

Abstract
It is a significant challenge to predict RNA secondary structures including pseudoknots. Here, a new algorithm capable of predicting pseudoknots of any topology, ProbKnot, is reported. ProbKnot assembles maximum expected accuracy structures from computed base-pairing probabilities in O(N2) time, where N is the length of the sequence. The performance of ProbKnot was measured by comparing predicted structures with known structures for a large database of RNA sequences with fewer than 700 nucleotides. The percentage of known pairs correctly predicted was 69.3%. Additionally, the percentage of predicted pairs in the known structure was 61.3%. This performance is the highest of four tested algorithms that are capable of pseudoknot prediction. The program is available for download at: http://rna.urmc.rochester.edu/RNAstructure.html.
Keywords: RNA partition function, RNA folding, RNA structure prediction
INTRODUCTION
There is a diverse world of functional RNA sequences. Originally in the central dogma of biology, RNA was considered to play a transient role in expressing inherited information as proteins. It was later discovered that, besides this role in generating proteins, RNA has a variety of other functions, such as regulating gene expression (Tucker and Breaker 2005; Storz and Gottesman 2006; Wu and Belasco 2008), catalyzing reactions (Nissen et al. 2000; Doudna and Cech 2002), and trafficking proteins (Walter and Blobel 1982). RNA sequences that do not code for proteins are referred to as noncoding RNA, or ncRNA (Eddy 2001). Many of these ncRNA sequences have well-defined structures, and to understand how these ncRNA sequences perform their functions it is important to know their structure.
Determination of RNA structure is challenging. Primary structure is an ordered sequence of nucleotides. Secondary structure consists of canonical base pairs, i.e., AU, GC, and GU pairs. Secondary structure prediction involves predicting the base pairs that occur in a specified sequence of nucleotides. RNA tertiary structure is the three-dimensional arrangement of atoms. Because RNA structure is generally hierarchical, the secondary structure can be largely determined without knowing the tertiary structure (Tinoco and Bustamante 1999).
Many secondary structure prediction methods are available. The most accurate method is comparative sequence analysis (Pace et al. 1999), which determines base pairs conserved among homologous sequences. The method is highly accurate (Gutell et al. 2002) but requires a large number of homologous sequences and significant human insight, and thus is limited in use. When a single sequence is available, the most popular approach for structure prediction is to predict the lowest free energy structure with a dynamic programming algorithm (Zuker 2003; Mathews et al. 2004; Mathews and Turner 2006; Gruber et al. 2008).
A more recent approach to predict RNA secondary structures is called maximum expected accuracy structure prediction (Knudsen and Hein 2003; Do et al. 2006; Hamada et al. 2009; Lu et al. 2009). Roughly, maximum expected accuracy structures are structures composed of pairs that provide the maximal sum of pairing probabilities. The pairing probabilities can be derived by machine learning methods or by thermodynamic methods using partition functions. Maximum expected accuracy structures have improved accuracy compared with free energy minimization because it has been observed that highly probable base pairs are more likely to be correctly predicted pairs (Mathews 2004).
One important topology for RNA secondary structures is a pseudoknot. This is a type of secondary structure that contains nonnested base pairs. Specifically, a pseudoknot is defined by at least two base pairs, i–j and i′–j′, such that nucleotide i appears before i′, i′ before j, and j before j′ in the sequence. Base pairs in pseudoknots represent a small fraction of base pairs in known RNA secondary structures, but pseudoknots occur in a number of functional RNA sequences (van Batenburg et al. 2001; Condon and Jabbari 2009).
The prediction of secondary structures including pseudoknots is a difficult task. For example, the most popular dynamic programming algorithms for finding low free energy structures do not allow pseudoknots. This allows those dynamic programming algorithms to run quickly and scale well, i.e., O(N3) in time where N is the length of the sequence. Including pseudoknots in the structure prediction requires higher-order scaling, the use of heuristics, and/or a compromise on the energy model.
