

Abstract
Wildtype p53-induced phosphatase 1 (Wip1) was identified as an oncogene amplified and overexpressed in a number of human cancers. Recent evidence suggested that Wip1 is a critical inhibitor in the ATM/ATR -p53 DNA damage signaling pathway. Wip1 dephosphorylates several key DNA damage-responsive proteins and reverses DNA damage-induced cell cycle checkpoints. Previous reports showed that Wip1 was transcriptionally induced by p53 at the early stage of the DNA damage response. To investigate the temporal and functional regulation of Wip1, we identified a microRNA, miR-16 that specifically targets the messenger RNA (mRNA) of Wip1 and thus negatively regulates the expression level of Wip1. Mir-16 itself is induced immediately after DNA damage. Therefore, the increase in Wip1 protein level is significantly postponed compared to that of its mRNA level, preventing a premature inactivation of ATM/ATR signaling and allowing a functional completion of the early DNA damage response. To better understand miR-16 biological functions in the context of cancer cells, we examined its expression in mammary tumor stem cells and found it to be markedly downregulated in mammary tumor stem cells. Overexpression of miR-16 or inhibition of Wip1 suppresses the self-renewal and growth of mouse mammary tumor stem cells, and sensitizes MCF-7 human breast cancer cells to the chemotherapeutic drug, doxorubicin. Together, our results suggest an important role of miR-16 in the regulation of Wip1 phosphatase in the DNA damage response and mammary tumorigenesis.
Keywords: miR-16, Wip1, Inhibition, Sensitization, Mammosphere
Introduction
Eukaryotic cells have evolved a multifaceted response to counteract the potentially deleterious effects of DNA damage. Upon sensing DNA damage or stalled replication forks, DNA damage checkpoints are activated to arrest cell cycle progression and repair damaged DNA. Once DNA repair is completed, DNA damage signaling pathways need to be inactivated when cells return to normal. The wildtype p53-induced phosphatase 1, Wip1 (or PPM1D) is a member of the type 2C serine/threonine phosphatases. Recent evidence indicates that Wip1 is a critical regulator of DNA damage signaling pathways(1;2).
A number of Wip1 dephosphorylation targets have been identified in the ATM/ATR-p53 DNA damage signaling pathway, including ATM, Chk1, Chk2, p38 MAP kinase, p53, Mdm2, and MdmX(3–8). Through dephosphorylation of those key kinases that initiate cell cycle checkpoints, Wip1 releases cells from cell cycle arrest. Furthermore, Wip1 negatively regulates p53 levels and activities by stabilizing Mdm2 and MdmX, and thus shuts down p53-mediated apoptosis and cell cycle checkpoints. Wip1-null mouse embryonic fibroblasts (MEFs) displayed reduced proliferation, enhanced p53 transcriptional activity, and enhanced DNA damage-induced checkpoints(9). The inhibitory roles of Wip1 on the DNA damage signaling and the tumor suppressor p53 suggested that the WIP1 gene is an oncogene. Consistent with an oncogenic function, the WIP1 gene is present in amplified copy numbers and is overexpressed in many human cancer types, including breast carcinomas, ovarian clear cell adenocarcinomas, neuroblastomas, pancreatic adenocarcinomas, gastric carcinomas, and medulloblastomas(10–16).
MicroRNAs (miRNAs) are small (~22 nucleotides) noncoding regulatory RNA molecules that are involved in diverse biological processes and various diseases. By virtue of sequence complementarity, miRNAs bind to the messenger RNAs of their target genes and then block translation or accelerate their degradation(17). Emerging evidence has shown that miRNA biogenesis is regulated upon DNA damage stresses. Pothof and colleagues reported that UV damage triggered a cell-cycle-dependent relocalization of Ago2 into stress granules and a change of microRNAs expression profiling(18). Recent work from the Miyazono group showed that the tumor suppressor p53 promoted the post-transcriptional processing of a subgroup of miRNAs. The interaction between p53 and the Drosha complex facilitates the processing of primary miRNAs to precursor miRNAs(19). MiRNAs also influence DNA damage response by regulating the expression levels of their target genes. Many genes involved in the DNA damage response can be targeted by their specific miRNAs. For instance, human miR-421 was shown to target Ataxia-telangiectasia mutated (ATM) transcripts and downregulate their protein expression. As a result, overexpression of miR-421 sensitized cells to ionizing radiation(20). Human miR-15a and miR-16 cluster targets Cyclin D1 (CCND1), BCL2 and WNT3A, which enhances G1/S cell cycle checkpoint and inhibits tumorigenic features such as survival, proliferation and invasion(21).
In the present study, we show that the transcripts of the WIP1 gene are specifically targeted by miR-16. Overexpression of miR-16 abolishes the DNA damage-responsive Wip1 induction while inhibition of miR-16 markedly accelerates and enhances the Wip1 induction. Deletion of the miR-16-targeted sequence in the 3’-UTR of WIP1 depleted miR-16 effects on Wip1. Previous studies reported that the 5' untranslated region (UTR) of the WIP1 gene includes a conserved p53 response element, facilitating a p53-dependent induction of the WIP1 transcripts. However, the induction of Wip1 proteins appears to have a delayed onset in contrast to an immediate induction of the WIP1 transcripts in response to DNA damage. We presented that the level of miR-16 is rapidly induced upon DNA damage stress, which postpones the accumulation of the Wip1 protein and thus allows cells to initiate functional cell cycle checkpoints in the early stage of DNA damage response. Interestingly, miR-16 is downregulated in mammospheres originated from mammary tumor stem cells. Overexpression of miR-16 in mammary tumor cells sensitizes them to doxorubicin treatment, and significantly reduces the proliferation of mammary tumor stem/progenitor cells, implicating miR-16 in the regulation of the self-renewal of mammary tumor stem cells.
