

Abstract
Main features of rheumatoid arthritis (RA), hyperplasia of fibroblast-like synoviocytes (FLS) and joint destruction are caused by inflammatory cytokines produced in chronic autoimmune inflammation. Cell-intrinsic acquisition of tumour-like phenotypes of RA-FLS could also be responsible for the aggressive proliferation and invasion, which are supported by the fact that in some cases RA-FLS has mutations of a tumour suppressor gene TP53. However, the underlying molecular mechanism for TP53 mutations in RA-FLS has not yet been clarified. Recently it has been reported that the non-lymphoid cells in the inflammatory tissues express ectopically the activation-induced cytidine deaminase (AID) gene that induces somatic hypermutations, not only at the immunoglobulin (Ig) gene variable regions in germinal centre B lymphocytes but also at coding regions in TP53. Real-time polymerase chain reaction (PCR) analyses revealed more than half (five of nine) of the RA-FLS lines we established showed the markedly increased expression of AID. AID transcription in RA-FLS was augmented by tumour necrosis factor (TNF)-α and even by physiological concentration of β-oestradiol that could not induce AID transcription in osteoarthritis-FLS. Furthermore, AID-positive RA-FLS presented a higher frequency of somatic mutations in TP53. Cytological and immunohistochemical analyses demonstrated clearly the ectopic expression of AID in the FLS at the RA synovium. These data suggested strongly a novel consequence of RA; the ectopic expression of AID in RA-FLS causes the somatic mutations and dysfunction of TP53, leading to acquisition of tumour-like properties by RA-FLS.
Keywords: human, rheumatoid arthritis, somatic mutation, synoviocyte
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease in which the joints are chronically inflamed and the cartilage and bone are destroyed by pannus formation, which is the invasion of cartilage and bone by proliferating fibroblast-like synoviocytes (FLS) [1–3]. The incidence of RA is correlated with certain human leucocyte antigen D-related (HLA-DR) haplotypes and the presence of autoantibodies, such as rheumatoid factor (RF) and anti-cyclic citrullinated peptide (CCP) antibody, suggesting strongly the involvement of the deregulated immune system; T helper type 17 (Th17), a novel helper T cell subset producing interleukin (IL)-17, has become a topic as a player in local inflammation driven by acquired immunity [4,5]. However, details of the pathophysiology of RA is not understood completely [6]. Local cytokines such as basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), transforming growth factor (TGF)-β, tumour necrosis factor (TNF)-α and IL-1β are considered to be responsible for the hyperplasia of FLS [7,8]. Upon activation, FLS produce TNF-α, IL-1β, IL-6 and matrix metalloproteinases, establishing the chronic and destructive inflammatory circuit [2]. The critical roles of these inflammatory cytokines have been shown by the effectiveness of cytokine-blockade therapies using anti-TNF-α or anti-IL-6 receptor antibodies for the treatment of RA [9,10]. However, it has also been recognized that certain RA subsets are resistant to these anti-cytokine therapies [11]. Such resistance may be explained partly by the intrinsic abnormality of RA-FLS independent of inflammation. Accumulated evidence has indicated that RA-FLS are stably activated and exhibit tumour-like characteristics. For example, they destroyed human cartilage when they were transplanted together into severe combined immunodeficiency (SCID) mice [12]. RA-FLS expressed high levels of proto-oncogenes such as c-myc and c-fos[13,14]. In addition to these findings, RA-FLS are reported to express the tumour suppressor gene TP53 with somatic mutations [15–19], and down-regulate the tumour suppressor PTEN, a protein phosphatase gene [20]. In particular, the somatic mutation of the TP53 gene in RA-FLS appears consistent not only with their increased resistance to apoptosis but also with their proinflammatory properties such as the production of IL-6 and matrix metalloproteinase (MMP)-1 [21–23]. However, little is known about the mechanism by which the somatic mutations are introduced into the TP53 gene in RA-FLS.
Activation-induced cytidine deaminase (AID) is a member of the APOBEC family, which is a cellular cytidine deaminase involved in protection from retrovirus infection or regulation of cholesterol metabolism [24]. AID was identified originally as an indispensable molecule for somatic hypermutation (SHM) at the immunoglobulin variable region and class-switch recombination in germinal centre B lymphocytes [25,26]. Recently, several papers have demonstrated that AID was up-regulated in non-lymphoid tumour cells such as breast cancer, cholangiocarcinoma, hepatoma and colorectal cancer cells [27–33]. Moreover, the somatic mutations of TP53 found in these cancer cells appeared to be a direct target of AID [29,30].
In our study, we demonstrated that AID was expressed selectively by a fraction of RA-FLS, and that it was associated with somatic mutations in TP53. This suggests a possible mechanism by which the aberrant expression of AID within certain RA-FLS induces somatic mutations in TP53, leading to the acquisition of proinflammatory or tumour-like phenotypes.
Materials and methods
Cells and cell culture
Transformed human FLS cell lines were established from the synovial tissues of RA (n = 9; five males and four females) and osteoarthritis (OA) (n = 9; nine females) patients at the time of joint replacement surgery (Table 1). The tissues were obtained with the informed consent of the patients. Briefly, the synovial tissues were minced into small pieces and dissociated with collagenase and hyarulonidase in DMEM at 37°C for 1 h with shaking. After passing through mesh and washing with Dulbecco's modified Eagle's medium (DMEM), the synovial cells were suspended in the culture medium [DMEM containing 10% fetal calf serum (FCS) and penicillin/streptomycin] and plated in dishes. On the next day, non-adherent cells were removed and the medium was refreshed. The synovial cells were cultured in 10% CO2 at 37°C with humidified air. After four passages, when it was ensured that no haematopoietic cells were present in the cell lines, plasmids pAct-SVT containing SV40T antigen were transduced with lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). All the cell lines showed fibroblast-like morphology and expressed CD44, CD106 and CD157/BST-1 on their surfaces, characteristic of synovial fibroblasts (data not shown). The transformed and original non-transformed primary FLS cells were maintained in DMEM supplemented with 10% heat-inactivated FCS with antibiotics. Materials for mRNA and protein analyses were obtained from the cell lines with passage numbers of <8 after thawing the frozen stocks. This study protocol was approved by the institutional review boards for ethics at the Faculty of Medicine, Osaka University (no. 340-1) and the Kawasaki Medical School (no. 291). For cell stimulation, FLS were cultured in phenol red-free DMEM (Sigma-Aldrich, St Louis, MO, USA) complete medium in the presence or absence of TNF-α (50 ng/ml; Pepro Tech Inc., Rocky Hill, NJ, USA), β-oestradiol (E2; 10−9 M; Sigma-Aldrich) or both for 24 h.