It has been proven that the prediction of lowest free energy secondary structures with pseudoknots is NP-hard (Lyngsø and Pederson 2000). In spite of this, a number of innovative and practical approaches have been developed to predict structures with pseudoknots. These approaches can roughly be summarized in six categories. One approach is to use a dynamic programming algorithm to predict structures with a limited topology (Rivas and Eddy 1999; Uemura et al. 1999; Akutsu 2000; Dirks and Pierce 2003; Reeder and Giegerich 2004). A classification of topologies and an explanation of topologies handled by several dynamic programming algorithms are available (Condon et al. 2004). A second approach to predicting pseudoknots is to construct structures using multiple iterations of algorithms that would otherwise not be capable of predicting pseudoknots (Ruan et al. 2004; Ren et al. 2005; Jabbari et al. 2008). One of these algorithms is also capable of using an alignment of multiple homologous sequences to improve its accuracy by finding a consensus structure (Ruan et al. 2004). A third approach is to either simulate a folding pathway or sample structures with a stepwise addition of helices (Abrahams et al. 1990; Gultyaev et al. 1995; Isambert and Siggia 2000; Dawson et al. 2007; Meyer and Miklos 2007). A fourth approach uses the maximum weight matching algorithm to construct structures composed of pairs that give a maximum score (Tabaska et al. 1998; Witwer et al. 2004). These algorithms use alignments of multiple homologous sequences and scoring functions that summarize free energies associated with pairs and covariation of pairs. Recently, a sixth approach, using constrained integer programming has also been applied to finding lowest free energy structures (Poolsap et al. 2009).
Many of the above algorithms use rules for predicting the free energy change—i.e., stability—of pseudoknots. Significant progress has been reported in this area as well using several approaches. Two sets of empirical rules were designed for use with dynamic programming algorithms (Rivas and Eddy 1999; Dirks and Pierce 2003). A set of parameters was developed using polymer theory and calibrated to experimentally measured stabilities (Wyatt et al. 1990; Nixon and Giedroc 1998; Theimer et al. 1998; Gultyaev et al. 1999; Theimer and Giedroc 1999, 2000). Another set of parameters was developed using lattice models and self-avoidant walks (Cao and Chen 2006, 2009). Additionally, a set of parameters was developed using polymer theory (Aalberts and Hodas 2005). A recent report provides a technique for refining parameters for predicting pseudoknot stability that utilizes experimental data and the database of sequences with known structure (Andronescu et al. 2010).
This contribution reports ProbKnot, a new RNA secondary structure prediction algorithm that is capable of predicting RNA secondary structures of any topology in O(N3) time. Base-pair probabilities are first predicted using a partition function (Mathews 2004), which does not include pseudoknotted structures, in O(N3) time (Xia et al. 1998; Mathews et al. 2004). ProbKnot then assembles a type of maximum expected accuracy structure in O(N2) time from the base-pairing probabilities, but does so without using a dynamic programming algorithm. By assembling structures from base-pair probabilities determined without pseudoknots, ProbKnot does not require a set of rules for predicting the stability of pseudoknots.
The performance of ProbKnot was benchmarked against other freely available programs that predict pseudoknots: pknotsRG-mfe (Reeder and Giegerich 2004), ILM (Ruan et al. 2004), and HotKnots (Ren et al. 2005); and programs that do not predict pseudoknots: MaxExpect, a maximum expected accuracy approach (Lu et al. 2009) and Free Energy Minimization (Mathews et al. 2004). ProbKnot was able to predict the largest fraction of known base pairs.
RESULTS
ProbKnot
ProbKnot is an algorithm that predicts RNA secondary structure by finding the structure with the most probable base pairs. It assembles structures composed of base pairs, i–j, where the probability of the i–j pair is higher than any i–k or j–k base pair, where k is any other nucleotide in the sequence. This is performed in O(N2) time by first calculating and storing the pairing probability of the most probable pair for each nucleotide, Pmax(i). Then each base pair is considered for inclusion in the structure. If the probability of the i–j pair is equal to both Pmax(i) and Pmax(j), that pair is included in the structure. The algorithm is additionally capable of supporting multiple iterations in a similar manner by finding the most probable i–j pair for nucleotides that remained unpaired after previous iterations. For benchmarks shown here, only a single iteration was performed.