Materials and Methods
Cell lines and cell culture
U2OS (human osteosarcoma line) and MCF-7 (human breast cancer line) cell lines were obtained from the American Type Culture Collection (ATCC) in 2007 and maintained in DMEM supplemented with 10% (V/V) fetal bovine serum (FBS). Cells were cultured and stored according to the supplier’s instructions and used at passage 5 to 20. Once resuscitated, cell lines obtained from ATCC are routinely authenticated (once every 6 months, cells were last tested in October, 2009) through cell morphology monitoring, growth curve analysis, species verification by isoenzymology and karyotyping, identity verification using short tandem repeat profiling analysis, and contamination checks. Expression of p53 and Wip1 in these two cell lines was confirmed by immunoblotting before they were used in the experiments. Primary Wip1+/+ and Wip1−/− mouse embryonic fibroblasts were harvested and cultured as previously described(9).
Plasmid Constructs, Cell transfection and infection
Chemically synthesized pre-miRNAs, control miRNAs and antagomirs were purchased from Ambion. WIP1 shRNA expression vector (#RHS3979-9571552) was purchased from Openbiosystems. Using PCR primers 5’ ACTCTAGA-AATGCATCTGGGAAATGAGG 3’ and 5’ TGTCTAGA-GCAGGCATGATGCTCAAAG 3’, the wildtype 3’ UTR of WIP1 (1.1 Kbp) was amplified from human cDNA library, and cloned into the XbaI site of pRL vector (Promega). Mutant WIP1 3’ UTR was generated based on the pRL-WIP1-3’UTR by deleting 6 nt that is recognized by miR-16. Lentiviral miR-16 (#MmiR3359-MR01) and miR-21 (#MmiR3355-MR03) expression constructs were obtained from Genecopoeia. Lentiviral packaging vectors (#FIV-PK-20, Genecopoeia) and miRNA expression vector were transfected into 293T cells by Fugene HD transfection reagent (#11633400, Roche) following the manufacturer’s manual. Two days after transfection, cell culture supernatants were collected, filtered and titrated using target cells. Control or pseudovirus particles expressing miRNAs were used to infect mouse or human cells at the multiplicity of infection (MOI) of 3 to ensure the complete infection of target cells.
Isolation of mouse mammary tumor cells and mammosphere culture
MMTV-ErbB2 transgenic mice (FVB/N-Tg(MMTVneu)202Mul/J) were purchased from Jackson Laboratory. Normal mammary tissues or mammary tumors from ErbB2-transgenic mice were mechanically dissociated and placed in a digestion medium (DMEM with 1 mM glutamine, 5% fetal bovine serum) supplemented with 200 U/ml collagenase (Sigma) and 100 U/ml hyaluronidase (Sigma) for 5 h at 37 °C. Cell suspensions were centrifuged at 80 g, resuspended in 0.2% NaCl to lyse red blood cells, and filtered through 20 μm mesh. Mammary tumor cells were plated onto ultralow attachment plates (Corning) at a density of 20,000 viable cell/ml (to obtain primary mammospheres) in a serum-free DMEM-F12 (Invitrogen), supplemented with 5 μg/ml insulin, 20 ng/ml EGF and 20 ng/ml bFGF (Sigma), and 0.4% bovine serum albumin (Sigma)(22). Mammospheres were harvested after 10 days for the analysis of gene and microRNAs expression. For the differentiation culture of mammospheres, collected mammospheres were treated with Trypsin-EDTA (Invitrogen) and mechanically dissociated by pippetting, and then cultured in DMEM-F12 supplemented with 5% fetal bovine serum (Atlanta Biologicals) without growth factors. Differentiated cells were harvested for microRNA analysis after 3 days.
Western Blot Analysis, Antibodies, and Purified Proteins
Immunoprecipitations, Western blot analysis, and immunoprecipitation-Western blot analyses were performed by standard methods described previously(4). Anti-actin (#1616), anti-p53 (#126), HRP-anti-goat IgG (#2020), HRP-anti-rabbit IgG (#2302), and HRP-anti-mouse IgG (#2302) were purchased from Santa Cruz; Anti-Wip1 (#AP-8437b) was purchased from Abgent; Anti-Bcl-2 (#2876) and anti-Caspase-3 (#9664) were purchased from Cell Signaling Technology.
Treatments with DNA damaging agents or Wip1 inhibitor
Cells were treated with 500 ng/ml neocarzinostatin (NCS, #N9162, Sigma-Aldrich) and harvested at the indicated time points after treatment for the analyses of mRNA and proteins. In cell viability assays, MCF-7 cells were seeded at the concentration of 1000 cells per well in flat-bottom 96-well microplates. After 24 h, the cells were cultivated with doxorubicin at the indicated concentration for 72h. At the end of incubation, the viability of cells was determined using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega), according to the manufacturer's instructions. In mammosphere culture, mouse mammary tumor cells were treated with the specific Wip1 inhibitor, CCT007093 (5 μM, #C9369, Sigma-Aldrich) or DMSO (mock treatment) as indicated.
