

Abstract
Testicular germ cell tumors (TGCTs) originate from germ cells. The 129-Ter and M19 (129.MOLF-Chr19 consomic) mouse strains have extremely high incidences of TGCTs. We found that the expression levels of Sf1 encoded Splicing factor 1 (SF1) can modulate the incidence of TGCTs. We generated mice with inactivated Sf1. Sf1 null mice (Sf1-/-) died before birth. Mice with one intact allele of Sf1 (Sf1+/-) were viable but expressed reduced levels of Sf1. When Sf1 deficient mice (Sf1+/-) were crossed to the 129-Ter and M19 strains, we observed decreased incidence of TGCTs in Sf1+/-;Ter and Sf1+/-;M19/+ mice compared to that in control cohorts. Therefore, Sf1 deficiency protects against TGCT development in both strains. Sf1 is expressed in the testes. We found that Sf1 levels vary significantly in the testes of inbred strains such as 129 and MOLF and as such Sf1 is an oncogenic tumor susceptibility factor from 129. Our results also highlight the complications involved in evaluating Sf1 levels and TGCT incidences. When a large number of tumor promoting factors are present in a strain, the protective effect of lower Sf1 levels is masked. However, when the dosage of tumor promoting factors is reduced, the protective effect of lower Sf1 levels becomes apparent. SF1 is involved in splicing of specific pre-mRNAs in cells. Alternate splicing generates the complex proteosome in eukaryotic cells. Our data indicates that Sf1 levels in mouse strains correlate with their incidences of TGCTs and implicate the importance of splicing mechanisms in germ cell tumorigenesis.
Introduction
TGCTs are the most common malignancy in young men. These tumors originate from germ cells at different stages of development (1, 2). Both genetic factors, such as ethnicity and family history, and environmental factors contribute to TGCT development (3, 4). Evidence indicates that a combination of multiple genetic factors contribute to susceptibility to TGCT development (5-8). Individually, each of these factors contributes with relatively modest effects towards tumor development. It has been a challenge to identify the factors that cause TGCTs particularly because the tumors initiate in utero even though the disease may become evident decades after birth.
In mice, TGCTs occur predominantly on the 129 strain background. About 10% of 129 males develop spontaneous TGCTs (9). The genetic factors from the 129 strain that support TGCT development have not been identified. However, a number of gene defects have been experimentally shown to increase (10-14) or suppress TGCT incidences (15). The tumors in mice originate from primordial germ cells (PGCs) and initiate development around embryonic day (E) 11.5 - E13.5. For reasons not well understood, some PGCs on the 129 strain background become transformed to embryonal carcinoma (EC) cells. EC cells proliferate rapidly in the embryonic gonads. Soon after birth, EC cells differentiate randomly into embryonic and adult cells that constitute the TGCTs in the testes. TGCTs in mice resemble the pediatric TGCTs of humans (16).
Two 129 derived mouse strains, the 129-Ter and M19, have extremely high rates of spontaneous TGCT development (Supplementary Fig.1). The Ter defect is due to inactivation of the function of the RNA-binding protein, Dead end 1 (Dnd1) (11). Dnd1 is essential for PGC viability (11, 17). Loss of Dnd1 results in progressive death of germ cells contributed to some extent by BAX-mediated apoptosis (18). This results in sterility in all Ter mice. However, 129 strain mice with inactivated Dnd1 (129-Ter mice) develop TGCTs in addition to being sterile due to germ cell loss (19, 20). Thus, some PGCs of the 129-Ter strain escape death to transform into EC cells and EC cells subsequently differentiate to form large tumors in the testes.
A second mouse strain with high incidence of spontaneous germ cell tumors is the consomic, 129.MOLF-Chr19, mouse strain (also referred to as M19, chromosome substitution strain or CSS) (21). M19 strain differs from the 129 only because chromosome (Chr) 19 of the MOLF strain replaces that of the 129 (Supplementary Fig.1). The M19 strain does not carry the Ter (inactivation of Dnd1) defect. Multiple TGCT susceptible loci have been mapped to Chr 19 of the M19 strain. These loci either independently or interact epistatically to contribute to testicular tumor development (22). Unlike in the 129-Ter strain, the TGCT causing genes in M19 do not cause germ cell death. Thus both normal and transformed germ cells are present in the M19 strain and M19 males can be fertile despite having testicular tumors.
We identified Splicing factor 1, Sf1, as a TGCT candidate gene from the M19 strain (23). Here, we report the role of Sf1 in TGCT development. Interestingly, our results indicate that Sf1 expression levels influence the incidence of germ cell tumor development.
SF1 (also known as Splicing factor 1, Mammalian branch point-binding protein (mBBP), Zinc finger gene in MEN1 locus (ZFM1), Zinc finger protein 162 (ZNF162 or ZFP162)) participates in the early spliceosome assembly step during pre-mRNA splicing (24, 25). SF1 is involved in the assembly of the earliest spliceosome complex (E' complex) committed to the splicing pathway (26, 27). Splice site recognition requires cross talk between multiple proteins that are involved in forming complexes that commit the pre-mRNA to splicing. SF1 interacts co-operatively with U2 snRNP auxiliary factor (U2AF65), and these proteins bind to the branch point site and the polypyrimidine tract in the intron of pre-mRNAs, respectively (28-30). SF1 is essential for viability of cells in culture. SF1 is not required for general splicing of all pre-mRNAs in cells but the premRNA substrates of SF1 that are necessary to maintain cell viability have not been identified (31). Thus, SF1 targets a sub-set of cellular pre-mRNAs for splicing and acts as an alternative splicing factor.
A previous study reported higher incidence of colon tumors when Sf1+/- mice were treated with an organotropic carcinogen (32). This indicated that lower levels of SF1 promote tumorigenesis. We report the results of genetic studies using Sf1+/- mice and contrary to the effects observed in the colon, we found that lower SF1 levels suppress germ cell tumor development. Therefore, our studies implicate SF1 and splicing mechanisms in germ cell tumor development.
