2 AM
fura-2 acetoxymethyl ester
- GLP
glucagon-like peptide
- [Ca2+]i
intracellular calcium
- KATP
ATP-sensitive potassium channel
- NPY
neuropeptide Y
- Ob-R
leptin receptor
- Ob-Rb
full-length leptin receptor
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- RT
reverse transcriptase
- SES
standard extracellular solution
REFERENCES
- 1.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 2.Stephens TW, Basinski M, Bristow PK, Bue-Valleskey JM, Burgett SG, Craft L, Hale J, Hoffmann J, Hsiung HM, Kriauciunas A, MacKellar W, Rosteck PR, Jr, Schoner B, Smith D, Tinsley FC, Zhang X-Y, Heiman M. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature. 1995;377:530–532. doi: 10.1038/377530a0. [DOI] [PubMed] [Google Scholar]
- 3.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir G, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Woolf EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;84:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 4.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 5.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse ob protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 7.Lee G-H, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 8.Levin N, Nelson C, Gurney A, Vandlen R, De Sauvage F. Decreased food intake does not completely account for adiposity reduction after ob protein infusion. Proc Natl Acad Sci USA. 1996;93:1726–1730. doi: 10.1073/pnas.93.4.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kieffer TJ, Heller RS, Habener JF. Leptin receptors expressed on pancreatic β-cells. Biochem Biophys Res Commun. 1996;224:522–527. doi: 10.1006/bbrc.1996.1059. [DOI] [PubMed] [Google Scholar]
- 10.Emilsson V, Liu Y-L, Cawthorne MA, Morton NM, Davenport M. Expression of the functional leptin receptor mRNA in pancreatic islets and direct inhibitory action of leptin on insulin secretion. Diabetes. 1997;46:313–316. doi: 10.2337/diab.46.2.313. [DOI] [PubMed] [Google Scholar]
- 11.Grynkiewicz G, Poenie M, Tsein RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 12.Muzzin P, Eisensmith RC, Copeland KC, Woo SLC. Correction of obesity and diabetes in genetically obese mice by leptin gene therapy. Proc Natl Acad Sci USA. 1996;93:14804–14808. doi: 10.1073/pnas.93.25.14804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen G, Koyama K, Yuan X, Lee Y, Zhou Y-T, O’Doherty R, Newgard CB, Unger RH. Disappearance of body fat in normal rats induced by adenovirus-mediated leptin gene therapy. Proc Natl Acad Sci USA. 1996;93:14795–14799. doi: 10.1073/pnas.93.25.14795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiraoka J, Hosoda K, Ogawa Y, Ikeda K, Nara Y, Masuzaki H, Takaya K, Nakagawa K, Mashimo T, Sawamura M, Koletsky RJ, Yamori Y, Nakao K. Augmentation of obese (ob) gene expression and leptin secretion in obese spontaneously hypertensive rats (obese SHR or Koletsky rats) Biochem Biophys Res Commun. 1997;231:582–585. doi: 10.1006/bbrc.1997.6145. [DOI] [PubMed] [Google Scholar]
- 15.Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Prog Biophys Molec Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 16.Leclercq-Meyer V, Considine RV, Sener A, Malaisse WJ. Do letpin receptors play a funtional role in the endocrine pancreas? Biochem Biophys Res Commun. 1997;229:794–798. doi: 10.1006/bbrc.1996.1882. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- 18.Cohen B, Novick D, Rubinstein M. Modulation of insulin activities by leptin. Science. 1996;274:1185–1188. doi: 10.1126/science.274.5290.1185. [DOI] [PubMed] [Google Scholar]
- 19.Murakami T, Yamashita T, Iida M, Kuwajima M, Shima K. A short form of leptin receptor performs signal transduction. Biochem Biophys Res Commun. 1997;231:26–29. doi: 10.1006/bbrc.1996.6030. [DOI] [PubMed] [Google Scholar]
- 20.Meissner HP, Schmidt H. The electrical activity of pancreatic β-cells of diabetic mice. Febs Lett. 1976;67:371–374. doi: 10.1016/0014-5793(76)80567-4. [DOI] [PubMed] [Google Scholar]
- 21.Berglund O, Sehlin J, Täljedal I-B. 86Rb+ fluxes and K+-stimulated nitrophenyl phosphatase activity in the pancreatic islets of genetically diabetic mice (C57BL/KsJ-db/db) Diabetologia. 1978;15:191–195. doi: 10.1007/BF00421238. [DOI] [PubMed] [Google Scholar]