Analysis of miRNAs by Northern blotting
Total RNA was extracted by Trizol ragents (Invitrogen) following the manufacturer’s instructions. An equalvolume of 4 M LiCl and 10 mM EDTA, pH 7.0, was added to theRNA solution and incubated overnight at 4°C. High molecularweight RNA was recovered by centrifugation at 12,000g for 15min. The supernatant, containing DNA and low molecular weightRNAs, was made 10% in polyethylene glycol 8000 and 0.5 M NaCland incubated for 30 min on ice to precipitate the DNA. Aftercentrifugation to remove DNA, low molecular weight RNA was concentratedby ethanol precipitation.RNA gel blot analysis of low molecular weight RNA wereperformed exactly as described using Ambion ULTRAhyb hybridization solution at 45°C. DNA probes used were: U6 RNA probe 5’ ATATGGAACGCTTCACGAATT 3’ and miR-16 probe 5’ CGCCAATATTTACGTGCTGCTA 3’.
Primers for quantitative RT-PCR
Primers for genes examined in the studies were: human WIP1: 5’ TTATACCTGAACCTGACT -GAC 3’ and 5’ GATCTTTTGAGGGTATGACTA 3’; mouse Wip1: 5’ TTGCCTCACACACC -ATTTGT 3’ and 5’ GGGTGTTAGCAGCACCATTT 3’; Oct4: 5’ GTGGAGGAAGCCGACA -ACAATG 3’ and 5’ GCCTCATACTCTTCTCG3'; Klf-4: 5’ AGAGGAGCCCAAGCCAAAGA -GG 3’ and 5’ CCACAGCCGTCCCAGTCACAGT3’; Keratin-14: 5’ GCTGAGGAATGGTTC -TTCAG 3’ and 5’ CAGGTTATTCTCCAGGGATG3’; Keratin-18: 5’ AGATCGACAATGCC -CGCCTT 3’ and 5’ CTTGCTGAGGTCCTGAGATT 3’. Primers for miRNA quantitative RT-PCR were obtained from Exiqon: U6 RNA control (#201510), miR-16 (#202004) and miR-21 (#204230).
Results
MiR-16 Inhibits Wip1 Expression by Targeting 3’UTR of Wip1
Previous studies showed that the WIP1 gene is transcriptionally induced by the tumor suppressor p53, but it remains unclear if post-transcriptional regulation affects the expression of Wip1 protein. To explore the possibility that microRNAs might regulate WIP1 expression, we searched the 3′UTR of the human WIP1 gene for microRNA-targeting motifs using the TargetScan 5.1, from which three microRNAs were predicted to bind to the 3’ UTR of WIP1, including miR-16, miR-217, and miR-153. Twelve nucleotides from miR-16 are complementary to the target sequence in the 3′UTR of WIP1 (Fig. 1A). To test the in silico prediction, we first examined the endogenous Wip1 protein level by immunoblotting after transient transfection of a control and the precursors of the three predicted miRNAs (pre-miRNAs) into human U2OS cells. As shown in Figure 1B, the WIP1 protein level was significantly decreased by ectopic miR-16 expression while the other two predicted miRNAs, miR-217 and miR-153 had only minor effects. MiRNAs that were randomly picked as negative controls, miR-145 and miR-203, had minimal or no effect on the Wip1 level. We next examined the levels of Wip1 in cells transfected with pre-miR-16 or specific inhibitory oligonucleotide against miR-16, antigomir-16 during 0 – 4 h after the treatment of a radiomimetic drug, neocarzinostatin (NCS). Ectopic miR-16 had a pronounced suppression against the induction of Wip1 proteins, whereas antagomir-16 promoted the expression of WIP1 (Fig. 1C).
Figure 1.

miR-16 suppresses Wip1 expression by targeting WIP1 3’UTR. (A) Predicted miRNA sequences and their putative recognition sites within 3’ UTR of WIP1. (B) Wip1 expression is suppressed by ectopic miR-16 transfection. U2OS cells were transfected with the indicated miRNAs. 72 h after transfection, Wip1 protein levels in each sample were detected by immunoblotting (upper panel), quantitated according to the intensity of Wip1 bands, and normalized with the control sample (bottom panel). (C) Immunoblots of endogenous Wip1 in U2OS cells transfected with control miRNA, miR-16 or antagomir-16, and treated with 500 ng/ml NCS at 72 h after transfection. (D) MiR-16 specifically targets the 3’ UTR of WIP1. A luciferase construct containing wildtype WIP1 3’UTR (left panel) or mutant WIP1 3’ UTR (right panel) was transfected into U2OS cells with the indicated miRNAs or antagomirs. Renilla luciferase activities were measured 48 h after transfection and normalized to firefly luciferase. Relative luciferase activity (luminescence) was obtained after normalizing with the control number.
To further validate Wip1 as a bona fide target of miR-16, we cloned the WIP1 3′UTR portion containing the miR-16 target site into a Renilla luciferase reporter construct and performed a luciferase reporter assay following cotransfection of reporter constructs with precursor miR-16 into U2OS cells. A significant reduction (~75%) in the luciferase activity was observed for the reporter construct containing the WIP1 3′UTR in the presence of pre-miR-16, but not in the presence of control or the other four pre-miRNAs (Fig.1D, left panel). Transfection of antagomir-16 increased the luciferase activity by ~50% for the WIP1 3’UTR reporter construct. Deletion of six nucleotides of seed sequence almost completely abolished the effects of pre-miR-16 and antagomir-16 on the luciferase transcripts with the WIP1 3’UTR, suggesting that the WIP1 gene is an authentic target of miR-16 in vivo (Fig. 1D, right panel).
