

Summary
The cyclin-dependent kinase inhibitor p16INK4a (CDKN2A) is an important tumor-suppressor gene frequently inactivated in human tumors. p16 suppresses the development of cancer by triggering an irreversible arrest of cell proliferation termed cellular senescence. Here, we describe another anti-oncogenic function of p16 in addition to its ability to halt cell cycle progression. We show that transient expression of p16 stably represses the hTERT gene, encoding the catalytic subunit of telomerase, in both normal and malignant breast epithelial cells. Short-term p16 expression increases the amount of histone H3 trimethylated on lysine 27 (H3K27) bound to the hTERT promoter, resulting in transcriptional silencing, likely mediated by polycomb complexes. Our results indicate that transient p16 exposure may prevent malignant progression in dividing cells by irreversible repression of genes, such as hTERT, whose activity is necessary for extensive self-renewal.
Keywords: INK4a, hTERT, histone methylation, heterochromatin, senescence
Introduction
The cyclin-dependent kinase (CDK) inhibitor p16INK4a is deleted in ~20% (Sharpless 2005) and inactivated epigenetically in an additional 20% (Tlsty et al. 2004) of human breast tumors. The binding of p16 to CDKs 4 and 6 induces conformational changes that disrupt the interaction of these kinases with D-type cyclins (Pavletich 1999), thus antagonizing activation of the CDKs. Through inactivation of CDKs 4 and 6, p16 prevents phosphorylation and inactivation of the pRb family of cell cycle regulators. This canonical description, while valid, obscures the p16 function(s) that result in irreversible senescence-associated changes rather than reversible cell cycle arrest. Using U2OS cells in which the transcription of p16 was regulated by tetracycline, Dai and Enders (Dai & Enders 2000) found that induction of p16 for one day arrested most cells in the G1 phase of the cell cycle. If the inducer was then removed, p16 levels returned to baseline and growth resumed within 3-5 days. If, however, p16 was induced for 6 days, DNA synthesis remained strongly inhibited and the cells acquired morphological features of senescence. These results demonstrated that sustained p16 expression is sufficient to impose a stable block to cell proliferation and that this state becomes independent of p16 expression and hypophosphorylation of pRb.
The stable effects of p16 may be mediated by chromatin changes. Recent studies have indicated that p16 can play an active role in the formation of senescence-induced heterochromatic foci (SAHF) in human cells. Originally characterized in senescent fibroblasts (Narita et al. 2003), these foci consist of reorganized DNA and are enriched for proteins normally associated with heterochromatin. The formation of SAHF is slow, taking several days or weeks, depending on the initiating stimulus, and has been reported to coincide with the enhanced association of E2F-target promoters with heterochromatin proteins. Senescent fibroblasts which poorly express p16 and are less stably arrested (Beausejour et al. 2003), display fewer SAHFs than those that express higher amounts of p16 (Narita et al. 2006).
In addition to senescence, stable effects of p16 may play a role in differentiation. Experiments with transgenic mice have demonstrated that p16 deficiency results in an increased number of hematopoietic stem cells, and is associated with more active cycling and decreased apoptosis of stem cells under stressful conditions (Janzen et al. 2006). p16 expression has also been linked to decreased self-renewal in neural stem cells (Molofsky et al. 2006). Increased p16 expression during stem cell differentiation is often associated with decreased levels of Bmi-1, a polycomb protein required for self-renewal of stem cells (Molofsky et al. 2003; Liu et al. 2006). Conversely, suppression of p16, either by expression of Bmi-1 or directly by shRNA or anti-sense methods, is sufficient to immortalize several types of human epithelial cells (Maurelli et al. 2006; Song et al. 2006), and to extend the life span of human fibroblasts (Itahana et al. 2003). This may be due, in part, to relaxation of p16-mediated repression of genes, such as hTERT – encoding the catalytic subunit of telomerase, necessary for extended or indefinite proliferation.
Results
p16 suppresses hTERT in human mammary epithelial cells (HMEC)
p16 increases progressively in adherent HMEC cultures established from non-malignant tissues in serum-free medium, resulting in p16-dependent senescence after 10 - 20 population doublings (Fig. 1A,B) (Yaswen & Stampfer 2002). Using sensitive quantitative PCR-based techniques, we detected significant levels of both telomerase activity and hTERT mRNA in early passage HMEC (Fig. 1C,D) in agreement with published reports demonstrating significant telomerase activity and hTERT mRNA in normal breast cells both in vitro (Belair et al. 1997) and in vivo (Hines et al. 2005). However, telomerase activity and hTERT mRNA levels decreased rapidly with each passage in culture, reaching negligible levels within 3 passages. At this stage, most cells in the cultures continued to proliferate, although, as previously reported (Yaswen & Stampfer 2002), cell proliferation ceased several population doublings later at passage 5.
Fig. 1.