- 22.Rosario LM, Atwater I, Rojas E. Membrane potential measurements in islets of Langerhans from ob/ob obese mice suggest an alteration in [Ca2+]i-activated K+ permeability. QJ Exp Physiol. 1985;70:137–150. doi: 10.1113/expphysiol.1985.sp002885. [DOI] [PubMed] [Google Scholar]
- 23.Fournier LA, Heick HMC, Bégin-Heick N. The influence of K+-induced membrane depolarization on insulin secretion in islets of lean and obese (ob/ob) mice. Biochem Cell Biol. 1990;68:243–248. doi: 10.1139/o90-033. [DOI] [PubMed] [Google Scholar]
- 24.Kukuljan M, Li MY, Atwater I. Characterization of potassium channels in pancreatic β cells from ob/ob mice. Febs Lett. 1990;266:105–108. doi: 10.1016/0014-5793(90)81518-s. [DOI] [PubMed] [Google Scholar]
- 25.Fournier LA, Bégin-Heick N, Whitfield JF, Schwartz J-L. Comparison of the properties of the ATP-sensitive K+ channels of pancreatic β-cells of lean and obese (ob/ob) C57BL/6J mice. J Membrane Biol. 1992;129:267–276. doi: 10.1007/BF00232908. [DOI] [PubMed] [Google Scholar]
- 26.Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, IV, Boyd AE, III, González G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the β cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–429. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- 27.Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- 28.Prevarskaya NB, Skryma RN, Vacher P, Daniel N, Djiane J, Dufy B. Role of tyrosine phosphorylation in potassium channel activation: functional association with prolactin receptor and JAK2 tyrosine kinase. J Biol Chem. 1995;270:24292–24299. doi: 10.1074/jbc.270.41.24292. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz MW, Baskin DG, Bukowski TR, Kuijper JL, Foster D, Lasser G, Prunkard DE, Porte D, Jr, Woods SC, Seeley RJ, Weigle DS. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes. 1996;45:531–535. doi: 10.2337/diab.45.4.531. [DOI] [PubMed] [Google Scholar]
- 30.Wilding JPH, Gilbey SG, Bailey CJ, Batt RAL, Williams G, Ghatei MA, Bloom SR. Increased neuropeptide-Y messenger ribonucleic acid (mRNA) and decreased neurotensin mRNA in the hypothalamus of the obese (ob/ob) mouse. Endocrinology. 1993;132:1939–1944. doi: 10.1210/endo.132.5.7682936. [DOI] [PubMed] [Google Scholar]
- 31.Erickson JC, Hollopeter G, Palmiter RD. Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science. 1996;274:1704–1707. doi: 10.1126/science.274.5293.1704. [DOI] [PubMed] [Google Scholar]
- 32.Dubuc PU. The development of obesity, hyperinsulinemia, and hyperglycemia in ob/ob mice. Metabolism. 1976;25:1567–1574. doi: 10.1016/0026-0495(76)90109-8. [DOI] [PubMed] [Google Scholar]
- 33.Herberg L, Coleman DL. Laboratory animals exhibiting obesity and diabetes syndromes. Metabolism. 1977;26:59–99. doi: 10.1016/0026-0495(77)90128-7. [DOI] [PubMed] [Google Scholar]
- 33a.Holz GG, IV, Kühltrelber WM, Habener JF. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-l(7-37) Nature. 1993;361:362–365. doi: 10.1038/361362a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LI, Bauer TL, Caro JF. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 35.Sinha MK, Ohannesian JP, Heiman ML, Kriauciunas A, Stephens TW, Magosin S, Marco C, Caro JF. Nocturnal rise of leptin in lean, obese, and non-insulin-dependent diabetes mellitus subjects. J Clin Invest. 1996;97:1344–1347. doi: 10.1172/JCI118551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dagogo-Jack S, Fanelli C, Paramore D, Brothers J, Landt M. Plasma leptin and insulin relationships in obese and nonobese humans. Diabetes. 1996;45:695–698. doi: 10.2337/diab.45.5.695. [DOI] [PubMed] [Google Scholar]
- 37.Saladin R, Vos PD, Guerre-Millo M, Leturque A, Girard J, Staels B, Auwerx J. Transient increase in obese gene expression after food intake or insulin administration. Nature. 1995;377:527–529. doi: 10.1038/377527a0. [DOI] [PubMed] [Google Scholar]
- 38.Malmström R, Taskinen M-R, Karonen S-L, Yki-Järvinen H. Insulin increases plasma leptin concentrations in normal subjects and patients with NIDDM. Diabetologia. 1996;39:993–996. doi: 10.1007/BF00403921. [DOI] [PubMed] [Google Scholar]
- 39.Wabitsch M, Jensen PB, Blum WF, Christoffersen CT, Englaro P, Heinze E, Rascher W, Teller W, Tornqvist H, Hauner H. Insulin and Cortisol promote leptin production in cultured human fat cells. Diabetes. 1996;45:1435–1438. doi: 10.2337/diab.45.10.1435. [DOI] [PubMed] [Google Scholar]