MiR-16 Regulates Wip1 Induction in the DNA Damage Response
Accumulating evidence supports a role of Wip1 as a critical negative regulator of DNA damage signaling pathways. Wip1 itself is also responsive to DNA damage. The Appella group reported that Wip1 levels were transcriptionally induced in a p53-dependent manner upon DNA damage(23;24). To explore the temporal regulation of Wip1 during DNA damage response, we measured the Wip1 mRNA and protein levels in U2OS cells at 0 – 8 h after NCS treatment. Consistent with the results from the Appella group, we observed that the Wip1 mRNA level was rapidly increased in response to NCS, reaching the peak level within 2 hours. However, the Wip1 protein level was induced at a slower pace, which reached a peak at 6 h post-treatment (Fig. 2A & B). We further explored whether there was post-transcriptional inhibition of WIP1 expression. By Northern blotting analysis, the level of mature miR-16 increased shortly after NCS treatment and hit its peak around 2–4 h post-treatment when the Wip1 protein levels still remained at a relatively low level (Fig. 2C). To exclude the possibility that the effects of miR-16 are limited to cellular response to NCS, we performed the same experiments in U2OS cells treated with 10 Gy of ionizing radiation. The similar phenomena were observed on the induction of miR-16, Wip1 mRNA and protein at the early stage of DNA damage response (Supplemental Fig. 1A and 1B). To further determine if altered levels of miR-16 change the temporal pattern of Wip1 induction, we examined Wip1 protein levels in the presence of ectopic pre-miR-16 or antagomir-16 over a 12-hour time course. Transfection of pre-miR-16 significantly suppressed the induction of Wip1 throughout the whole time course. Inhibiting miR-16 by antagomir boosted Wip1 expression as early as 2h after NCS treatment, and maintained a stable high level of Wip1 up to 6 hours (Fig. 2D). These results suggest that miR-16 is an important regulator for Wip1 induction in the early stage of DNA damage response.
Figure 2.

miR-16 inhibits the DNA damage-mediated induction of Wip1. (A) Wip1 protein is induced in response to DNA damage. U2OS cells were treated with 500 ng/ml NCS and cell lysates were harvested at indicated time points after NCS treatment. Protein levels were determined by immunoblotting. (B) Induction of Wip1 protein has a delayed onset compared to the induction of Wip1 mRNA. Levels of Wip1 mRNA and protein in the above NCS-treated cells were determined by quantitative RT-PCR and the intensity of Wip1 immunoblots. (C) miR-16 has a rapid induction in response to DNA damage. U2OS cells were treated with 500 ng/ml NCS and total RNAs were harvested at indicated time points. miR-16 levels were determined by Northern blots (left panel) and quantitative RT-PCR (from 3 sets of samples, right panel). (D) miR-16 counteracts the induction of Wip1 in the DNA damage response. U2OS cells were transfected with control miRNA, miR-16 or antagomir-16. 48 h after transfection, cells were treated with 500 ng/ml NCS and cell lysates were harvested for immunoblotting analysis (left panel). The intensity of Wip1 blots in each sample was quantified by phosphorimager and normalized with the control sample.
MiR-16 Regulates Cell Proliferation and Sensitizes MCF-7 Cells to Doxorubicin
To assess whether miR-16 modulates cell proliferation, we examined the cell growth of the littermate Wip1+/+ and Wip1−/− mouse embryonic fibroblasts isolated from midgestation. Consistent with a previous report(9), Wip1−/− MEFs exhibited a much slower growth in contrast to their Wip1+/+ counterparts.Growth curve analyses of Wip1+/+ and Wip1−/− MEFs showed that the proliferation rates of early-passage Wip1−/− MEFs weresubstantially retarded compared to Wip1+/+ MEFs (Fig. 3A). Fourdays after plating, Wip1−/− MEFs almost stopped proliferation while Wip1+/+ MEFs were still in the doubling stage. Lentivirus particles expressing the precursor of mmu-miR-16 (mouse miR-16) were transduced into both Wip1+/+ and Wip1−/−MEFs and the growth curves of these fibroblasts were analyzed. Ectopic overexpression of miR-16 dramatically inhibited the cell proliferation of Wip1+/+ MEFs to a level similar to that of Wip1−/− MEFs. Moreover, the absence of Wip1 largely diminished the effects of miR-16 on cell proliferation. No significant effects were observed on the growth of Wip1−/− MEFs in the presence or absence of the lentiviral miR-16, suggesting that Wip1 may be one of the major miR-16 targets in the regulation of cell cycle checkpoints and cell proliferation.
Figure 3.

MiR-16 suppresses cell proliferation and sensitizes MCF-7 cells to doxorubicin. (A) Growth curves of Wip1+/+ and Wip1−/− MEFs infected with control or miR-16 expressing pseudoviruses. (B) MiR-16 inhibits the expression of Wip1 in MCF-7 cells. MCF-7 cells were transfected with control or miR-16 expression vector DNA. 36 h after transfection, cell lysates were harvested or total RNA was extracted for the analyses of miRNAs (by quantitative PCR, left panel) and proteins (by Western blotting, right panel). (C) MiR-16 sensitizes MCF-7 cells to doxorubicin. MFC-7 cells were transfected with control, Wip1 shRNA, miR-16 expression vector DNA, or miR-16 and Wip1 shRNA expression vector DNA. 48 h after transfection, cells were incubated with increasing doses of doxorubicin as indicated. Cell viability was determined 3 days after incubation.