Table 1.
Characteristics of the patients in the study of transformed fibroblast-like synoviocytes.
| OA (n = 9) | RA (n = 9) | |
|---|---|---|
| Age, mean (range) years | 70·7 (64–80) | 61·3 (35–75) |
| Sex, no. female/male | 9/0 | 4/5 |
| Disease duration, mean (range) years | n.a. | 9·9 (4–19) |
| Medications, no. taking/no. assessed | ||
| NSAIDs | 9/9 | 6/9 |
| DMARDs | 0/9 | 7/9 |
| Plus steroids | 0/9 | 5/9 |
| Soluble TNFR | 0/9 | 1/9 |
| No. RF+(>20IU)/no. assessed | n.a. | 7/8 |
| CRP, mean (range) mg/dl | n.a. | 2·8 (0·7–5·3) |
| MMP-3, mean (range) ng/ml | n.a. | 424·1 (49·7–973) |
CRP, C-reactive protein; DMARDs, disease-modifying anti-rheumatic drugs; MMP, matrix metalloproteinase; n.a., not assessed; NSAID, non-steroidal anti-inflammatory drugs; OA, osteoarthritis; RF, rheumatoid factor; TNF, tumour necrosis factor.
Non-quantitative reverse transcription–polymerase chain reaction (RT–PCR) and quantitative real-time RT–PCR
Total RNAs were extracted using TRIzol® (Invitrogen), according to the manufacturer's instructions. Random hexamer-primed cDNAs were prepared using the ReverTra Ace® RT kit (Toyobo, Osaka, Japan) with 1 µg of total RNA and amplified by PCR using KOD-FX (Toyobo). The synthetic oligonucleotide primers for amplification of AID, APOBEC1 and APOBEC3G were as follows: AID, 5′-AAATGTCCGCTGGGCTAAGG-3′ (forward) and 5′-GGAGGAAGAGCAATTCCACGT-3′ (reverse) [28]; APOBEC1, 5′-GGGACCTTGTTAACAGTGGAGT-3′ (forward) and 5′-CCAGGTGGGTAGTTGACAAAA-3′ (reverse); APOBEC3G, 5′-GAGCGCATGCACAATGAC-3′ (forward) and 5′-GCCTTCAAGGAAACCGTGT-3′ (reverse). The latter two primer sets were synthesized using Primer-BLAST (NCBI). The PCR for CD19 to exclude the contamination of B cells in FLS lines was conducted with the following primers: 5′-GACCTCACCATGGCCCCTGG-3′ (forward) and 5′-CAGCCAGTGCCATAGTAC-3′ (reverse) [34]. The specific primers for human AID for quantitative real-time RT–PCR were designed using the Universal Primer Design Tools available on the Roche website. The 6-carboxyfluorescein-labelled probe for human AID was obtained from Roche (Universal Probe Library, probe no. 69; Roche Diagnostics, Indianapolis, IN, USA). The expression levels of target cDNAs were normalized to the endogenous transcription levels of human GAPDH. Gene expression was quantified using the 7500 real-time PCR system (PE Applied Biosystems, Foster City, CA, USA).
Western blot analysis
Whole-cell lysates (equivalent to 25 µg) in 1% TNE lysis buffer [1% Nonidet-P40, 150 mM NaCl, 10 mM Tris-HCl, pH 7·5, 1 mM ethylenediamine tetraacetic acid (EDTA)], in the presence of a protease inhibitor cocktail (Sigma-Aldrich) and protein kinase/phosphatase inhibitor cocktail (Roche) were subjected to Western blot analysis using polyvinylidine fluoride membrane and signal-enhancing kit (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). The membrane was probed with rabbit anti-human AID antibody (Abcam, Cambridge, MA, USA) and reprobed with anti-β-actin antibody (Santa Cruz).
Immunocytochemistry and immunohistochemistry
We cultured 2 × 104 cells on the culture slide chamber (BD Falcon, Franklin Lakes, NJ, USA) for 12 h. After fixation with chilled acetone, the cells were washed and then blocked with 3% bovine serum albumin (BSA) in PBS. The cells were immunostained with either rabbit anti-human AID antibody (Abcam) combined with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin (Ig)G antibody (Southern Biotech, Birmingham, AL, USA) or with rat anti-human AID monoclonal antibody (mAb) (EK2 5G9; Cell Signaling Tech. Inc., Danvers, MA, USA) combined with biotinylated donkey anti-rat IgG antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) and vectastain elite ABC kit (Vector Laboratories Inc., Burlingame, CA, USA). The bound antibodies were visualized using diaminobenzidine (DAB) and counterstaining with haematoxylin. The images were obtained using a bright-field microscope (Axiophot, Carl Zeiss, Munich, Germany).
Cryosections with 6 µm thickness of the synovial tissues from RA patients were also stained with rat anti-human AID mAb after fixation with chilled acetone and detected as described above. The endogenous peroxidase activity was inactivated by treatment with 0·01% H2O2/PBS for 10 min at room temperature. Non-specific binding of second antibodies was blocked with PBS containing 5% of either normal goat or donkey serum for 30 min. To identify B cell follicles, mouse anti-human CD20 (Dako, Glostrup, Denmark) mAb and Envision goat anti-mouse and rabbit IgG antibody conjugated with horseradish peroxidase (Dako) were used. Rat anti-mouse CD4 mAb (GK1·5) was used as an isotype-matched control. For immunofluorescence staining, cryosections with 6 µm thickness of the synovial tissues from RA patients were fixed and stained with rat anti-human AID mAb and mouse anti-human CD20, the same as above. Either donkey anti-rat IgG antibody conjugated with Alexa 488 or donkey anti-mouse IgG antibody conjugated with Alexa 594 were used as second antibodies. Fluorescence images were obtained under the fluorescence microscope (Eclipse E800, Nikon, Tokyo, Japan).