As a post-processing step, after the structure is obtained, the algorithm removes helices composed of two or fewer stacked base pairs. For these calculations, single nucleotide bulges are considered stacked and therefore do not interrupt helical stacking. So, for example, two pairs separated by a single bulged nucleotide would be considered stacked.
Structure prediction accuracy
The accuracy of ProbKnot was evaluated by predicting structures for sequences with known structure as determined by comparative sequence analysis. Both sensitivity and positive predictive value (PPV) were determined. Sensitivity is the percent of known pairs correctly predicted and PPV is the percent of predicted pairs in the known structure.
For a diverse set of sequences with known secondary structure, ProbKnot had an average of 69.3% sensitivity (Table 1). The performance was compared against three other programs that are capable of pseudoknot prediction and freely available for download. These programs were demonstrated to be among the top performers in structure prediction accuracy in a previous benchmark (Ren et al. 2005). The programs are ILM version 1.0 (Ruan et al. 2004), HotKnots version 1.2 (Ren et al. 2005), and pknotsRG version 1.3 (Reeder and Giegerich 2004). Each was run using default parameters. Additionally, the performance was compared against two other algorithms from RNAstructure, which predicts structures without pseudoknots, free energy minimization (Mathews et al. 2004), and maximum expected accuracy structure prediction (Lu et al. 2009). Overall, ProbKnot had the highest average sensitivity for all methods and the highest PPV among methods that are capable of predicting pseudoknots.
TABLE 1.
Sensitivities of prediction methods

ProbKnot had an average PPV of 61.3% (Table 2), performing best in six out of 10 RNA families including two families with pseudoknots, and performing on the same level with pknotsRG-mfe on the group I intron family that is also known to have pseudoknots. This was the best performance among algorithms that predict pseudoknots, but not as high as MaxExpect, which does not predict pseudoknots. This is consistent with previous observations. Algorithms that predict pseudoknots consider a larger space of possible structures, which leads to a tendency for lower fidelity of structure prediction.
TABLE 2.
Positive predictive value of prediction methods

Pseudoknot prediction
The accuracy of pseudoknot prediction was evaluated separately. First, the number of predicted pseudoknotted pairs was tabulated (Table 3). These pairs were found using the method of Smit et al. (2008) to identify the fewest pairs that need to be removed to remove the pseudoknots from a structure. The number of pseudoknotted pairs reported in Table 3 is the sum of the number of pairs that are removed to remove the pseudoknot. Then, the number of these predicted pseudoknotted pairs that are both in the known structure and pseudoknotted in the known structure was determined and reported as the number of correctly predicted pseudoknotted pairs (Table 3).
TABLE 3.
Base-pair statistics: Evaluation of methods in terms of predicted pseudoknotted base pairs

The accuracy of pseudoknot prediction for structures was also tabulated (Table 4). The number of predicted structures with pseudoknotted pairs was determined. The number of the predicted structures with pseudoknots that were correct was then tabulated as correctly predicted pseudoknotted structures. A predicted pseudoknotted structure was considered correct if it contained at least one correctly predicted pseudoknotted pair. For structures with multiple pseudoknots, such as tmRNA sequences, a structure can be considered correctly predicted if only one pseudoknot is correctly predicted.
TABLE 4.
Pseudoknot prediction statistics on structures

ILM has the highest number of correctly predicted pseudoknotted structures and the highest number of correctly predicted pseudoknotted base pairs. Of the predicted pseudoknotted pairs, pknotsRG-mfe has the highest portion of correctly predicted pairs. All algorithms, however, correctly predict only a small fraction of the pseudoknotted base pairs that are in the known structure.