Materials and Methods
Mouse strains
The 129.MOLF-Chr19 (21), 129 (129S1/SvImJ; JR002448, Jackson Laboratory, Bar Harbor, ME) and 129-Ter (11) strains have been described.
Generation of Sf1 knockout mice
The gene trap 129/Ola strain embryonic stem (ES) cell clone, XD130, was purchased from BayGenomics (University of California, Davis). The trap vector insertion was validated by BayGenomics to contain gene trap inactivated Sf1 by 5'-RACE (Rapid amplification of 5' complementary DNA ends) and sequencing which detected correct splicing between exon 1 of Sf1 and the trap vector. XD130 was injected into C57BL/6J (B6) mice-derived blastocysts, which were then transferred into pseudopregnant recipient female mice and this yielded chimera mice. The chimeras were crossed to 129. Founder males which showed germline transmission of the gene-trap, β-geo allele, were crossed to 129 mice.
Germline transmission was screened by PCR genotyping for the targeted allele. Primers for genotyping were: gt1F, gt4R, v1F, v1R (Supplementary Fig.2). gt1F/i5R identify the wild-type allele. All animals were genotyped using the three sets of primers. Mice genotyped to be positive for both v1F/v1R and gt1F/gt4R were selected to maintain the Sf1 gene trap line. Inactivation of Sf1 by the β-geo encoding gene trap was further verified by Southern blotting and by sequencing of the genomic sequences flanking the vector and the vector insertion site.
Southern blotting
Genomic DNA extracted from the spleen of 129.Sf1+/- and +/+ mice, of G1 and G2 generations, were digested with Bgl II or EcoR V and used for Southern blotting. Blots were hybridized with a 32P-labelled 626 bp Pst I/Xba I fragment derived from the β-geo cassette (Supplementary Fig.2). A 10.4 kb Bgl II fragment or a 8.9 kb EcoR V fragment is expected when one copy of trap vector is inserted in the genome. However, 8.6 kb and 10.4 kb Bgl II and 8.6 kb and 8.9 kb EcoR V fragments are expected when two copies of the trap vector are tandemly inserted into the Sf1 gene.
X-gal staining and in situ hybridization was performed as described (33). Immunohistochemistry and western blotting for detection of SF1 used anti-SF1 antibody (SC-21157, Santa Cruz Biotechnology). Additional antibodies included β-actin (A5316 Sigma); anti-SSEA1-Alexa Fluor® 647 (SC-21702, Santa Cruz Biotechnology) and Sox9 (AB5535, Chemicon).
RT-PCR and quantitative real-time PCR was carried out as described (23). Primers used for qRT-PCR of SF1 were : 5'-ttcttcgttctgctgctttgc-3' and 5'-gtgaaaagaacgcgcattacac-3'; GAPDH: 5'-ttgtctcctgcgacttcaaca-3' and 5'-accaggaaatgagcttgacaaa-3'. Primers used for RT-PCR to amplify Sf1 were: 5'-cacagcttcccacacccattaccc-3' and 5'-gctgctttgccgaaccatcc-3'; Oct4: 5'-ggaggaagccgacaacaatga-3' and 5'-tccacctcacacggttctcaa-3', Dnd1: 5'-gccctggtagaaggtcagtcac-3' and 5'-gccctgttcctaaacacttggtc-3'; Sox9: 5'-gcggagctcagcaagactctg-3' and 5'-atcggggtggtctttcttgtg-3'; HPRT: 5'-gttgagagatcatctccacc-3' and 5'-agctatgatgaaccaggtta-3'.
Results
Sf1 is essential during embryonic development
We used a gene trap, ES cell clone XD130, to generate Sf1+/- mice. Southern blotting and PCR determined that XD130 harbored tandem insertion of two copies of the gene trap in the first intron of Sf1 and 261 bp downstream of exon 1 (Supplementary Fig 2). This disrupts expression of normal Sf1. Instead, β-geo (fusion of LacZ encoding β-galactosidase and neo resistance gene) is fused with the first ten amino acids of Sf1 and expressed from the targeted allele.
We intercrossed heterozygous Sf1+/- (or Sf1β-geo/+) mice, but did not obtain any homozygous null mice, Sf1-/- (Sf1β-geõ β-geo), at weaning or at E11.5 (Supplementary Table 1). This result is similar to that reported for genetically modified Sf1 mice made by Shitashige et. al. (32). Thus, Sf1 is critical during embryonic development.
Next, we examined the levels of Sf1 transcripts in Sf1+/- mice using quantitative RT-PCR (qRT-PCR). Total RNA from post-natal day 1 (PN 1) testes was used for qRTPCR. We found that Sf1 mRNA levels were significantly lower (2 to 4-fold lower) in the testes of Sf1+/- mice compared to that in wild-type mice (Fig.1). SF1 protein levels had also decreased in PN 1 testes of Sf1+/- mice as determined by immunoblotting using anti-SF1 antibodies (Fig.1B and C).
Fig.1. Reduction of Sf1 mRNA and protein levels in Sf1+/- mice.