Mutation analysis of TP53
The coding region of TP53 spanning from exons 4–11 was amplified using high-fidelity DNA polymerase (iProof™, Bio-Rad, Hercules, CA, USA) with forward and reverse primers containing the SalI and EcoRI sequences, respectively, as follows: forward, 5′-GCGTCGACCTACCAGGGCAGCTACGGTTTC-3′ and reverse, 5′-GGAATTCTTATGGCGGGAGGTAGACTGACC-3′. The amplicon was digested using restriction enzymes and subcloned into the corresponding cloning sites of the pBluescript vector. The insertion was sequenced with both the T3 and T7 primers located upstream and downstream of the cloning site, respectively, in pBluescript using sequence detector (Applied Biosystems). From the amplicons prepared from one FLS line, more than 35 colonies were picked up after bacterial transformation and each PCR product were sequenced. The sequence data were compared with those of the TP53 nucleotide sequence obtained from GenBank (Accession number: NM_000546).
Results
Aberrant expression of AID in RA-FLS
First, we assessed the expression of the AID gene in transformed FLS cell lines obtained from OA and RA patients by RT–PCR. Surprisingly, AID was transcribed in more than half the RA-FLS lines (five of nine) and in none of the OA-FLS lines (Fig. 1a). In the RA-FLS lines APOBEC1 was not expressed, whereas APOBEC3G was expressed ubiquitously, suggesting that AID transcription in RA-FLS is selective (Fig. 1a). Then, we used real-time PCR to quantify AID transcription among FLS. The same sets of RA-FLS showed approximately 7–18-fold higher AID transcription compared to the FLS from OA2 (Fig. 1b) that expressed a low but detectable level of AID transcription. Due probably to the higher sensitivity of probe-based real-time PCR, negligible to very low levels of AID transcription were detectable by real-time PCR in the other four RA-FLS, as well as in some OA-FLS. Here, five RA-FLS with the expression levels of AID higher than fivefold compared with the level of OA2 are defined as AIDhigh FLS (RA1, 4, 5, 7, 9) and the other four RA-FLS (RA2, 3, 6, 8) with the equivalent levels to OA-FLS are defined as AIDlow FLS. This aberrant expression of AID is not likely to have resulted by contamination of B cells expressing AID, because the synovial cell culture was occupied with adherent and morphologically fibroblast-like cells after four passages. The possibility of B cell contamination was clearly excluded because no transcription of CD19, a pan B cell marker, was detectable in the AIDhigh FLS (data not shown, but the primer pairs are described in Materials and methods). Next, we confirmed the endogenous expression of the AID protein by Western blot analysis with the two representative cell lines from OA, AIDlow RA and AIDhigh RA (Fig. 1c).
Fig. 1.
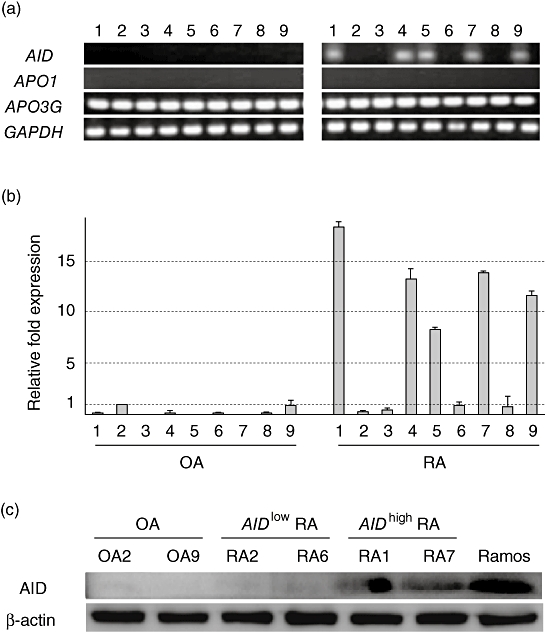
Endogenous expression of activation-induced cytidine deaminase (AID) in rheumatoid arthritis-fibroblast-like synoviocytes (RA-FLS) lines. (a) The ectopic transcription of AID was detected in RA-FLS lines by reverse transcription–polymerase chain reaction (RT–PCR). The expression of two other APOBEC family members (APO1: APOBEC1, APO3G: APOBEC3G) was also examined by RT–PCR. (b) AID mRNA was quantified (relative quantification) by real-time PCR and normalized to housekeeping gene, GAPDH and fold expression over that of OA-FLS (OA2) was calculated by ΔΔCT method in triplicate. The grey bars and error bars indicate the averages and standard deviations, respectively, of two independent experiments. (c) Crude protein extracts from the representative FLS lines were analysed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE)/immunoblotting using anti-human AID polyclonal antibody (upper panel) or anti-β-actin (lower panel).