WIP1 gene amplification occurs in many cancer types, including 11–18% of primary human breast cancer samples nearly all of which express wildtype p53(10;25–29). This observation has led to the hypotheses that aberrantly high levels of Wip1 are a causative factor in tumorigenesis. Inhibiting Wip1 by small interfering RNA or specific inhibitor CCT007093 reduced the cell viability selectively on cells with WIP1 amplification. Furthermore, loss of Wip1 sensitized cells to stress- or DNA damage- induced apoptosis. To determine if miR-16 increases sensitivity of cells to chemotherapeutic drugs that cause DNA damage, we examined the viability of human breast cancer MCF-7 cells in which the WIP1 gene is substantially amplified and overexpressed. We delivered miR-16 to MCF-7 cells by lentiviral infection. Lentival expression of miR-16 increased the level of total miR-16 by 7-fold in MCF cells, while the level of the control microRNAs, miR-21 was unchanged (Fig. 3B). MiR-16 overexpression dramatically promoted apoptosis, indicated by a higher level of cleaved caspase-3 in cells expressing lentiviral miR-16, as compared to the control cells. Reduced Wip1 levels led to higher levels of DNA damage-induced phosphorylation of ATM (Ser1981) and Chk2 (Thr68) that are two of the identified Wip1 dephosphorylation substrates(3;5). As expected, ectopic miR-16 knocked down the expression of Bcl-2, another known target of miR-16(30) (Fig. 3B). To examine whether miR-16 modulates the chemosensitivity of MCF-7 cells in a Wip1-dependent manner, we treated MCF-7 cells with increasing doses of doxorubicin. Cell survival was assessed by measuring the proliferation of viable cells. As shown in Fig. 3C, MCF-7 cells overexpressing miR-16 displayed a significantly higher sensitivity to doxorubicin compared with the control MCF-7 cells (IC50: 0.037μM vs 0.13μM). Knockdown of Wip1 by chemically synthesized small interfering RNA had no additional effect on the sensitivity of MCF-7 cells overexpressing miR-16 (IC50: 0.037 μM vs 0.032 μM) (Fig. 3C). Our results indicated that miR-16 sensitizes MCF-7 cells to the treatment of doxorubicin by inhibiting Wip1.
MiR-16 is Down-regulated in Mammary Tumor Stem/Progenitor Cells
The WIP1 gene is frequently amplified in human breast cancers that contain wildtype p53. Deletion of Wip1 in mice bearing mouse mammary tumor virus (MMTV) promoter-driven oncogenes Erbb2 or Hras1 impaired mammary carcinogenesis(26). However, no spontaneous mammary tumor formation was observed in MMTV-Wip1 transgenic mice(31). This suggested that overexpression of Wip1 phosphatase alone is not sufficient to cause breast cancer in mice. Nevertheless, Wip1 can complement in vitro other oncogenes, such as Ras and ErbB2, in their ability to transform mouse embryo fibroblasts(26). We hypothesized that up-regulation of Wip1 probably provides a selective advantage for the clonal expansion of cancer cells. It is now becoming clear that a subgroup of cells in mammary tumors possesses tumor-initiating cell function and contributes to tumor expansion and growth(32–35). The tumor-initiating cells, or cancer stem cells, can be isolated using cell surface markers or can be enriched in non-adherent mammospheres.
Dontu and colleagues developed a culture system in which cells derived from reduction mammoplasties were seeded in non-adherent non-differentiating culture conditions. Cells capable of surviving and proliferating in such conditions formed discrete clusters of cells termed mammospheres(34). Such spheroids were enriched in progenitor cells capable of differentiating along multiples lineages. To determine if Wip1 plays a role in the proliferation of mammary tumor stem cells, we isolated primary tumor cells from the mammary tumors in MMTV-Erbb2 transgenic mice. Amplification of the Wip1 gene was not observed in any of the three studied MMTV-Erbb2 mammary tumors. Levels of Wip1 protein and mRNA in two tumor samples were slightly increased over those in the corresponding normal tissues, while the level of miR-16 was 10–30% lower in tumor samples than in normal tissues (Fig. 4A). The whole population of primary tumor cells was cultured in regular cell culture medium or in mammosphere culture medium. As shown in Fig 4B, about 2.5 % of total cells formed mammospheres containing ~300 cells after 12 d of cultivation in non-adherent dishes. In contrast to the whole cell population, stem cell markers, Oct4 and KLF-4 genes were highly expressed exclusively in mammosphere cells, while differentiation markers, Keratin-14 and Keratin-18 expressions were suppressed, which are typical characteristics of tumor stem cells. Quantitated by real-time PCR, the levels of miR-16 were observed to have a marked reduction of 70%–80% in all mammospheres tested in our experiments, compared to that of the whole population of tumor cells. Accordingly, the Wip1 protein levels increased by 3–5 fold in mammospheres (Fig. 4C). We isolated three mammospheres under the microscope and then cultured them in the regular culture medium. The progenitor cells in the mammospheres re-differentiated and proliferated. Levels of miR-16 in these recovered cancer cells from each mammospheres were similar to the original cancer cells (Figure 4D). The results suggested that Wip1 may be differentially expressed in normal cancer cells and cancer stem cells, and implicated that Wip1 is likely involved in the maintenance and proliferation of mammary tumor stem/progenitor cells.