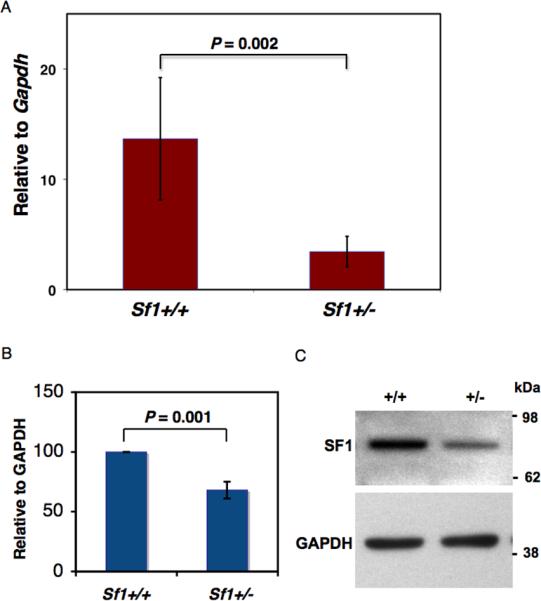
(A) qRT-PCR performed on total RNA from PN1 testis of wild-type and Sf1+/- mice. Sf1 expression is normalized against Gapdh expression in the testes. Error bar is the standard deviation derived from two independent experiments. (B) SF1 protein levels in PN1 testes of wild-type and Sf1+/- mice as determined by immunoblotting using anti-SF1 antibodies as shown in (C). Protein levels are normalized against GAPDH expression in the testes. Error bar is the standard deviation derived from four independent experiments. (C) A representative immunoblot using anti-SF1 antibody for PN1 testis of wild-type and Sf1+/- mice. The blots were re-probed with anti-GAPDH as controls.
Sf1 expression in the testes
We examined the expression pattern of Sf1 in mouse testes. First, we examined PN 1 testes for LacZ activity from the endogenous Sf1 promoter in Sf1+/- (Sf1β-geo/+) mice. X-gal staining of Sf1+/- mice revealed that the germ cells were positive (Supplementary Fig.2) but the supporting cells of the seminiferous tubules were not positive for LacZ activity.
We sought to verify if the X-gal expression pattern in the testes of Sf1+/- mice was a true reflection of endogenous SF1 expression. We therefore performed in situ hybridization using antisense Sf1 probes as well as immunohistochemistry using anti-SF1 antibody on PN 1 testes from normal, wild-type (+/+) mice. In contrast to the LacZ expression pattern, both in situ hybridization (Supplementary Fig.2) and immunostaining indicated (Fig.2) that SF1 is present in both the germ and Sertoli cells. Immunostaining indicated that SF1 was present in the nucleus as would be expected for a splicing factor.
Fig.2. Expression of Sf1 in the testes.
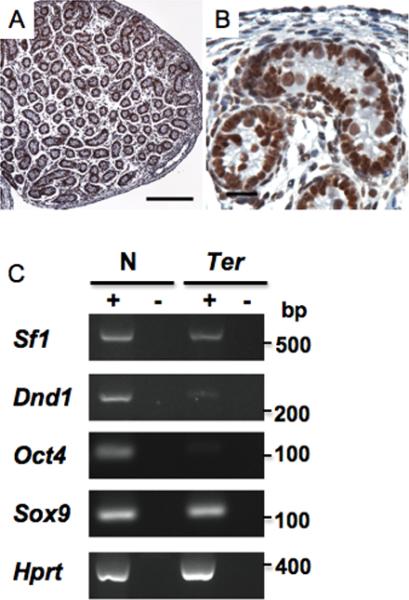
(A) Immunostaining of PN1 testes from wild-type mice using anti-SF1 antibody at low (bar represents 200 um) and (B) higher magnifications (bar represents 20 um). (C) RT-PCR for Sf1, Dnd1, Oct4, Sox9 and Hprt using total RNA from PN1 testes of wild-type (N) and B6-Ter/Ter (Ter) mice. PCR was performed on equal amounts of cDNA. + indicates presence of Superscript during cDNA preparation. - are control lanes and indicates no Superscript was added.
Because we obtained contradictory data regarding LacZ expression as opposed to in situ hybridization and immunostaining, we used RT-PCR to determine whether Sf1 transcripts are indeed expressed in both the germ and supporting cells. Therefore, we compared Sf1 expression in the testes of Ter and wild-type mice. Mice homozygous for the Ter defect, and on a C57BL/6J background (B6-Ter/Ter), completely lack germ cells (11, 19). RT-PCR indicated that Sf1 was present in the testes of B6-Ter/Ter mice (Fig.2C). This indicates that Sf1 transcripts are indeed present in the supporting cells of the testes. RT-PCR also indicated lack of expression of germ cell markers (Dnd1 and Oct4) and presence of Sertoli cell markers (Sox9) in testes of Ter strain (18).
To determine why LacZ activity was absent in somatic cells, we examined lacZ transcript (Sf1-geo fusion transcript) in Sf1+/-;Ter/Ter testes. Sf1+/-;Ter/Ter mice also lack germ cells (strain described below). However, RT-PCR indicated that lacZ transcript is expressed in the somatic cells of the testes (Supplementary Fig. 2K). One reason we did not detect LacZ activity in the somatic cells of Sf1+/- testes could be because of lower levels of LacZ transcript and protein levels in the somatic cells. The LacZ reporter system is reported to be not as sensitive as RT-PCR or antibody staining (34).
Taking together the results of the above studies, we conclude that SF1 is expressed in both germ and Sertoli cells of the testes. However, in our Sf1+/- (Sf1β-geo/+) mice, for reasons not known, lacZ activity is strong only in the germ cells.
Deficiency of Sf1 reduces TGCT incidence
Next, we examined the effect of Sf1 deficiency on germ cell tumorigenesis. We were unable to assess TGCT incidence in Sf1-/- adult mice because of their embryonic lethality. We therefore crossed Sf1+/- mice to mouse strains with inherent genetic predisposition to TGCT. Males of the Ter strain develop testicular germ cell tumors at an exceptionally high rate. 94% of male mice homozygous for 129-Ter (Ter/Ter) develop tumor in at least one testes (19). In cases where testes of 129-Ter mice are tumor free, it remains small in size because of lack of germ cells.