The aberrant expression of AID in a fraction of RA-FLS prompted us to investigate its correlation with the clinical characteristics in nine RA patients from whom FLS were established. The frequencies of AIDhigh RA-FLS were 75% (three of four) and 40% (two of five) in RA-FLS derived from female and male RA patients, respectively. No correlation was observed in the ages, serum MMP-3 levels or medicines prescribed to the patients. Laboratory data revealed the tendency of higher C-reactive protein (CRP) levels in the AIDhigh RA-FLS than in the AIDlow RA-FLS (3·9 ± 1·7 versus 1·5 ± 0·6, respectively), although the sample size of our study is too small to detect these correlations adequately. These data suggested that AID expression in FLS might be facilitated under conditions of inflammation in female patients. Because inflammatory cytokines and oestrogen have been reported to induce AID transcription [28,30,35], we investigated whether this is also the case in FLS. In OA-FLS with low levels of AID expression, TNF-α but not physiological concentration of β-oestradiol (E2) augmented the transcription of AID, and simultaneous stimulation with E2 and TNF-α exhibited synergistic effects (Fig. 2). In AIDlow RA-FLS, E2 or TNF-α alone enhanced the transcription of AID much higher than OA-FLS, although the synergistic effect was not obvious. Even in AIDhigh RA-FLS, E2 or TNF-α alone augmented the transcription of AID, which resulted in more than 20-fold higher levels of AID transcription compared with basal levels those in OA-FLS. It is of note that physiological concentration of β-oestradiol up-regulated AID transcription only in RA-FLS, suggesting intrinsic RA-FLS abnormality in the susceptibility of AID induction to female sex steroids. As another molecular mechanism for AID induction, negative regulatory roles for the microRNA miR-155 on AID transcription have been reported in previous studies on germinal centre B cells [36,37], but there was no significant down-regulation of miR-155 in AIDhigh RA-FLS (data not shown). Real-time PCR analyses of RA-FLS did not show TNF-α transcription or correlation between the transcription levels of IL-6 or IL-1β with that of AID (data not shown), suggesting that AID transcription is induced passively by external stimuli present most commonly in RA rather than that provided by inflammatory cytokines in an autocrine fashion.
Fig. 2.
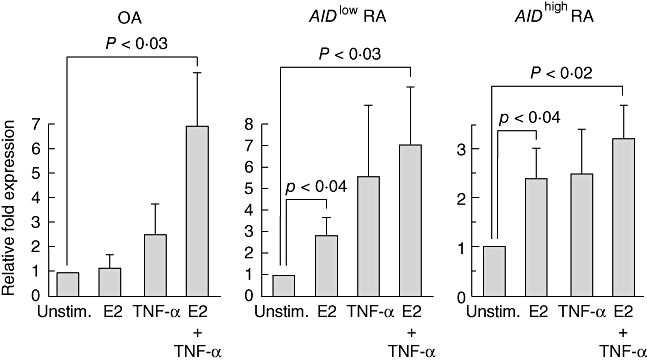
Inducible expression of AID in transformed fibroblast-like synoviocytes (FLS) lines by stimulation with tumour necrosis factor (TNF)-α and oestrogen. One million osteoarthritis (OA)-FLS (OA2, OA6 and OA9), AIDlow RA-FLS (RA2, RA3 and RA6) and AIDhigh RA-FLS (RA1, RA4 and RA7) were stimulated in phenol red-free Dulbecco's modified Eagle's complete medium (DMEM) with either TNF-α (50 ng/ml) or β-oestradiol (E2; 10−9 M) for 24 h. The AID transcription was quantified (relative quantification) by real-time polymerase chain reaction (PCR) and normalized to the housekeeping gene, GAPDH and the relative fold induction over the unstimulated cells (Unstim.) was monitored by the ΔΔCT method. The grey bars and error bars indicate the averages and standard deviations, respectively, of three cell lines. P-values calculated by Student's t-test are indicated.
Accumulation of TP53 gene mutations in AID-expressing RA-FLS
Detailed studies have established that the mutations of the p53 tumour suppressor gene found in RA-FLS could contribute to both the tumour-like and proinflammatory properties of RA-FLS, such as aggressive growth, invasion and destruction of cartilage and bone [15–19,21–23]. Although genotoxic and oxidative stresses have been speculated to be the causative candidates for the somatic mutation in the TP53 gene in RA-FLS, the molecular mechanism has not yet been elucidated. Recently, a clear relationship between AID expression and the frequency of TP53 somatic mutations has been demonstrated in some non-B lymphocytes, such as hepatocytes and colon epithelial cells [29–33]. To investigate whether this is the case for RA-FLS, the coding region of TP53 was amplified by RT–PCR with high-fidelity polymerase, sequenced and then compared among OA-FLS, AIDlow RA-FLS and AIDhigh RA-FLS. The frequency of TP53 somatic mutations was elevated significantly in AIDhigh RA-FLS rather than in OA-FLS and AIDlow RA-FLS (summarized in Table 2). Although sporadic mutations were detected even in the latter two FLS, AIDhigh RA-FLS exhibited approximately two- to 3·5-fold more mutations than the other two FLS subsets. In addition, the frequency of non-silent mutations was three times greater than that of silent mutations. Notably, the dominant base substitution pattern is the transition type, which is very similar to the mutations found at the variable region of the immunoglobulin gene, introduced exclusively by AID (Fig. 3a) [26]. Introduction of somatic mutations at the Ig variable region by AID is coupled with the transcription level of the Ig gene [38] because AID attacks the single-strand DNA that appears at transcription. Thus, the somatic mutation requires both the expression of AID and the transcription of the target gene. Among the representative FLS lines, TP53 was transcribed constitutively and the amounts of TP53 mRNA were comparable, indicating that the increased frequency of mutation in TP53 resulted mainly from AID expression rather than the transcription levels of TP53 (data not shown). Compared with TP53 mutations in previous reports [15,18,19,22], 17% of the mutations that we identified resulted in the changes of amino acids identical with those and 33% of those in different amino acids, but were located at the same codon. The non-sense mutation in RA7 in this study occurred at the same position as that reported by Yamanishi et al. [22].
Table 2.