Figure 4.

MiR-16 is downregulated in mammary tumor stem cells. (A) Levels of Wip1 and miR-16 in mouse mammary tumors are not significantly different from those of normal mammary tissues. N1-3: normal mammary gland tissues; T1-3: mammary tumors. Levels of Wip1 mRNA and miR-16 were determined by quantitative RT-PCR. (B) Micrographs of mammospheres originated from mammary tumor stem cells. TC-1, 2: mammary tumor cells; MS-1, 2: mammospheres from the corresponding mammary tumor cells. (C) Expression levels of stem cell markers and differentiation markers in mammospheres and their original tumors. Stem cell markers (Oct-4, KLF-4), differentiation markers (Keratin-14 and Keratin-18), miR-16, and β-actin (as control) were analyzed by semi-quantitative RT-PCR in starting tumor cells and mammospheres. NC: normal tissue cells. (D) miR-16 is upregulated in cells differentiated from mammospheres. Wip1 protein levels were increased in mammospheres, but were downregulated when mammospheres were differentiated to non-stem cells (Rec: recovered cells) (upper panels). In contrast, miR-16 was downregulated in each of the individual mammospheres, but was upregulated in re-differentiated cells. Levels of miR-16 were determined by quantitative RT-PCR (bottom panel).
To further determine the roles of Wip1 in mammary tumor stem cells, we inhibited the activity of Wip1 by its specific inhibitor CCT007093(36), lentiviral shRNA, or ectopically overexpressed lentiviral miR-16 in mouse mammary tumor cells. In each sample, we counted the number of mammospheres that were larger than 50 μm in size. Mammosphere forming ability of tumor cells was sharply reduced when Wip1 was inhibited or silenced (Fig. 5A). As much as 2.54% of tumor cells in the control sample formed mammospheres with an average size of 215 μm. Inhibiting Wip1 by CCT007093, shRNA or miR-16 decreased the numbers of mammospheres to 0.17%, 0.58%, and 0.26% of total cells respectively and the mammospheres were significantly smaller with the size of 80–105μm. As a negative control, overexpression of miR-21 had no noticeable effects on the size and number of mammospheres. Restoration of Wip1 in the miR-16 overexpressed cells remarkably reversed the inhibitory effect of miR-16 on blocking mammosphere formation in culture, suggesting that Wip1 is a primary target of miR-16 in the regulation of mammary tumor stem cells (Fig. 5B & 5C).
Figure 5.
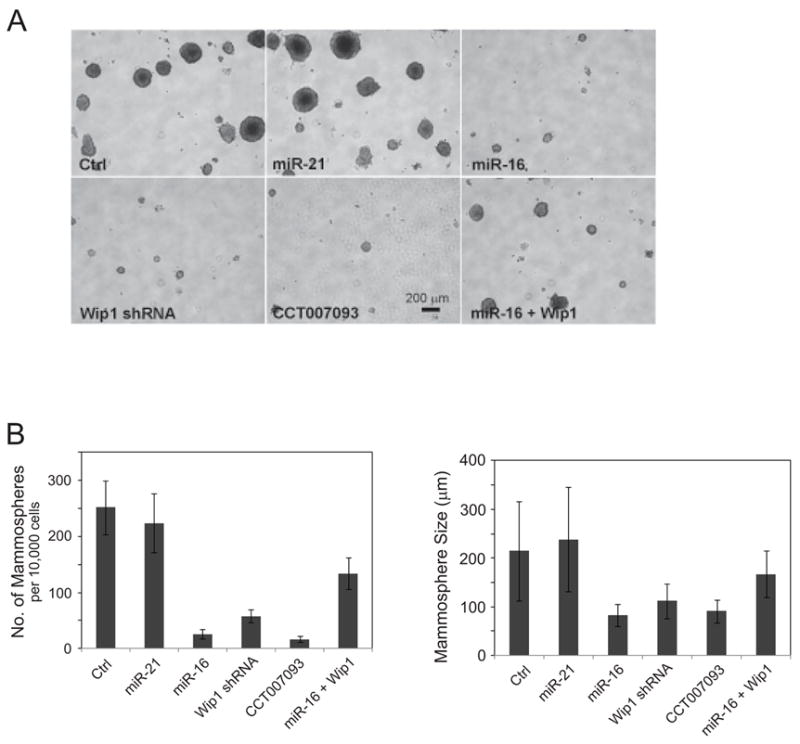
MiR-16 inhibits the maintenance and proliferation of mammary tumor stem cells. (A) miR-16 and the Wip1 inhibitor reduce the number of mammary tumor stem cells. Cells isolated from mammary tumors in transgenic MMTV-erbB2 mice were infected by control viruses or pseudoviruses expressing miR-21, miR-16, and/or mouse Wip1, and cultured in the mammosphere-forming medium with or without 5 μM of the Wip1 inhibitor CCT007093. 10 days after incubation, mammospheres were observed and their sizes and numbers were determined. (B) Mammosphere formation is inhibited by miR-16, the Wip1 inhibitor, or Wip1 shRNA, and restoration of Wip1 reversed the effects of miR-16 on blocking mammosphere formation in culture. Mammosphere numbers (left panel) and sizes (right panel) were measured under each condition.