We crossed Sf1+/- with Ter/+ mice and examined tumor incidence in the double mutant progeny (crosses are illustrated in Supplementary Fig.3). We found that 40% of Sf1+/-;Ter/Ter male mice have testicular tumors (Table 1). The tumor incidence of the control cohort of siblings of Ter/Ter genotype was 77%. Thus, tumor incidence had decreased in Sf1+/-;Ter/Ter mice and this change was statistically significant (P=0.0174). The incidence of bilateral testicular tumors had also decreased in Sf1+/-;Ter/Ter mice. 40% of Sf1+/-;Ter/Ter compared to 71% of Ter/Ter tumor bearing mice developed bilateral tumors. However, there was a corresponding increase in the number of Sf1+/-;Ter/Ter males with sterile, bilateral small testes (60%) compared to that in Ter/Ter males (23%). Mice of Ter/Ter genotypes with unilateral tumors had contralateral small testes. Thus, neither Ter/Ter nor Sf1+/-;Ter/Ter cohorts had normal testes.
Table 1. Sf1 deficiency reduces incidence of germ cell tumors. TGCT incidence in Ter mice deficient for Sf1.
Ter/+; Sf1+/- mice were intercrossed with Ter/+ mice (illustrated in Supplementary Fig.3). Progeny were of genotypes listed in the table. Progeny were genotyped and the testes phenotypes examined. The significance of the difference in tumor incidences between the Ter/Ter and Ter/Ter;Sf1+/- strains is P = 0.0174. Differences in the tumor incidences of Ter/+ compared to Ter/+;Sf1+/- and +/+ compared to Sf1+/- were not statistically significantly.
| Genotype | No. with tumors | Bilateral tumors | Unilateral tumors | Sterile testes | Normal testes | No. of males examined |
|---|---|---|---|---|---|---|
| Ter/Ter | 17 (77%) | 12 (71%) | 5 | 5 (23%) | 0 | 22 |
| Ter/Ter; Sf1+/- | 10 (40%) | 4 (40%) | 6 | 15 (60%) | 0 | 25 |
| Ter/+ | 23 (45%) | 5 | 18 | 0 | 28 | 51 |
| Ter/+; Sf1+/- | 21 (40%) | 4 | 17 | 0 | 32 | 53 |
| +/+ | 0 | 0 | 0 | 0 | 20 | 20 |
| +/+; Sf1+/- | 1 (5%) | 0 | 1 | 0 | 21 | 22 |
Next, we examined Sf1 levels in Sf1+/-;Ter/Ter testes. qRT-PCR indicated that Sf1 transcript levels were lower in Sf1+/-;Ter/Ter compared to that in Ter/Ter testes (Supplementary Fig.4). In addition, SF1 protein levels were also decreased in Sf1+/-;Ter/Ter testes (Fig.3A).
Fig.3. Variation of Sf1 levels in different mouse strains.

(A) (top) SF1 levels in Sf1 deficient mice. PN1 testes, from M19/+, Ter/Ter, Ter/+ and wild-type (+/+) mice and corresponding Sf1 deficient (Sf1: +/-) mice, were used for immunoblotting using anti-SF1 antibody. The blots were re-probed with anti-β-actin as controls. (bottom) Quantitation of SF1 levels in the mouse strains. The results are the average from 2 experiments. (B) qRT-PCR was used to determine Sf1 levels in PN 1 testes of M19, M19/+ and M19/+;Sf1+/- strains. (C) Comparing Sf1 levels in 129, M19 and MOLF strains. Total RNA from PN1 testes was used. The expression levels are normalized against Gapdh expression in the same sample. Error bar is the standard deviation derived from two independent experiments.
The incidence of tumors in Ter/Ter mice in this study was 77% and not the reported incidence of 94% (19). This could be because of the smaller sample size that was collected for this study (22 Ter/Ter and 25 Sf1+/-;Ter/Ter males were examined) (Table 1). An alternate possibility may be that 129 sub-strain differences influenced the tumor incidence. The Sf1 gene trap was on 129/Ola sub-strain ES cells, founder mice were crossed to 129S1/SvImJ and Ter mice are another substrain, (129T1/Sv-+pTyrc-chTer/+@Na) (35). This genetic variability between 129 substrains could have influenced the tumor incidences in Ter/Ter mice in our crosses.
Overall, although Sf1 deficiency lowers the incidence of testicular tumors in Ter/Ter males, it does not restore germ cells in the testes of Ter/Ter males. This suggests that Sf1 deficiency does not rescue the germ cell death in Ter (due to lack of DND1) but more likely affects germ cell transformation. Because SF1 mediates splicing of pre-mRNAs, it is likely that deficiency of SF1 causes reduced production of ‘oncogenic’ spliced variants, which leads to attenuation of germ cell transformation processes and an overall decreased level of germ cell tumors. This also suggests that SF1 functions downstream to DND1. The identities of the pre-mRNAs that are targets of SF1 in germ cells are at present unknown.
In conclusion, lack of one allele of Sf1 results in decreased expression of SF1 and is correlated with a significant reduction of TGCT incidence in Ter mice. Thus, Sf1 is haploinsufficient and Sf1 levels influence germ cell transformation.
Deficiency of Sf1 reduces tumor incidence of M19 strain males
To determine how Sf1 affects TGCT incidence of the M19 mouse strain, we crossed Sf1+/- to M19 mice and examined tumor incidences in the male progeny, Sf1+/-;M19/+ and M19/+ males (Supplementary Fig.3). These males carry one MOLF chromosome as signified by M19/+ and one allele of Sf1β-geo in Sf1+/-;M19/+. We found reduced TGCT incidence in Sf1+/-;M19/+ males (17%) compared to that in M19/+ males (27%) (Table 2). This reduction in tumor incidence was modest but statistically significant at P < 0.04. The incidence of bilateral tumors had also decreased in the Sf1+/-;M19/+ (18% bilateral tumors) compared to that in M19/+ males (31% bilateral tumors).
Table 2. TGCT incidence in M19 mice deficient for Sf1.