Mutation frequency of TP53.
| Mutation |
||||||
|---|---|---|---|---|---|---|
| Number of base substitution | Total base pair of sequenced | Frequency/10 000 | Silent | Missense | Non-sense | |
| OA2 | 0 | 23 587 | 0 | 0 | 0 | 0 |
| OA6 | 0 | 25 150 | 0 | 0 | 0 | 0 |
| OA9 | 1 | 27 146 | 0·368 | 1 | 0 | 0 |
| RA2 | 2 | 22 456 | 0·891 | 2 | 0 | 0 |
| RA3 | 1 | 29 459 | 0·339 | 0 | 1 | 0 |
| RA6 | 2 | 24 631 | 0·812 | 1 | 1 | 0 |
| RA1 | 8 | 26 187 | 3·054 | 2 | 6 | 0 |
| RA4 | 6 | 26 655 | 2·251 | 2 | 4 | 0 |
| RA7 | 4 | 23 657 | 1·691 | 0 | 3 | 1 |
cDNA was prepared from representative fibroblast-like synoviocyte (FLS) lines in each group. Osteoarthritis (OA)-FLS: OA2, OA6 and OA9. Activation-induced cytidine deaminase (AID)low rheumatoid arthritis (RA)-FLS: RA2, RA3 and RA6. AIDhigh RA-FLS: RA1, RA4 and RA7. The coding region of TP53 ranging from exons 4–11 was amplified using high-fidelity DNA polymerase. Mutation frequency was analysed by sequencing of polymerase chain reaction product.
Fig. 3.
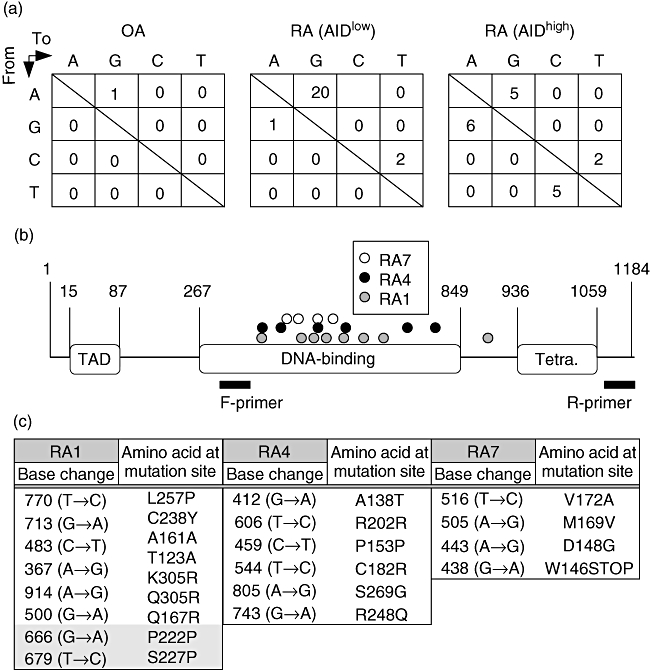
TP53 mutations in fibroblast-like synoviocytes (FLS). (a) Gene mutation profiles of TP53 in FLS lines. Base substitution patterns seen in TP53 extracted from the same data sets as those used for the mutation frequency analysis in Table 1 are shown. The numbers in the box are the sum of three FLS lines in each group. (b) Distribution of gene mutations in TP53 from FLS lines. Each number in the figure corresponds to the position of nucleotide on the coding sequence of TP53. Open circles are from RA7, filled circles are from RA4 and grey circles are from RA1. The location of polymerase chain reaction (PCR) primers used for sequencing is also depicted. TAD: transactivation domain; Tetra. tetramerization domain. (c) Base and amino acids substitution in TP53 from three of AIDhigh RA-FLS lines. Amino acids are described in a single letter. The two shaded mutations in RA1 were double mutations found in one clone.
While the sequencing range did not cover the whole coding sequence of TP53, the mutations in the TP53 gene were obviously concentrated at the DNA-binding domain, which is the hotspot of somatic mutations found in some malignant tumours (Fig. 3b). In particular, the Arg248 mutation found in RA4 has been reported as one of the cancer hotspot mutations [39]. The details of both base and amino acid substitutions are listed in Fig. 3c. These data indicate that AIDhigh RA-FLS have higher frequencies of TP53 mutations, some of which could result in the loss of function of TP53.
AID is expressed by non-transformed RA-FLS and in the RA-synovium outside of the B cell follicles
To address the issue of whether the aberrant expression of AID is affected by the transformation with SV40 large T antigen, first we examined the endogenous AID expression in non-transformed primary cell lines of FLS. AID transcription was detectable in four of 11 primary RA-FLS at three to eight times higher levels compared to transformed OA-FLS, but in none of six primary OA-FLS (Fig. 4a, and data not shown), indicating that increased expression of AID in RA-FLS is independent of transformation with SV40 large T antigen.
Fig. 4.
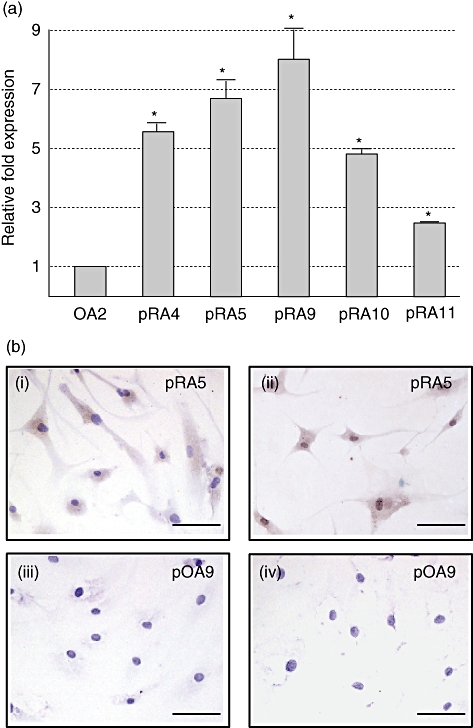
Endogenous expression of activation-induced cytidine deaminase (AID) in non-transformed primary rheumatoid arthritis-fibroblast-like synoviocytes (RA-FLS). The identical number of primary FLS corresponding to that of transformed FLS indicates that both are derived from the same patient, except for pRA10 and pRA11, which have no corresponding transformants. (a) The expression level of AID was estimated and compared between transformed osteoarthritis (OA2) as control and non-transformed primary RA-FLS by real-time polymerase chain reaction (PCR). Relative fold expression was determined by the AID expression in OA2 being defined as 1. The grey bars and error bars indicate the averages and standard deviations, respectively, of two independent experiments. Asterisks indicate the statistically significant difference by Student's t-test. P-value < 0·01. Uncapitalized p at the head of RA means ‘primary’. (b) Primary FLS grown on the glass slide were stained with rabbit anti-AID polyclonal antibody used in Western blot analysis (i and iii) or rat anti-AID monoclonal antibody (mAb) (ii and iv) combined with horseradish peroxidase-conjugated second antibodies. Diaminobenzidine was used as chromogenic substrate. Primary FLS cells from the same source of RA5 (i and ii, pRA5) and of OA9 (iii and iv, pOA9) are shown. Nuclear staining was performed with haematoxylin (violet). Scale bar indicates 100 µm.