Discussion
The WIP1 gene was recently identified as an oncogene that is aberrantly regulated in a number of human cancers (10–16). In particular, the WIP1 gene at 17q23 is amplified and overexpressed in 18% of human aggressive primary breast tumors that exclusively express wildtype p53(29). Wip1 removal significantly inhibited mammary tumorigenesis induced by other breast cancer oncogenes, such as Hras1 or Erbb2(26). Recent studies from our laboratory and other groups have revealed the mechanisms for Wip1’s oncogenecity. Wip1 is a master inhibitor in the initiation and maintenance of DNA damage signaling and repair pathways through dephosphorylating H2AX, ATM, Chk1, Chk2 and UNG2(4;5;36–39). Due to its wide-spectrum functioning in the control of DNA damage responses, Wip1 stands out from other DNA damage responsive proteins as a very attractive target for potential breast cancer therapeutics.
Upon DNA damage stress, Wip1 is transcriptionally induced in p53-procificent cells, but not in p53-null cells(23). Recent studies identified WIP1 as a p53- and CREB- regulated gene. A cyclic AMP response element (CRE) and a p53 response element are located in the 5' untranslated region (UTR) of the WIP1 gene. CREB binding to the CRE contributes to the regulation of basal expression of Wip1 and directs transcription initiation at upstream sites, while the p53 response element is required for the p53-dependent induction of transcription(24;40). Our results showed that the level of Wip1 transcripts was immediately induced at the very early stage of the DNA damage response, but Wip1 protein levels increased at a much slower pace. The delayed onset for Wip1 protein induction allows cells to activate a DNA damage-triggered signaling transduction cascade without early inhibition by Wip1.
MiR-16 appears to be a major regulatory factor in suppressing Wip1 protein expression. The level of miR-16 rapidly increased after DNA damage and then slowly returned to normal. Altered levels of miR-16 led to dramatically different patterns of Wip1 induction. Inhibition of miR-16 resulted in greater abundance and accelerated induction of Wip1 proteins, whereas overexpression of miR-16 almost abolished the DNA damage-mediated induction of Wip1. Our studies provide strong evidence that miR-16 is a critical regulator for the induction of Wip1 phosphatase in the stress response pathway. Given the important role of Wip1 in mammary tumorigenesis, we further demonstrated a lower abundance of miR-16 in mammospheres that initiated from mammary tumor stem/progenitor cells. Accordingly, Wip1 levels were markedly up-regulated in spheroid cells. Inhibiting Wip1 or overexpressing miR-16 dramatically reduced the number and size of mammospheres, but no cumulative effects from these two treatments were achieved in our assays. This observation implicates that miR-16 may regulate the proliferation and differentiation of mammary tumor stem cells at least partly through inhibiting Wip1. Two recent publications suggest that loss of p53 permits expansion of presumptive cancer stem cells in mouse mammary tumors and in human breast cell lines. These results add restriction of cancer stem cells as a new tumor suppressor activity attributed to p53(41;42). Given the important roles of Wip1 in the regulation of p53 signaling, it is likely that Wip1 promotes mammary tumor stem cells through p53. The most interesting part of the miR-16-Wip1 regulatory loop is that miR-16 is responsive to DNA damage. A recent study from the Miyazono group showed that p53 enhances the post-transcriptional maturation of several miRNAs with growth-suppressive function including miR-16. We showed here that miR-16 was induced in the DNA damage response. Putting together, miR-16 is a novel player in the ATM-p53-Wip1 autoregulatory feedback loop.
Supplementary Material
Acknowledgments
We are grateful to the members in the Lu Laboratory and Dr. Lawrence Donehower for helpful discussions. This research was supported by NIH grants to XL (R01CA136549 and 5P20RR017698-08) and by an NSF grant to VV (#0646540).
Reference List
- 1.Lu X, Nguyen TA, Donehower LA. Reversal of the ATM/ATR-mediated DNA damage response by the oncogenic phosphatase PPM1D. Cell Cycle. 2005;4:1060–4. [PubMed] [Google Scholar]
- 2.Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008;27:123–35. doi: 10.1007/s10555-008-9127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shreeram S, Demidov ON, Hee WK, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23:757–64. doi: 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–74. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujimoto H, Onishi N, Kato N, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006;13:1170–80. doi: 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- 6.Takekawa M, Adachi M, Nakahata A, et al. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000;19:6517–26. doi: 10.1093/emboj/19.23.6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu X, Ma O, Nguyen TA, Jones SN, Oren M, Donehower LA. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell. 2007;12:342–54. doi: 10.1016/j.ccr.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Lin L, Guo H, et al. Phosphorylation and degradation of MdmX is inhibited by Wip1 phosphatase in the DNA damage response. Cancer Res. 2009;69:7960–8. doi: 10.1158/0008-5472.CAN-09-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi J, Nannenga B, Demidov ON, et al. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol Cell Biol. 2002;22:1094–105. doi: 10.1128/MCB.22.4.1094-1105.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Yang Y, Peng Y, et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet. 2002;31:133–4. doi: 10.1038/ng888. [DOI] [PubMed] [Google Scholar]
- 11.Saito-Ohara F, Imoto I, Inoue J, et al. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003;63:1876–83. [PubMed] [Google Scholar]