M19 (or M19/M19) was crossed to Sf1+/- mice (Supplementary Fig.3). The progeny were genotyped to determine the M19/+ and M19/+;Sf1+/- males. Testes phenotypes of all the males were examined. The significance of the difference in tumor incidences between the strains is P = 0.0375.
| Genotype | No. with tumors | Bilateral tumors | Unilateral tumors | Normal testes | Total number of males examined |
|---|---|---|---|---|---|
| M19/+ | 36 (27 %) | 11 (31%) | 25 | 95 | 131 |
| M19/+;Sf1+/- | 22 (17%) | 4 (18%) | 18 | 111 | 133 |
We found that SF1 levels had decreased, by about 50%, in Sf1+/-;M19/+ compared to the M19/+ strain (Fig.3A). This also correlated with lower Sf1 levels in the Sf1+/-;M19/+ compared to that in M19/+ gonads and lower TGCT incidence in Sf1+/-;M19/+ mice. Next, we compared the Sf1 mRNA levels in the PN1 testes of M19 to that of M19/+ and M19/+;Sf1+/- mice. The level of Sf1 transcript was lower (by about 4-fold) in the M19 compared to that in the M19/+ gonads (Fig.3B). The M19/+ has one chromosome 19 from MOLF and one from 129 (Fig.4) whereas M19 is homozygous and both chromosome 19 are MOLF derived.
Fig.4. Comparing Sf1 levels and germ cell tumor incidences of different mouse strains.
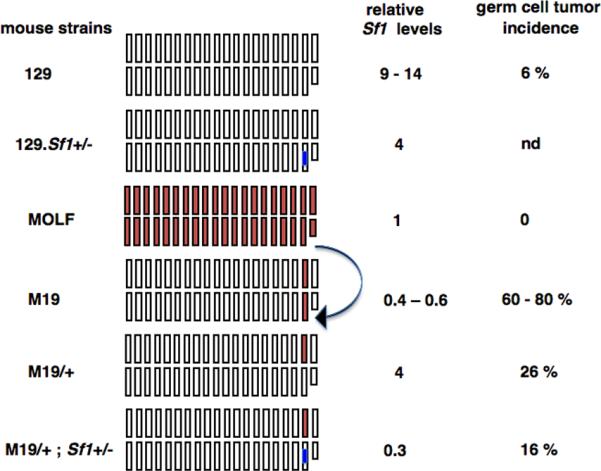
The boxes represent the 19 autosomal Chrs and Chr X and Y (smaller box) of the mouse strains. White boxes represent 129 chromosomes. MOLF chromosomes are in red. The Sf1β-geo allele is represented by the blue mark on Chr 19. M19 strain carries Chr 19 of MOLF (indicated by arrow) and all other chromosomes are of 129 origin. Therefore, Sf1 levels of M19 are similar to that in MOLF although tumor incidences of the two strains differ. M19/+ is heterozygous for MOLF Chr 19 and has higher levels of Sf1. Introduction of Sf1β-geo allele to generate M19/+;Sf1+/- mice results in lowering of Sf1 levels and concomitant reduction of tumors compared to that in M19/+. The relative Sf1 levels are the median values from the graphs in Figs.1 and 3.
Overall, the genetic analysis using both the Ter and M19 strains indicates that lowering of Sf1 levels in the testes protects against TGCT development. This indicates a common mechanism of action of Sf1 in these two strains that develop TGCTs due to different genetic defects.
Sf1 levels in the 129 and MOLF inbred mouse strains
We had initially identified Sf1 from the M19 strain and a comparative microarray screen had found a 2–8 reduced expression of Sf1 in M19 compared to 129 (23). Because M19 males have higher TGCT incidence compared to 129 (21), we expected that decreasing Sf1 levels further would increase TGCT incidence in Sf1+/-;Ter or Sf1+/-;M19 mice. Surprisingly, the results from our genetic data were opposite to that expected. We have verified that Sf1 levels are indeed reduced in Sf1+/-, Sf1+/-;Ter and Sf1+/-;M19/+ mouse gonads. This raised the question as to why Sf1 levels are low in the M19 strain, which has high tumor incidence. One explanation could be because Chr 19 of M19 strain is derived from and identical to that of the MOLF strain (Fig.4) (21). As Sf1 is located on Chr 19, it is possible that the expression level of Sf1 in M19 is inherited from MOLF.
Therefore, we examined Sf1 expression of the MOLF inbred mouse strain. qRTPCR indicated that Sf1 levels in the PN1 testes of MOLF are indeed similar to that in M19 (Fig.3C) and Sf1 levels in both MOLF and M19 is significantly lower than in the 129 strain. Therefore, MOLF has inherently low Sf1 expression compared to 129. Also to note is the fact that, unlike the 129, the MOLF strain does not develop spontaneous TGCTs.
Thus, Sf1 expression levels in M19 are similar to that in the MOLF strain although the testicular tumor frequency of M19 is extremely high and that of MOLF is extremely low.
Taking together all the above observations, we conclude that high testicular tumor incidence of M19 strain likely occurs in spite of the lower Sf1 levels. Tumor development in M19 strain is due to multiple TGCT susceptibility loci from Chr 19 (22, 23). The presence of multiple TGCT promoting genes likely overrides the protective effect of lower Sf1 levels in M19. Thus the M19 strain is an exception to the rule that lower Sf1 correlates with lower TGCT incidence.
However, when we reduce the dosage of the TGCT causing genes from that in M19 (homozygous for MOLF Chr 19) to M19/+ (heterozygous for MOLF Chr 19), the tumor incidence decreases in M19/+ to 26% (Fig.4). Sf1 levels are higher in M19/+ (Fig.4). Only when we further lower Sf1 levels, as in the Sf1+/-;M19/+ strain, both Sf1 levels and tumor incidences decrease (Fig.4). Thus, the protective effect of lower Sf1 levels becomes apparent when a single MOLF Chr 19 is present as in Sf1+/-;M19/+ mice.