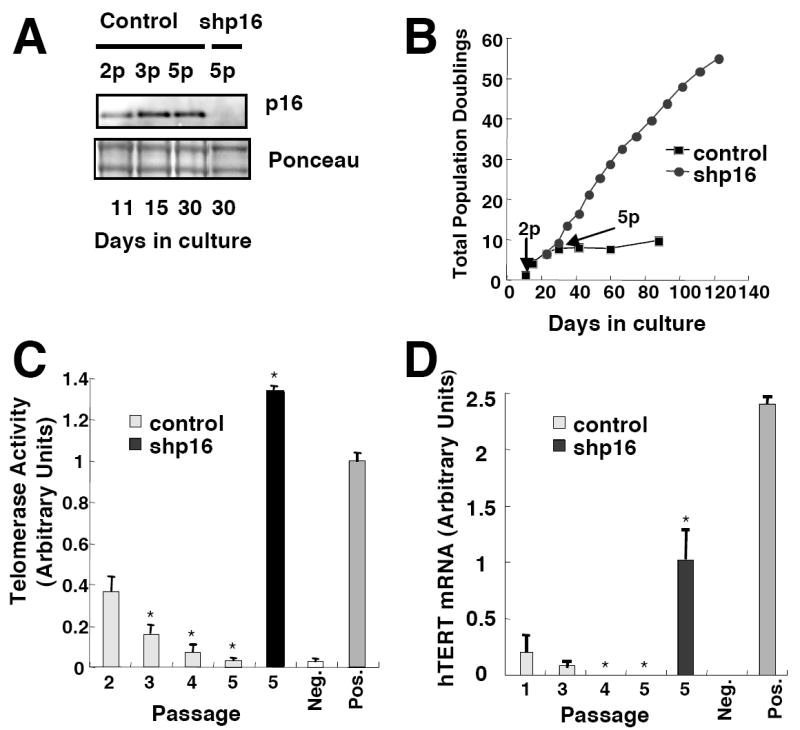
p16INK4a is responsible for telomerase repression in proliferating HMEC. (A) Control and corresponding cultures expressing p16shRNA were harvested at indicated passages (p). Total p16 levels were determined by immunoblotting. (B) Total population doublings of control HMEC or HMEC expressing shp16 are plotted versus time. Results shown are representative of 4 non-malignant specimens. (C) Telomerase activity was assayed at indicated passages. Mean values and SD (n=3) are representative of two non-malignant specimens. Values significantly different from telomerase activity at passage 2 (p < 0.05, Student’s t-test) are denoted with a *. (D) hTERT mRNA levels were assayed at indicated passages by Taqman PCR. Mean values and SD (n=6) are representative of two non-malignant specimens and presented relative to each other after normalization using mRNA levels of TATA binding protein (TBP). Values significantly different from hTERT mRNA levels at passage 1 (p < 0.05, Student’s t-test) are denoted with a *. Values obtained for normal human fibroblasts (WI38) and malignant human breast cancer cells (MCF7) are presented as negative and positive controls.
To understand the inverse relationship between p16 and hTERT expression, we silenced p16 expression by RNA interference. We infected HMEC with lentiviruses encoding p16 shRNAs (Beausejour et al. 2003), and achieved significant suppression of p16 expression. As expected, cultures in which p16 expression was suppressed bypassed the p16-mediated arrest of cell proliferation, and continued to proliferate exponentially for more than 120 days. Strikingly, cultures with silenced p16 retained significant telomerase activity and hTERT mRNA, suggesting that p16 is responsible for the passage-dependent decline in telomerase expression. We verified that this effect of p16 was mediated through pRb family proteins by employing a retrovirus containing shRNAs directed against pRb, p107, and p130 (Fig. 2). Although compensatory mechanisms (Zhu et al. 1995) prevented repression of p107 mRNA levels, pRb and p130 mRNAs and protein were significantly reduced using this construct (Fig 2 A,B), while hTERT mRNA levels and proliferation were maintained (Fig 2 C,D).
Fig. 2.

Telomerase repression in HMEC is mediated by pRb-family proteins. (A) Relative levels of pRb, p107, and p130 mRNAs in HMEC stably transduced at passage 2 with a control vector or a vector encoding shRNAs against these three transcripts were determined by quantitative RT-PCR. Mean values and SD (n=3) are shown. Values significantly different from controls (p < 0.05, Student’s t-test) are denoted with a *. (B) HMEC expressing shRb/107/130, shp16, or vector controls were harvested and pRb, p107, p130 and p16 levels were determined by immunoblotting. (C) Total population doublings of HMEC expressing shRB/107/130, shp16, or control viruses are plotted versus time. (D) hTERT mRNA levels were assayed at passage 5 by Taqman PCR. Mean mRNA levels ± SD (n=3) are presented relative to each other after normalization using mRNA levels of TATA binding protein (TBP).