Structure prediction example
Figure 1 shows an example of predicted structure, the Tetrahymena thermophila group I intron structure predicted by ProbKnot. Thick lines between the base pairs represent correctly predicted pairs. As can be seen from Figure 1, ProbKnot correctly predicts almost all base pairs with probabilities >70%. Helixes S1 and S2 that form pseudoknots in the structure are correctly predicted by ProbKnot.
FIGURE 1.

Predicted secondary structure of group I intron from T. thermophila by ProbKnot. Thick lines represent correctly predicted base pairs; thin lines represent incorrectly predicted base pairs. The boxed helices, labeled S1 and S2, are the two helices that define the pseudoknot.
Time benchmarks
Time trials were performed on sequences ranging in length from 77 to 2904 nucleotides (nt) (Table 5). On the longest sequence, ProbKnot showed the second best time performance, requiring 63 min of runtime to predict both the base-pair probabilities and to assemble the predicted structure. ILM had the best time performance and the dynamic programming algorithm (pknotsRG-mfe) had the slowest time performance.
TABLE 5.
Comparison of time performances of different algorithms

DISCUSSION
ProbKnot assembles maximum expected accuracy structures using base-pairing probabilities determined from a partition function calculation. Previous approaches for predicting maximum expected accuracy structures used dynamic programming algorithms that do not allow pseudoknots (Do et al. 2006; Hamada et al. 2009; Lu et al. 2009), but ProbKnot is not limited in the topology of structures it can predict. Although the partition function algorithm does not account for pseudoknotted structures, each of the helices in the pseudoknot can occur in different structures (Mathews 2004). ProbKnot takes advantage of this fact to assemble both helices into a single structure.
ProbKnot has some similarities with the maximum weight matching (MWM) methods previously explored to find secondary structures conserved among multiple sequences (Tabaska et al. 1998; Hofacker et al. 2004). The MWM algorithm takes pairing weights as input, where weights are a function of folding free energy change and covariation, and outputs a structure with the greatest sum of these weights. MWM runs in O(N3) time and is also not limited in topology. It has been noted that MWM methods tend to have poor PPV because the structures are saturated with pairs, but post-processing can remove pairs and improve performance. ProbKnot is distinct because it uses pair probabilities and not folding free energy changes as input. Additionally, the requirement that the pairs included in the structure be the highest pairing probability for pairs possible by either nucleotide provides a stopping rule so that structures are not oversaturated with pairs.
Based on the benchmarks in Tables 1 and 2, ProbKnot has the highest average accuracy for RNA secondary structure prediction among algorithms that predict pseudoknots. It performs on average 2%–4% better in sensitivity and 2%–3% better in PPV. These improvements are considerable, but they leave room for improvement. For example, the average performance for structure prediction on tmRNA, with four pseudoknots, is only 47.2% in sensitivity.
The performance results for ProbKnot were also compared with the performance of two algorithms, MaxExpect (Lu et al. 2009) and free energy minimization (Mathews et al. 2004), which are unable to predict pseudoknots. This comparison was performed to evaluate the benefit for increasing the range of topologies predicted to include pseudoknots. In sensitivity, ProbKnot outperformed both algorithms by ∼0.5%–1%. This was expected because ProbKnot has a wider predicting range of possible topologies, and thus it should predict more correct base pairs than other algorithms. Because of the wider range of possible prediction topologies, however, there is wider latitude for incorrectly predicting base pairs and, because of this, PPV decreases compared with MaxExpect.
Given the poor performance of the methods benchmarked here on tmRNA and telomerase RNA, including ProbKnot, there is a need for continued research in predicting pseudoknotted structures. One possible approach for improving ProbKnot is to use a partition function that explicitly includes pseudoknots to predict the base-pairing probabilities. For example, the algorithm reported by Dirks and Pierce is O(N4) in time and includes a restricted set of pseudoknots (Dirks and Pierce 2003, 2004; Condon et al. 2004). These pair probabilities could be used by ProbKnot to assemble structures of any topology and may yield more accurate structures.