Discussion
Our results demonstrate that Sf1 levels modulate the incidence of TGCT in two different mouse models of TGCTs. Haploinsufficiency of Sf1 correlated with decreases in tumor incidences in Sf1+/-;Ter/Ter and Sf1+/-;M19/+ mice. This argues for a common role of SF1 in germ cell transformation in both strains considering that different genetic defects are responsible for germ cell tumorigenesis in the two strains. Reduction of Sf1 levels likely reduces germ cell transformation rates and results in an overall reduced number of testicular tumors. Another possibility is that lower Sf1 levels in germ cells may result in reduced viability of germ cells. Thus fewer germ cells survive to transform. However, PN1 testes of Sf1+/- mice did not appear to have fewer germ cells compared to wild-type mice and Sf1+/- mice are fertile.
Sf1 levels in both MOLF and M19 strains were found to be significantly lower than in the 129 strain. Thus, different mouse inbred strains inherently express varying Sf1 levels in their tissues and the Sf1 levels could likely influence the tumorigenic potential of cells from these strains. Because the 129 strain has higher Sf1 levels and is permissive for TGCT development, we propose that Sf1 is an ‘oncogenic’ genetic susceptibility factor from 129 that promotes TGCT development.
Interestingly, Sf1 levels are observed to inherently vary in tissues of different inbred strains (NCBI Geo Profiles). This inherent variation in Sf1 levels could likely influence normal and disease phenotypes in mice.
We note that no significant tumor incidences have been observed for the MOLF inbred strain, which has inherently low Sf1 levels. Of the reported studies, TGCT development is found in the M19 (129.MOLF Chr 19) consomic strain (21) and suppression of mammary tumorigenesis has been reported in FVB/N-Tg (MMTV-PyMT) and MOLF F1 hybrid mice (36).
The difference in SF1 levels between the 129 and MOLF strains may be due to a number of factors such as differences in the sequence and activity of the promoters between strains, differences in the nature of alternate spliced Sf1 variants or SNPs (single nucleotide polymorphisms) in the transcripts that affect stability.
Contrary to other observations, we found that Sf1 levels are low in M19 but TGCT incidence is high in this strain. In this case, it appears that the protective effect of low Sf1 levels does not overcome the effects of multiple tumor promoting loci present in M19. However, when we reduce the dosage of tumor promoting loci in M19 to M19/+, then we clearly observe the protective effect of lower Sf1 levels in M19/+;Sf1+/-. TGCT incidences correlate with Sf1 levels in M19/+ and M19/+;Sf1+/- mice. Thus, the protective effect of Sf1 deficiency can be overshadowed by the presence of multiple tumor promoting loci in the genome.
An earlier study reported a gene trap inactivated Sf1 mouse line in which the gene trap was inserted in the promoter region of Sf1 (37). Treatment of Sf1+/- mice with an organotropic carcinogen resulted in higher number of colon tumors (32). This indicated that lower Sf1 levels are oncogenic and this is contradictory to our data. One explanation for this discrepancy could be that Sf1 functions as an oncogene or tumor-suppressor depending on the cell type. In colon cells, Sf1 may be responsible for generating a majority of splice variants with tumor-suppressor function whereas in the testes, Sf1 may generate mostly oncogenic splice variants. Thus lowering Sf1 levels in the colon enhances tumorigenesis whereas lowering Sf1 levels in the testes attenuates tumorigenesis. Another possibility is that application of DNA damaging carcinogen likely induces additional oncogenic mutations in colon cells, and the effects of multiple oncogenes outweigh the protective effect of Sf1 deficiency. A third possibility is that because the Sf1+/- mouse backgrounds in the two studies are different, this influences the differences in results.
SF1 has been shown to be critical for HeLa cell viability (31). We detected SF1 expression in developmentally important organs, such as the heart and brain (Supplementary Fig.5). Presumably, there are cell-type specific pre-mRNA targets of SF1 that are critical for cell and embryonic viability.
In the testes, SF1 is found in both the germ cells and supporting cells of the seminiferous tubules. It is possible that SF1 function in the supporting Sertoli cells of the testes may also be important for germ cell tumorigenesis. Overall, alternative splicing events are most prevalent in the testis and brain (38), implying that splicing regulation is especially important in these tissues.
Transformation of cells frequently occurs in conjunction with dysregulation in alternative splicing (39-42). Because splicing factors usually regulate production of a variety of different mRNAs, changes in either the level or function of splicing factors can cause global changes in RNA levels and splicing variants. Specific splicing factors have been found to be overexpressed or downregulated in cancer tissues (42, 43). Studies on human testicular tumors have found novel testicular cancer associated splice isoforms (44, 45) which could serve as potential diagnostic or prognostic markers.
Interestingly, in humans, SF1 has also been implicated in immune responses (46). SF1 interacts with the branchpoint sequence within the intron of the histocompatibility leukocyte antigen, HLA-DQBβ. Naturally occurring and disease associated branchpoint mutations in DQβ1 intron 3 show impaired SF1 binding and altered splicing which influences their gene expression (46). A recent global analysis of the human transcriptosome revealed that many genes have subtle genetic changes, such as SNPs in splice site usage (47). Splicing differences due to SNPs were found to be frequent in human populations, affect disease-causing genes and likely contribute to phenotypic diversity and susceptibility to complex diseases.
In another study, oncogenic alternate splice variants were generated when SF1 was transiently transfected into human colon cancer cell lines and this correlated with cell transformation (37). This again indicates a proto-oncogenic capability of SF1 in human cells.
In summary, we provide genetic evidence showing that deficiency of SF1 suppresses testicular tumorigenesis. SF1, an RNA binding protein (48, 49) necessary for spliceosome assembly of specific pre-mRNAs (24, 25) likely regulates alternative splicing of pre-mRNAs in the testes. Thus, higher Sf1 levels are oncogenic in the testes. Our data leads us to propose that SF1 is a tumor susceptibility factor for germ cell tumorigenesis.
Supplementary Material
Acknowledgements
We thank Gilbert Cote for critical reading of the manuscript; Pierre McCrea and DongMin Gu for invaluable discussions. Funding for these studies were provided by CA75056 to JHN and CA093754 and Texas ARP to AM.