We then examined the production of AID protein in non-transformed RA-FLS. Cytostaining with two different anti-AID antibodies clearly exhibited the production of AID protein in AID+ primary RA-FLS (Fig. 4b, i and ii corresponding to pRA5) but not in AID− primary OA-FLS (Fig. 4b, iii and iv, corresponding to pOA9). These data demonstrated clearly that the ectopic production of AID in RA-FLS is not a secondary effect by SV40 transformation.
Next, to examine whether AID production by RA-FLS is not the artefact in vitro, but the real event in the RA synovium in situ, we conducted immunohistochemical staining on the serial sections of the synovium from AID+ RA patients with anti-CD20, anti-AID and control antibodies. The localization of CD20+ B cells clearly isolated the follicles in the synovium (Fig. 5a, d), with a low frequency of AID-positive B cells. However, the majority of cells reacted by anti-AID antibody localized clearly in the lining and sublining outside the follicles (Fig. 5b, e). Isotype-matched irrelevant antibody towards mouse CD4 showed no signal on the serial specimen (Fig. 5c, f). Larger magnification revealed that AID signals were raised mainly from CD20− non-B cells that are morphologically compatible with FLS (Fig. 5g, h, black arrows), and some signals are originated from CD20+ B cells (Fig. 5g, h, red arrows). To ensure this finding, double immunofluorescence staining with anti-AID and -CD20 mAbs was performed. The reactivity of anti-AID mAb (green) was obvious only in the AID-positive synovial tissue (Fig. 6a, c) but not in the AID-negative tissue (Fig. 6b, d). In higher magnification (Fig. 6e), the distinct area consisting of the cells reactive only to anti-AID mAb (green) spread well outside the area containing CD20+ cells (red). The cells expressing both molecules are yellow and marked with an arrowhead in the figure.
Fig. 5.
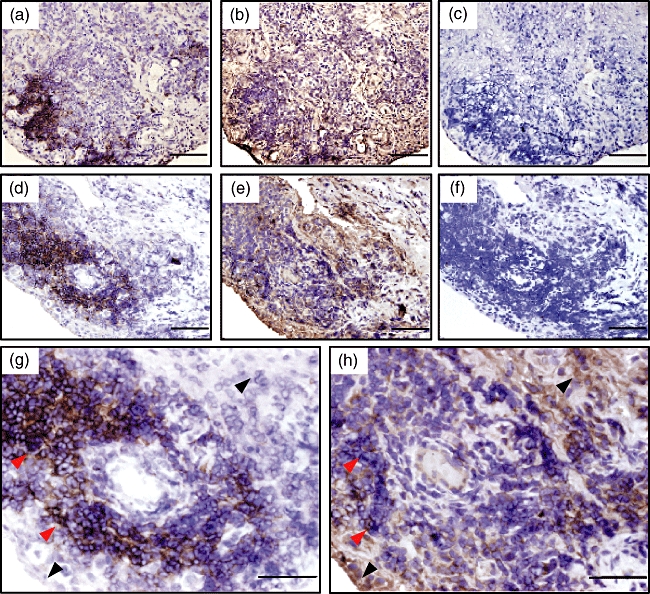
Immunohistochemical studies of activation-induced cytidine deaminase (AID) expression on synovial tissue section from the representative rheumatoid arthritis (RA) patient. The (a) (d) and (g) sections were stained with anti-CD20 monoclonal antibody (mAb) to identify the location of the B cell-containing follicle in synovial tissue. The (b) (e) and (h) sections were with the rat mAb for AID, and (c) and (f) were with isotype-matched control antibody. The serial sections of (a) (b) and (c) were from RA4, and of (d) (e) and (f) were from RA5. The (g) and (h) sections were high-power fields corresponding to (d) and (e), respectively. Red and black arrows indicate B cells and non-B cells, respectively. Nuclear staining was performed with haematoxylin (violet). Scale bar indicates 100 µm.
Fig. 6.
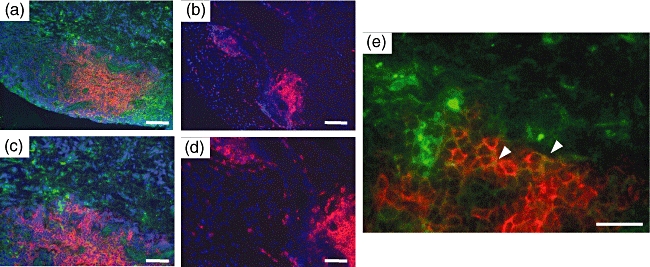
Immunofluorescence staining of activation-induced cytidine deaminase (AID) on synovial tissue sections from the representative rheumatoid arthritis (RA) patient. All sections were stained with rat monoclonal antibody (mAb) for AID and anti-CD20 mAb simultaneously. AID was visualized with Alexa 488 fluoro-dye-conjugated anti-rat secondary antibody (green) and CD20 was visualized with Alexa 594 fluoro-dye-conjugated anti-mouse secondary antibody (red) and observed under fluoresence microscope. The sections of (a) were from an AID-positive RA sample corresponding to RA4, and of (b) was from another AID-negative RA sample from which no transformed cell line had been established. The sections of (c) and (e) were high-power fields corresponding to (a), and (d) were high-power fields corresponding to (b). White arrowheads indicate the cells expressing both AID and CD20. Nuclear staining was performed with 4,6-diamidino-2-phenylindole (violet). Scale bar indicates 100 µm in (a) and (b), 50 µm in (c) (d) and (e).