ProbKnot is available in the RNAstructure package (Reuter and Mathews 2010). This includes the source code in C++; text interfaces for Linux, Unix, and Windows; a JAVA graphical interface for Linux and Mac OS-X; and a graphical interface for Microsoft Windows.
MATERIALS AND METHODS
Prediction of base-pairing probabilities
Base-pair probabilities were predicted using a partition function algorithm that includes coaxial stacking (Mathews 2004). This program uses the thermodynamic parameters assembled by Xia et al. (1998) and Mathews et al. (2004) to predict the stabilities of secondary structures. Similar to Lu et al. (2009), the multibranch loop parameter bonus for each branching helix was not optimized as done by Mathews et al. (2004) but was kept at −0.6 kcal/mol, the value suggested by optical melting experiments (Diamond et al. 2001; Mathews and Turner 2002).
Accuracy
All algorithms were tested on 1550 RNA sequences from 10 different families: small subunit rRNA (Gutell 1994), large subunit rRNA (Gutell et al. 1993; Schnare et al. 1996), 5S rRNA (Szymanski et al. 1998), group I intron (Waring and Davies 1984; Damberger and Gutell 1994), group II intron (Michel et al. 1989), RNase P RNA (Brown 1998), SRP RNA (Larsen et al. 1998), tRNA (Sprinzl et al. 1998), tmRNA (Zwieb et al. 1999), and telomerase RNA (Chen et al. 2000). This database is an expansion of a database of structures assembled previously (Mathews et al. 1999) to include the telomerase RNA and the tmRNA, which are pseudoknotted RNA structures. Vertebrate telomerase RNA secondary structure alignments were obtained from the Rfam 9.1 database (Griffiths-Jones et al. 2003, 2005; Daub et al. 2008; Gardner et al. 2009). tmRNA secondary structures were obtained from the tmRDB database (Zwieb et al. 2003). Structures with unknown nucleotides were omitted from the full list of structures in the tmRDB database. Small and large subunit rRNA sequences were divided into domains of ≤700 nt as previously reported (Mathews et al. 1999).
The performance of secondary structure prediction algorithms was evaluated by calculating sensitivity and PPV. Sensitivity measures the percent of known base pairs correctly predicted:
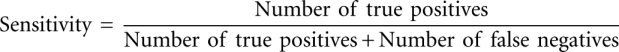 |
PPV measures percent of predicted base pairs that are correctly predicted:
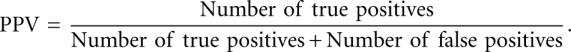 |
Both sensitivity and PPV were evaluated with an allowance for incomplete knowledge of the exact pairing in the known structure. A predicted base pair between nucleotides i and j was considered correctly predicted if i was paired to j, j − 1, or j + 1, or if j was paired to i − 1 or i + 1 (Mathews et al. 1999). Average values were calculated per RNA family and then overall averages were calculated as the mean of the values reported for each family.
Tabulation of pseudoknot content
The number of base pairs in pseudoknots was counted using an implementation of the optimization approach of Smit et al. (2008) as implemented in the RNA class component of RNAstructure (Reuter and Mathews 2010). In this implementation, the scoring function is pairs, so the algorithm counts the fewest number of pairs that would need to be removed to remove the pseudoknot.
ACKNOWLEDGMENTS
We thank A.O. Harmanci, T.D. Romo, G. Sharma, and R. Tyagi for discussions. This work was supported by National Institutes of Health grant no. R01GM076485 to D.H.M. Computer time was provided by the University of Rochester Center for Research Computing.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2125310.