- 12.Hirasawa A, Saito-Ohara F, Inoue J, et al. Association of 17q21-q24 gain in ovarian clear cell adenocarcinomas with poor prognosis and identification of PPM1D and APPBP2 as likely amplification targets. Clin Cancer Res. 2003;9:1995–2004. [PubMed] [Google Scholar]
- 13.Loukopoulos P, Shibata T, Katoh H, et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 2007;98:392–400. doi: 10.1111/j.1349-7006.2007.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehrbrecht A, Muller U, Wolter M, et al. Comprehensive genomic analysis of desmoplastic medulloblastomas: identification of novel amplified genes and separate evaluation of the different histological components. J Pathol. 2006;208:554–63. doi: 10.1002/path.1925. [DOI] [PubMed] [Google Scholar]
- 15.Castellino RC, De BM, Lu X, et al. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neuroonco. 2008;86:245–56. doi: 10.1007/s11060-007-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuku T, Semba S, Yutori H, Yokozaki H. Increased wild-type p53-induced phosphatase 1 (Wip1 or PPM1D) expression correlated with downregulation of checkpoint kinase 2 in human gastric carcinoma. Pathol Int. 2007;57:566–71. doi: 10.1111/j.1440-1827.2007.02140.x. [DOI] [PubMed] [Google Scholar]
- 17.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–14. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pothof J, Verkaik NS, van IW, et al. MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J. 2009;28:2090–9. doi: 10.1038/emboj.2009.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–33. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 20.Hu H, Du L, Nagabayashi G, Seeger RC, Gatti RA. ATM is down-regulated by N-Myc-regulated microRNA–421. Proc Natl Acad Sci U S A. 2010;107:1506–11. doi: 10.1073/pnas.0907763107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonci D, Coppola V, Musumeci M, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–7. doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- 22.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6:R605–R615. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fiscella M, Zhang H, Fan S, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A. 1997;94:6048–53. doi: 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossi M, Demidov ON, Anderson CW, Appella E, Mazur SJ. Induction of PPM1D following DNA-damaging treatments through a conserved p53 response element coincides with a shift in the use of transcription initiation sites. Nucleic Acids Res. 2008;36:7168–80. doi: 10.1093/nar/gkn888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bulavin DV, Demidov ON, Saito S, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–5. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 26.Bulavin DV, Phillips C, Nannenga B, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 27.Natrajan R, Weigelt B, Mackay A, et al. An integrative genomic and transcriptomic analysis reveals molecular pathways and networks regulated by copy number aberrations in basal-like, HER2 and luminal cancers. Breast Cancer Res Treat. 2010;121:575–89. doi: 10.1007/s10549-009-0501-3. [DOI] [PubMed] [Google Scholar]
- 28.Natrajan R, Lambros MB, Rodriguez-Pinilla SM, et al. Tiling path genomic profiling of grade 3 invasive ductal breast cancers. Clin Cancer Res. 2009;15:2711–22. doi: 10.1158/1078-0432.CCR-08-1878. [DOI] [PubMed] [Google Scholar]
- 29.Rauta J, Alarmo EL, Kauraniemi P, Karhu R, Kuukasjarvi T, Kallioniemi A. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res Treat. 2006;95:257–63. doi: 10.1007/s10549-005-9017-7. [DOI] [PubMed] [Google Scholar]
- 30.Cimmino A, Calin GA, Fabbri M, et al. miR–15 and miR–16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demidov ON, Kek C, Shreeram S, et al. The role of the MKK6/p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene. 2007;26:2502–6. doi: 10.1038/sj.onc.1210032. [DOI] [PubMed] [Google Scholar]
- 32.Kim ND, Oberley TD, Yasukawa-Barnes J, Clifton KH. Stem cell characteristics of transplanted rat mammary clonogens. Exp Cell Res. 2000;260:146–59. doi: 10.1006/excr.2000.5013. [DOI] [PubMed] [Google Scholar]
- 33.Al-Hajj M, Wicha MS, ito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–70. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36 (Suppl 1):59–72. doi: 10.1046/j.1365-2184.36.s.1.6.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rayter S, Elliott R, Travers J, et al. A chemical inhibitor of PPM1D that selectively kills cells overexpressing PPM1D. Oncogene. 2008;27:1036–44. doi: 10.1038/sj.onc.1210729. [DOI] [PubMed] [Google Scholar]
- 37.Macurek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene. 2010;29:2281–91. doi: 10.1038/onc.2009.501. [DOI] [PubMed] [Google Scholar]
- 38.Moon SH, Lin L, Zhang X, et al. Wildtype p53-induced phosphatase 1 dephosphorylates histone variant {gamma}-H2AX and suppresses DNA double strand break repair. J Biol Chem. 2010;285:12935–47. doi: 10.1074/jbc.M109.071696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu X, Bocangel D, Nannenga B, Yamaguchi H, Appella E, Donehower LA. The p53-induced oncogenic phosphatase PPM1D interacts with uracil DNA glycosylase and suppresses base excision repair. Mol Cell. 2004;15:621–34. doi: 10.1016/j.molcel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 40.Song JY, Han HS, Sabapathy K, Lee BM, Yu E, Choi J. Expression of a homeostatic regulator, Wip1 (wild-type p53-induced phosphatase), is temporally induced by c-Jun and p53 in response to UV irradiation. J Biol Chem. 2010;285:9067–76. doi: 10.1074/jbc.M109.070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M, Behbod F, Atkinson RL, et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008;68:4674–82. doi: 10.1158/0008-5472.CAN-07-6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Godar S, Ince TA, Bell GW, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.