These results demonstrated clearly that the FLS in the RA synovial tissues produce AID, providing strong evidence that ectopic and aberrant expression of AID occurs in RA.
Discussion
We have demonstrated that AID is expressed preferentially in transformed FLS cell lines from RA patients and that AID expression was correlated with accumulation of mutations in p53 gene. TNF-α enhanced the transcription of AID in both RA- and OA-FLS, whereas physiological concentration of β-oestradiol enhanced it only in RA-FLS. The selective and enhanced expression of AID was also confirmed in primary, non-transformed RA-FLS. Furthermore, localization of AID production outside the B cell follicles was demonstrated in the RA synovium tissue, providing definitive evidence that AID is expressed aberrantly and ectopically in certain cases of RA synovium. Thus, AID is a novel and intriguing candidate for the cause of the p53 mutation that has been reported to contribute the tumour-like phenotypes of RA-FLS.
To see the effects of TP53 mutations on SV40-transformed RA-FLS, we examined the cell number after 4 days' culture and the transcription levels of IL-6 and MMP-1 by real-time PCR analysis, both of which did not demonstrate any correlation with expression of AID. Despite the concomitant detection of TP53 mutations with AID transcription in a fraction of RA-FLS, the cell lines did not exhibit any tumour-like growth advantages or proinflammatory characteristics. However, this is plausible, because our sequence analyses could detect the generation of TP53 mutations in the very low-frequent clones that are not sufficiently predominant to affect the characteristics of the AIDhigh RA-FLS line as a whole. Although not definitive, there was an informative and supportive RA case from which we obtained the two FLS lines from different joint lesions at distinct times of operation with a 6-month interval. Interestingly, AID expression was detectable only in the cell line established after the second operation, suggesting the possibility that ectopic expression of AID was acquired during the course of RA (unpublished observation). These intriguing possibilities should be tested using a larger sample size of clinical cases and/or quantitative histological analyses.
Xu et al. reported that B cells in the follicle of synovium or in the peripheral blood from RA patients show a higher expression level of AID in comparison with OA patients [40]. Our analyses in this paper demonstrated selective expression of AID in both transformed and non-transformed FLS from RA. Furthermore, immunohistochemical analysis of our RA cases revealed that major AID-producing cells with FLS-like morphology localized in the area outside B cell follicles; B cell follicles without production of AID were also described by them. When these results by us and others are integrated, we hypothesize that both B cells and non-B cells in RA patients are liable to express AID as an intrinsic or secondary abnormality under inflammatory milieu, which is responsible for the various aspects in the pathophysiology of RA. Higher expression of AID in B cells causes the SHM at variable regions in Ig gene and involvement in the generation of autoreactive B cells, which produce autoantibodies such as RF and anti-CCP antibody. Conversely, ectopic expression of AID in non-B cells, such as FLS per se, introduces point mutations in some genes, including tumour suppressor genes, and alters the phenotype of the cells including FLS.
It may be imagined that AID could be a potential therapeutic target for RA; however, it would be difficult to know when AID expression has begun in FLS in the synovium of each RA case. Although we did not study in detail the triggers for the ectopic expression of AID in FLS of RA, it is reasonable to speculate that anti-inflammatory cytokine therapy starting at earlier stages of RA suppresses the induction of AID expression in FLS and might prevent the malignant transformation of FLS by AID-dependent random mutagenesis.
Acknowledgments
We thank Ms Reina Tanaka for her technical assistance. We also thank Dr Kazuhiko Kuwahara at Kumamoto University for providing us with the SW480 colorectal cancer cell line, which was used for the positive control on APOBEC1 amplification. This work was supported by Research Project Grants (20-418I to H.I. and 19-406M to K.I.) from Kawasaki Medical School and Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, Culture and Technology of Japan (19590390 to K.I.).
Disclosure
None.
References
- 1.Lipsky PE. Rheumatoid arthritis. In: Fauci AS, Braunwald E, Kasper DL, et al., editors. Harrison's principles of internal medicine. 17th edn. New York: McGraw-Hill Professional; 2008. pp. 2083–91. [Google Scholar]
- 2.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–61. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 3.Mor A, Abramson SB, Pillinger MH. The fibroblast-like synovial cell in rheumatoid arthritis: a key player in inflammation and joint destruction. Clin Immunol. 2005;115:118–28. doi: 10.1016/j.clim.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Kirkham BW, Lassere MN, Edmonds JP, et al. Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the damage study cohort) Arthritis Rheum. 2006;54:1122–31. doi: 10.1002/art.21749. [DOI] [PubMed] [Google Scholar]
- 5.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper t cells. N Engl J Med. 2009;361:888–98. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 6.Andersson AK, Li C, Brennan FM. Recent developments in the immunobiology of rheumatoid arthritis. Arthritis Res Ther. 2008;10:204. doi: 10.1186/ar2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 8.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–42. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 9.Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. 2006;2:619–26. doi: 10.1038/ncprheum0338. [DOI] [PubMed] [Google Scholar]
- 10.Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–45. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Firestein GS. Biomedicine. Every joint has a silver lining. Science. 2007;315:952–3. doi: 10.1126/science.1139574. [DOI] [PubMed] [Google Scholar]