REFERENCES
- Aalberts DP, Hodas NO 2005. Asymmetry in RNA pseudoknots: Observation and theory. Nucleic Acids Res 33: 2210–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams JP, van den Berg M, van Batenburg E, Pleij C 1990. Prediction of RNA secondary structure, including pseudoknotting, by computer simulation. Nucleic Acids Res 18: 3035–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akutsu T 2000. Dynamic programming algorithms for RNA secondary structure prediction with pseudoknots. Discrete Appl Math 104: 45–62 [Google Scholar]
- Andronescu MS, Pop C, Condon AE 2010. Improved free energy parameters for RNA pseudoknotted secondary structure prediction. RNA 16: 26–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JW 1998. The ribonuclease P database. Nucleic Acids Res 26: 351–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen SJ 2006. Predicting RNA pseudoknot folding thermodynamics. Nucleic Acids Res 34: 2634–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen SJ 2009. Predicting structures and stabilities for H-type pseudoknots with interhelix loops. RNA 15: 696–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Blasco MA, Greider CW 2000. Secondary structure of vertebrate telomerase RNA. Cell 100: 503–514 [DOI] [PubMed] [Google Scholar]
- Condon A, Jabbari H 2009. Computational prediction of nucleic acid secondary structure: Methods, applications, and challenges. Theor Comput Sci 410: 294–301 [Google Scholar]
- Condon A, Davy B, Rastegari B, Tarrant F, Zhao S 2004. Classifying RNA pseudoknotted structures. Theor Comput Sci 320: 35–50 [Google Scholar]
- Damberger SH, Gutell RR 1994. A comparative database of group I intron structures. Nucleic Acids Res 22: 3508–3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub J, Gardner PP, Tate J, Ramsköld D, Manske M, Scott WG, Weinberg Z, Griffiths-Jones S, Bateman A 2008. The RNA WikiProject: Community annotation of RNA families. RNA 14: 2462–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson WK, Fujiwara K, Kawai G 2007. Prediction of RNA pseudoknots using heuristic modeling with mapping and sequential folding. PLoS ONE 2: e905 doi: 10.1371/journal.pone.0000905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JM, Turner DH, Mathews DH 2001. Thermodynamics of three-way multibranch loops in RNA. Biochemistry 40: 6971–6981 [DOI] [PubMed] [Google Scholar]
- Dirks RM, Pierce NA 2003. A partition function algorithm for nucleic acid secondary structure including pseudoknots. J Comput Chem 24: 1664–1677 [DOI] [PubMed] [Google Scholar]
- Dirks RM, Pierce NA 2004. An algorithm for computing nucleic acid base-pairing probabilities including pseudoknots. J Comput Chem 25: 1295–1304 [DOI] [PubMed] [Google Scholar]
- Do CB, Woods DA, Batzoglou S 2006. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 22: e90–e98 [DOI] [PubMed] [Google Scholar]
- Doudna JA, Cech TR 2002. The chemical repertoire of natural ribozymes. Nature 418: 222–228 [DOI] [PubMed] [Google Scholar]
- Eddy SR 2001. Noncoding RNA genes and the modern RNA world. Natl Rev 2: 919–929 [DOI] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, et al. 2009. Rfam: Updates to the RNA families database. Nucleic Acids Res 37: D136–D140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR 2003. Rfam: An RNA family database. Nucleic Acids Res 31: 439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A 2005. Rfam: Annotating noncoding RNAs in complete genomes. Nucleic Acids Res 33: D121–D124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL 2008. The Vienna RNA websuite. Nucleic Acids Res 36: W70–W74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gultyaev AP, van Batenburg FHD, Pleij CWA 1995. The computer simulation of RNA folding pathways using a genetic algorithm. J Mol Biol 250: 37–51 [DOI] [PubMed] [Google Scholar]
- Gultyaev AP, van Batenburg FHD, Pleij CWA 1999. An approximation of loop free energy values of RNA H-pseudoknots. RNA 5: 609–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR 1994. Collection of small subunit (16S- and 16S-like) ribosomal RNA structures. Nucleic Acids Res 22: 3502–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR, Gray MW, Schnare MN 1993. A compilation of large subunit (23S- and 23S-like) ribosomal RNA structures. Nucleic Acids Res 21: 3055–3074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutell RR, Lee JC, Cannone JJ 2002. The accuracy of ribosomal RNA comparative structure models. Curr Opin Struct Biol 12: 301–310 [DOI] [PubMed] [Google Scholar]