References
- 1.Skakkebaek NE, Berthelsen JG, Giwercman A, Muller J. Carcinoma-in-situ of the testis: possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int J Androl. 1987;10:19–28. doi: 10.1111/j.1365-2605.1987.tb00161.x. [DOI] [PubMed] [Google Scholar]
- 2.Oosterhuis JW, Looijenga LHJ. Testicular germ-cell tumours in a broader perspective. Nature Rev Cancer. 2005;5:210–22. doi: 10.1038/nrc1568. [DOI] [PubMed] [Google Scholar]
- 3.Dieckmann KP, Skakkebaek NE. Carcinoma in situ of the testis: review of biological and clinical features. Int J Cancer. 1999;83:815–22. doi: 10.1002/(sici)1097-0215(19991210)83:6<815::aid-ijc21>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 4.Lutke Holzik MF, Sijmons RH, Sleijfer DT, et al. Syndromic aspects of testicular carcinoma. Cancer. 2003;97:984–92. doi: 10.1002/cncr.11155. [DOI] [PubMed] [Google Scholar]
- 5.Crockford GP, Linger R, Hockley S, et al. Genome-wide linkage screen for testicular germ cell tumour susceptibility loci. Hum Mol Genet. 2006;15:443–51. doi: 10.1093/hmg/ddi459. [DOI] [PubMed] [Google Scholar]
- 6.Linger R, Dudakia D, Huddart R, et al. A physical analysis of the Y chromosome shows no additional deletions, other than Gr/Gr, associated with testicular germ cell tumour. Br J Cancer. 2007;96:357–61. doi: 10.1038/sj.bjc.6603557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanetsky PA, Mitra N, Vardhanabhuti S, et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet. 2009;41:811–5. doi: 10.1038/ng.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rapley EA, Turnbull C, Olama AAA, et al. A genome-wide association study of testicular germ cell tumor. Nat Genet. 2009;41:807–10. doi: 10.1038/ng.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stevens LC. Embryology of testicular teratomas in strain 129 mice. J Natl Cancer Inst. 1959;23:1249–95. [PubMed] [Google Scholar]
- 10.Krentz AD, Murphy MW, Kim S, et al. The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. Proc Natl Acad Sci USA. 2009;106:22323–8. doi: 10.1073/pnas.0905431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Youngren KK, Coveney D, Peng X, et al. The Ter mutation in the dead end gene causes germ cell loss and testicular germ cell tumours. Nature. 2005;435:360–4. doi: 10.1038/nature03595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura T, Suzuki A, Fujita Y, et al. Conditional loss of PTEN leads to testicular teratoma and enhances embryonic germ cell production. Development. 2003;130:1691–700. doi: 10.1242/dev.00392. [DOI] [PubMed] [Google Scholar]
- 13.Heaney JD, Lam M-YJ, Michelson MV, Nadeau JH. Loss of the transmembrane but not the soluble Kit ligand isoform increases testicular germ cell tumor susceptibility in mice. Cancer Res. 2008;68:5193–7. doi: 10.1158/0008-5472.CAN-08-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donehower LA, Harvey M, Vogel H, et al. Effects of genetic background on tumorigenesis in p53-deficient mice. Mol Carcinog. 1995;14:16–22. doi: 10.1002/mc.2940140105. [DOI] [PubMed] [Google Scholar]
- 15.Heaney JD, Michelson MV, Youngren KK, Lam M-YJ, Nadeau JH. Deletion of eIF2beta suppresses testicular cancer incidence and causes recessive lethality in agouti-yellow mice. Hum Mol Genet. 2009;18:1395–404. doi: 10.1093/hmg/ddp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rescorla FJ. Pediatric germ cell tumors. Semin Surg Oncology. 1999;16:144–58. doi: 10.1002/(sici)1098-2388(199903)16:2<144::aid-ssu6>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 17.Weidinger G, Stebler J, Slanchev K, et al. dead end, a novel vertebrate germ plasm component, is required for zebrafish primordial germ cell migration and survival. Curr Biol. 2003;13:1429–34. doi: 10.1016/s0960-9822(03)00537-2. [DOI] [PubMed] [Google Scholar]
- 18.Cook MS, Coveney D, Batchvarov I, Nadeau JH, Capel B. BAX-mediated cell death affects early germ cell loss and incidence of testicular teratomas in Dnd1(Ter/Ter) mice. Dev Biol. 2009;328:377–83. doi: 10.1016/j.ydbio.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noguchi T, Noguchi M. A recessive mutation (ter) causing germ cell deficiency and a high incidence of congenital testicular teratomas in 129/Sv-ter mice. J Natl Cancer Inst. 1985;75:385–92. [PubMed] [Google Scholar]
- 20.Noguchi T, Stevens LC. Primordial germ cell proliferation in fetal testes in mouse strains with high and low incidences of congenital testicular teratomas. J Natl Cancer Inst. 1982;69:907–13. [PubMed] [Google Scholar]
- 21.Matin A, Collin GB, Asada Y, Varnum D, Nadeau JH. Susceptibility to testicular germ-cell tumours in a 129.MOLF-Chr 19 chromosome substitution strain. Nat Genet. 1999;23:237–40. doi: 10.1038/13874. [DOI] [PubMed] [Google Scholar]
- 22.Youngren KK, Nadeau JH, Matin A. Testicular cancer susceptibility in the 129.MOLF-Chr 19 mouse strain: additive effects, gene interactions and epigenetic modifications. Hum Mol Genet. 2003;12:389–98. doi: 10.1093/hmg/ddg036. [DOI] [PubMed] [Google Scholar]
- 23.Zhu R, Ji Y, Xiao L, Matin A. Testicular germ cell tumor susceptibility genes from the consomic 129.MOLF-Chr 19 mouse strain. Mamm Genome. 2007;18:584–95. doi: 10.1007/s00335-007-9036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z, Luyten I, Bottomley MJ, et al. Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science. 2001;294:1098–102. doi: 10.1126/science.1064719. [DOI] [PubMed] [Google Scholar]