- 12.Pierer M, Muller-Ladner U, Pap T, Neidhart M, Gay RE, Gay S. The SCID mouse model: novel therapeutic targets – lessons from gene transfer. Springer Semin Immunopathol. 2003;25:65–78. doi: 10.1007/s00281-003-0126-2. [DOI] [PubMed] [Google Scholar]
- 13.Roivainen A, Pirila L, Yli-Jama T, Laaksonen H, Toivanen P. Expression of the myc-family proto-oncogenes and related genes max and mad in synovial tissue. Scand J Rheumatol. 1999;28:314–18. doi: 10.1080/03009749950155517. [DOI] [PubMed] [Google Scholar]
- 14.Aikawa Y, Morimoto K, Yamamoto T, et al. Treatment of arthritis with a selective inhibitor of c-fos/activator protein-1. Nat Biotechnol. 2008;26:817–23. doi: 10.1038/nbt1412. [DOI] [PubMed] [Google Scholar]
- 15.Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 1997;94:10895–900. doi: 10.1073/pnas.94.20.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reme T, Travaglio A, Gueydon E, Adla L, Jorgensen C, Sany J. Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clin Exp Immunol. 1998;111:353–8. doi: 10.1046/j.1365-2249.1998.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kullmann F, Judex M, Neudecker I, et al. Analysis of the p53 tumor suppressor gene in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 1999;42:1594–600. doi: 10.1002/1529-0131(199908)42:8<1594::AID-ANR5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Inazuka M, Tahira T, Horiuchi T, et al. Analysis of p53 tumour suppressor gene somatic mutations in rheumatoid arthritis synovium. Rheumatology (Oxf) 2000;39:262–6. doi: 10.1093/rheumatology/39.3.262. [DOI] [PubMed] [Google Scholar]
- 19.Yamanishi Y, Boyle DL, Green DR, et al. P53 tumor suppressor gene mutations in fibroblast-like synoviocytes from erosion synovium and non-erosion synovium in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R12–R18. doi: 10.1186/ar1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pap T, Franz JK, Hummel KM, Jeisy E, Gay R, Gay S. Activation of synovial fibroblasts in rheumatoid arthritis: lack of expression of the tumour suppressor pten at sites of invasive growth and destruction. Arthritis Res. 2000;2:59–64. doi: 10.1186/ar69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han Z, Boyle DL, Shi Y, Green DR, Firestein GS. Dominant-negative p53 mutations in rheumatoid arthritis. Arthritis Rheum. 1999;42:1088–92. doi: 10.1002/1529-0131(199906)42:6<1088::AID-ANR4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 22.Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 2002;99:10025–30. doi: 10.1073/pnas.152333199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Y, Zeng XR, Wenger L, Firestein GS, Cheung HS. P53 down-regulates matrix metalloproteinase-1 by targeting thecommunications between ap-1 and the basal transcription complex. J Cell Biochem. 2004;92:258–69. doi: 10.1002/jcb.20044. [DOI] [PubMed] [Google Scholar]
- 24.Goila-Gaur R, Strebel K. Hiv-1 vif, apobec, and intrinsic immunity. Retrovirology. 2008;5:51. doi: 10.1186/1742-4690-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honjo T, Muramatsu M, Fagarasan S. Aid: how does it aid antibody diversity? Immunity. 2004;20:659–68. doi: 10.1016/j.immuni.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 27.Babbage G, Ottensmeier CH, Blaydes J, Stevenson FK, Sahota SS. Immunoglobulin heavy chain locus events and expression of activation-induced cytidine deaminase in epithelial breast cancer cell lines. Cancer Res. 2006;66:3996–4000. doi: 10.1158/0008-5472.CAN-05-3704. [DOI] [PubMed] [Google Scholar]
- 28.Endo Y, Marusawa H, Kinoshita K, et al. Expression of activation-induced cytidine deaminase in human hepatocytes via nf-kappab signaling. Oncogene. 2007;26:5587–95. doi: 10.1038/sj.onc.1210344. [DOI] [PubMed] [Google Scholar]
- 29.Kou T, Marusawa H, Kinoshita K, et al. Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int J Cancer. 2007;120:469–76. doi: 10.1002/ijc.22292. [DOI] [PubMed] [Google Scholar]
- 30.Endo Y, Marusawa H, Kou T, et al. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008;135:889–98. doi: 10.1053/j.gastro.2008.06.091. 98 e1-3. [DOI] [PubMed] [Google Scholar]
- 31.Komori J, Marusawa H, Machimoto T, et al. Activation-induced cytidine deaminase links bile duct inflammation to human cholangiocarcinoma. Hepatology. 2008;47:888–96. doi: 10.1002/hep.22125. [DOI] [PubMed] [Google Scholar]
- 32.Morisawa T, Marusawa H, Ueda Y, et al. Organ-specific profiles of genetic changes in cancers caused by activation-induced cytidine deaminase expression. Int J Cancer. 2008;123:2735–40. doi: 10.1002/ijc.23853. [DOI] [PubMed] [Google Scholar]
- 33.Chan-On W, Kuwahara K, Kobayashi N, et al. Cholangiocarcinomas associated with long-term inflammation express the activation-induced cytidine deaminase and germinal center-associated nuclear protein involved in immunoglobulin v-region diversification. Int J Oncol. 2009;35:287–95. [PubMed] [Google Scholar]
- 34.Szczepek AJ, Bergsagel PL, Axelsson L, Brown CB, Belch AR, Pilarski LM. CD34+ cells in the blood of patients with multiple myeloma express CD19 and igh mrna and have patient-specific igh vdj gene rearrangements. Blood. 1997;89:1824–33. [PubMed] [Google Scholar]
- 35.Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates aid transcription and function. J Exp Med. 2009;206:99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dorsett Y, McBride KM, Jankovic M, et al. Microrna-155 suppresses activation-induced cytidine deaminase-mediated myc-igh translocation. Immunity. 2008;28:630–8. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teng G, Hakimpour P, Landgraf P, et al. Microrna-155 is a negative regulator of activation-induced cytidine deaminase. Immunity. 2008;28:621–9. doi: 10.1016/j.immuni.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen HM, Poirier MG, Allen MJ, et al. The activation-induced cytidine deaminase (aid) efficiently targets DNA in nucleosomes but only during transcription. J Exp Med. 2009;206:1057–71. doi: 10.1084/jem.20082678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ko LJ, Prives C. P53: puzzle and paradigm. Genes Dev. 1996;10:1054–72. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 40.Xu X, Hsu HC, Chen J, et al. Increased expression of activation-induced cytidine deaminase is associated with anti-ccp and rheumatoid factor in rheumatoid arthritis. Scand J Immunol. 2009;70:309–16. doi: 10.1111/j.1365-3083.2009.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]