- Hamada M, Kiryu H, Sato K, Mituyama T, Asai K 2009. Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics 25: 465–473 [DOI] [PubMed] [Google Scholar]
- Hofacker IL, Priwitzer B, Stadler PF 2004. Prediction of locally stable RNA secondary structures for genome-wide surveys. Bioinformatics 20: 186–190 [DOI] [PubMed] [Google Scholar]
- Isambert H, Siggia ED 2000. Modeling RNA folding paths with pseudoknots: Application to hepatitis delta virus ribozyme. Proc Natl Acad Sci 97: 6515–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbari H, Condon A, Zhao S 2008. Novel and efficient RNA secondary structure prediction using hierarchical folding. J Comput Biol 15: 139–163 [DOI] [PubMed] [Google Scholar]
- Knudsen B, Hein J 2003. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res 31: 3423–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen N, Samuelsson T, Zwieb C 1998. The signal recognition particle database (SRPDB). Nucleic Acids Res 26: 177–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZJ, Gloor JW, Mathews DH 2009. Improved RNA secondary structure prediction by maximizing expected pair accuracy. RNA 15: 1805–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyngsø RB, Pederson CN 2000. RNA pseudoknot prediction in energy-based models. J Comput Biol 7: 409–427 [DOI] [PubMed] [Google Scholar]
- Mathews DH 2004. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA 10: 1178–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Turner DH 2002. Experimentally derived nearest neighbor parameters for the stability of RNA three- and four-way multibranch loops. Biochemistry 41: 869–880 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Turner DH 2006. Prediction of RNA secondary structure by free energy minimization. Curr Opin Struct Biol 16: 270–278 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH 1999. Expanded sequence dependence of thermodynamic parameters provides improved prediction of RNA secondary structure. J Mol Biol 288: 911–940 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH 2004. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci 101: 7287–7292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer IM, Miklos I 2007. SimulFold: Simultaneously inferring RNA structures including pseudoknots, alignments, and trees using a Bayesian MCMC framework. PLoS Comput Biol 3: e149 doi: 10.1371/journal.pcbi.0030149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Umesono K, Ozeki H 1989. Comparative and functional anatomy of group II catalytic introns—a review. Gene 82: 5–30 [DOI] [PubMed] [Google Scholar]
- Nissen P, Hansen J, Ban N, Moore PB, Steitz TA 2000. The structural basis of ribosomal activity in peptide bond synthesis. Science 289: 920–930 [DOI] [PubMed] [Google Scholar]
- Nixon PL, Giedroc DP 1998. Equilibrium unfolding (folding) pathway of a model H-type pseudoknotted RNA: The role of magnesium ions in stability. Biochemistry 37: 16116–16129 [DOI] [PubMed] [Google Scholar]
- Pace NR, Thomas BC, Woese CR 1999. Probing RNA structure, function, and history by comparative analysis. In The RNA world, 2nd ed (ed. Gesteland RF et al. ), pp.113–141 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Poolsap U, Kato Y, Akutsu T 2009. Prediction of RNA secondary structure with pseudoknots using integer programming. BMC Bioinformatics Suppl 110: S38 doi: 10.1186/1471-2105-10-S1-S38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder J, Giegerich R 2004. Design, implementation, and evaluation of a practical pseudoknot folding algorithm based on thermodynamics. BMC Bioinformatics 5: 104 doi: 10.1186/1471-2105-5-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Rastegari B, Condon A, Hoos HH 2005. HotKnots: Heuristic prediction of RNA secondary structures including pseudoknots. RNA 11: 1494–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS, Mathews DH 2010. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11: 129 doi: 10.1186/1471-2105-11-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas E, Eddy SR 1999. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J Mol Biol 285: 2053–2068 [DOI] [PubMed] [Google Scholar]