- 25.Selenko P, Gregorovic G, Sprangers R, et al. Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol Cell. 2003;11:965–76. doi: 10.1016/s1097-2765(03)00115-1. [DOI] [PubMed] [Google Scholar]
- 26.Das R, Reed R. Resolution of the mammalian E complex and the ATP-dependent spliceosome complexes on native agarose mini-gels. RNA. 1999;5:1504–8. doi: 10.1017/s1355838299991501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michaud S, Reed R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes Dev. 1991;5:2534–46. doi: 10.1101/gad.5.12b.2534. [DOI] [PubMed] [Google Scholar]
- 28.Berglund JA, Chua K, Abovich N, Reed R, Rosbash M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell. 1997;89:781–7. doi: 10.1016/s0092-8674(00)80261-5. [DOI] [PubMed] [Google Scholar]
- 29.Kramer A. Purification of splicing factor SF1, a heat stable protein that functions in the assembly of a presplicing complex. Mol Cell Biol. 1992;12:4545–52. doi: 10.1128/mcb.12.10.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berglund JA, Abovich N, Rosbash M. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev. 1998;12:858–67. doi: 10.1101/gad.12.6.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanackovic G, Kramer A. Human splicing factor SF3a, but not SF1, is essential for pre-mRNA splicing in vivo. Mol Biol Cell. 2005;16:1366–77. doi: 10.1091/mbc.E04-11-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shitashige M, Satow R, Honda K, Ono M, Hirohashi S, Yamada T. Increased susceptibility of Sf1+/- mice to azoxymethane-induced colon tumorigenesis. Cancer Sci. 2007;98:1862–7. doi: 10.1111/j.1349-7006.2007.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagy A, Gertsenstein M, Vintersten K, Behringer R, editors. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press; New York: 2003. pp. 687–9. [Google Scholar]
- 34.Dejosez M, Kreumenacker JS, Zitur LJ, et al. Ronin is essential for embryogenesis and the pluripotency of mouse embryonic stem cells. Cell. 2008;133:1162–74. doi: 10.1016/j.cell.2008.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- 36.Qiu TH, Chandramouli GV, Hunter KW, Alkharouf NW, Green JE, Liu ET. Global expression profiling identifies signatures of tumor virulence in MMTV-PyMT-transgenic mice: correlation to human disease. Cancer Res. 2004;64:5973–81. doi: 10.1158/0008-5472.CAN-04-0242. [DOI] [PubMed] [Google Scholar]
- 37.Shitashige M, Naishiro Y, Idogawa M, et al. Involvement of splicing factor-1 in bcatenin/T-cell factor-4-mediated gene transactivation and pre-mRNA splicing. Gastroenterology. 2007;132:1039–54. doi: 10.1053/j.gastro.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 38.Yeo G, Holste D, Kreiman G, Burge CB. Variation in alternative splicing across human tissues. Genome Biol. 2004;5:R74. doi: 10.1186/gb-2004-5-10-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci. 2006;119:2635–41. doi: 10.1242/jcs.03053. [DOI] [PubMed] [Google Scholar]
- 40.Kim E, Goren A, Ast G. Insights into the connection between cancer and alternative splicing. Trends Genet. 2008;24:7–10. doi: 10.1016/j.tig.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 41.Ritchie W, Granjeaud S, Puthier D, Gautheret D. Entropy measures quantify global splicing disorders in cancer. PLoS Comput Biology. 2008;4:e1000011. doi: 10.1371/journal.pcbi.1000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grosso AR, Martins S, Carmo-Fonseca M. The emerging role of splicing factors in cancer. EMBO reports. 2008;9:1087–93. doi: 10.1038/embor.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zerbe LK, Pino I, Pio R, et al. Relative amounts of antagonistic splicing factors, hnRNP A1 and ASF/SF2, change during neoplastic lung growth: implications for pre-mRNA processing. Mol Carcinog. 2004;41:187–96. doi: 10.1002/mc.20053. [DOI] [PubMed] [Google Scholar]
- 44.He C, Zuo Z, Chen H, et al. Genome-wide detection of testis- and testicular cancer-specific alternative splicing. Carcinogenesis. 2007;28:2484–90. doi: 10.1093/carcin/bgm194. [DOI] [PubMed] [Google Scholar]
- 45.Kempkensteffen C, Hinz S, Krause H, et al. Expression of splicing variants of the inhibitor of apoptosis Livin in testicular germ cell tumors. Tumor Biol. 2008;29:76–82. doi: 10.1159/000135687. [DOI] [PubMed] [Google Scholar]
- 46.Kralovicova J, Houngninou-Molango S, Kramer A, Vorechovsky I. Branch site haplotypes that control alternative splicing. Hum Mol Genet. 2004;24:3189–202. doi: 10.1093/hmg/ddh334. [DOI] [PubMed] [Google Scholar]
- 47.Coulombe-Huntington J, Lam KCL, Dias C, Majewski J. Fine-scale variation and genetic determinants of alternative splicing across individuals. PLoS Genet. 2009;5:e1000766. doi: 10.1371/journal.pgen.1000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toda T, Iida A, Miwa T, Nakamura Y, Imai T. Isolation and characterization of a novel gene encoding nuclear protein at a locus (D11S636) tightly linked to multiple endocrine neoplasia type 1 (MEN1). Hum Mol Genet. 1994;3:465–70. doi: 10.1093/hmg/3.3.465. [DOI] [PubMed] [Google Scholar]
- 49.Wrehlke C, Wiedemeyer WR, Schmitt-Wrede HP, Mincheva A, Lichter P, Wunderlich F. Genomic organization of mouse gene zfp162. DNA Cell Biol. 1999;18:419–28. doi: 10.1089/104454999315303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.