- Ruan J, Stormo GD, Zhang W 2004. An iterated loop matching approach to the prediction of RNA secondary structures with pseudoknots. Bioinformatics 20: 58–66 [DOI] [PubMed] [Google Scholar]
- Schnare MN, Damberger SH, Gray MW, Gutell RR 1996. Comprehensive comparison of structural characteristics in Eukaryotic cytoplasmic large subunit (23S-like) ribosomal RNA. J Mol Biol 256: 701–719 [DOI] [PubMed] [Google Scholar]
- Smit S, Rother K, Heringa J, Knight R 2008. From knotted to nested RNA structures: A variety of computational methods for pseudoknot removal. RNA 14: 410–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S 1998. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 26: 148–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Gottesman S 2006. Versatile roles of small RNA regulators in bacteria. In The RNA world, 3rd ed (ed. Gesteland RF et al. ), pp.567–594 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Szymanski M, Specht T, Barciszewska MZ, Barciszewski J, Erdmann VA 1998. 5S rRNA data bank. Nucleic Acids Res 26: 156–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabaska JE, Cary RB, Gabow HN, Stormo GD 1998. An RNA folding method capable of identifying pseudoknots and base triples. Bioinformatics 14: 691–699 [DOI] [PubMed] [Google Scholar]
- Theimer CA, Giedroc DP 1999. Equilibrium unfolding pathway of an H-Type RNA pseudoknot which promotes programmed −1 ribosomal frameshifting. J Mol Biol 289: 1283–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theimer CA, Giedroc DP 2000. Contribution of the intercalated adenosine at the helical junction to the stability of the gag-pro frameshifting pseudoknot from mouse mammary tumor virus. RNA 6: 409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theimer CA, Wang Y, Hoffman DW, Krisch HM, Giedroc DP 1998. Non-nearest neighbor effects on the thermodynamics of unfolding of a model mRNA pseudoknot. J Mol Biol 279: 545–564 [DOI] [PubMed] [Google Scholar]
- Tinoco I Jr, Bustamante C 1999. How RNA folds. J Mol Biol 293: 271–281 [DOI] [PubMed] [Google Scholar]
- Tucker BJ, Breaker RR 2005. Riboswitches as versatile gene control elements. Curr Opin Struct Biol 15: 342–348 [DOI] [PubMed] [Google Scholar]
- Uemura Y, Hasegawa A, Kobayashi S, Yokomori T 1999. Tree joining grammars for RNA structure prediction. Theor Comput Sci 210: 1330–1348 [Google Scholar]
- van Batenburg FHD, Gultyaev AP, Pleij CWA 2001. PseudoBase: Structural information on RNA pseudoknots. Nucleic Acids Res 29: 194–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Blobel G 1982. Signal recognition particle contains a 7S RNA essential for protein translocation across the endoplasmic reticulum. Nature 299: 691–698 [DOI] [PubMed] [Google Scholar]
- Waring RB, Davies RW 1984. Assessment of a model for intron RNA secondary structure relevant to RNA self-splicing—a review. Gene 28: 277–291 [DOI] [PubMed] [Google Scholar]
- Witwer C, Hofacker IL, Stadler PF 2004. Prediction of consensus RNA secondary structures including pseudoknots. IEEE/ACM Trans Comput Biol Bioinformatics 1: 66–77 [DOI] [PubMed] [Google Scholar]
- Wu L, Belasco JG 2008. Let me count the ways: Mechanisms of gene regulation by miRNAs and siRNAs. Mol Cell 29: 1–7 [DOI] [PubMed] [Google Scholar]
- Wyatt JR, Puglisi JD, Tinoco I Jr 1990. RNA pseudoknots, stability and loop size requirements. J Mol Biol 214: 455–470 [DOI] [PubMed] [Google Scholar]
- Xia T, SantaLucia J Jr, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, Cox C, Turner DH 1998. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson–Crick pairs. Biochemistry 37: 14719–14735 [DOI] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwieb C, Wower I, Wower J 1999. Comparative sequence analysis of tmRNA. Nucleic Acids Res 27: 2063–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwieb C, Gorodkin J, Knudsen B, Burks J, Wower J 2003. tmRDB (tmRNA database). Nucleic Acids Res 31: 446–447 [DOI] [PMC free article] [PubMed] [Google Scholar]