Transient expression of p16 leads to stable suppression of telomerase
Because telomerase is a cell cycle regulated enzyme (Belair et al. 1997), the decline in telomerase expression could be a direct consequence of p16 expression or an indirect consequence of p16-induced cell cycle arrest. This ambiguity also pertains to reports of suppressed telomerase activity after enforced expression of p16 in cancer cell lines, such as MCF7, in which endogenous p16 genes are mutated or non-functional, but the rest of the pRb pathway is intact (Duan et al. 2004; Saito et al. 2004). To determine whether the ability of p16 to repress telomerase expression is separable from its growth-inhibitory functions, we transiently expressed p16 in the telomerase-positive, p16-negative human breast cancer cell line MDA-MB-231. Cells expressing high levels of tetracycline responsive repressor (tet-R) protein were infected with lentiviruses expressing p16 under the control of tet-operator sequences (tet-p16). We induced p16 expression by adding doxycycline (DOX) for 24 h, after which the cells were extensively washed to remove DOX (Fig 3A). The rise and fall in p16 expression (Fig. S1) caused transient pRb hypophosphorylation and arrest of cell proliferation, from which some of the cells recovered and resumed exponential growth (Fig 3B,C). Strikingly, in the cells that recovered growth, transient p16 exposure caused a stable reduction in telomerase activity that persisted for 29 days (p < 0.084, Student’s t-test) (Fig. 3D), despite exponential growth and the absence of detectable p16 at this time.
Fig. 3.

Transient expression of p16 leads to stable repression of telomerase in MDA-MB-231 cells. (A) Scheme of experimental design. (B) Following 24 hr incubation with or without doxycycline, MDA-MB231-tetR-tet-p16 cells were maintained in culture and propagated when confluent. Total population doublings are plotted versus days in culture. (C) p16 and phospho-pRb expression at times indicated were determined by immunoblotting. Results shown for “No Induction” and “p16 Pulse” were obtained from the same gel. (D) Telomerase activity was determined in control and doxycycline-treated MDA-MB231-tetR-tet-p16 cells harvested at times indicated. Mean values and SD (n=3) are shown.
Similar results were obtained using another telomerase-positive, p16-negative human breast cancer cell line, MCF-7. Treatment with DOX for 6, 12, or 24 h caused a rapid rise in p16 expression (Fig. 4A) and a time-dependent, highly significant, repression of telomerase expression, which then persisted for 55 days (Fig. 4B). Interestingly, p16 exposure resulted in significant reduction, but not complete suppression of telomerase activity in the MCF7 and MDA-MB-231 breast cancer cells that recovered proliferative potential after DOX removal, and these mass populations did not undergo telomere crisis. The decrease in hTERT transcripts in those cells that recovered proliferative potential may not have been sufficient to suppress the levels of telomerase activity below that required for telomere maintenance in these cells. Since the majority of the p16-exposed cells did not recover proliferative potential, it is possible that those tumor cells in which hTERT transcription was repressed below levels required for telomere maintenance also underwent additional changes incompatible with continued proliferation.
Fig. 4.

Time-dependent repression of telomerase activity following p16 induction is not due to loss or gain of diffusible transcriptional activators/repressors. (A) MCF7-tetR-tetp16 cells were treated with doxycycline for times indicated and harvested. p16 levels were determined by immunoblotting. (B) MCF7-tetR-tetp16 cells were treated with doxycycline for times indicated and grown under normal conditions. The cultures were divided when sub-confluent. At days 1 and 55 of the experiment, the cells were harvested and telomerase activity was determined. Mean values and standard deviations are shown (n=3). Values significantly different from the untreated control (p < 0.05, Student’s t-test) are denoted with a *. (C) MCF7-tetR-tetp16 cells were treated with doxycycline for 26 hours, seeded at clonal densities and expended into cell lines. Six doxycycline treated clones and 6 control clones were analyzed for telomerase activity. Arrows indicate telomerase activity in clones used in subsequent experiments. (D) p16 expression levels in MCF7-tetR-tetp16 clones were determined by immunoblotting. Negative control: parental MCF7; positive control: MDA-MB468. (E) Indicated cells were transfected with plasmids encoding luciferase under the control of hTERT promoter sequences of indicated lengths. hTERT promoter activity was measured and normalized to that of a co-transfected promoterless Renilla construct. Mean values and standard deviations are shown (n=3).
The above experiments were performed using mass populations of transduced cells, raising the possibility that the reduction in telomerase activity was due to delayed p16-mediated cell cycle exit by some cells after DOX removal. We therefore obtained single-cell clones from an untreated cell population and a population that was exposed to DOX for 26 h. We expanded the clones for >20 population doublings, and then harvested the cells to measure telomerase activities (Fig. 4C). While telomerase activity varied in individual clones, clones from the population exposed to DOX for 26 h collectively expressed significantly less telomerase activity than clones from the control population (p < 0.045, Wilcoxon test). At the time of harvest, no obvious differences in growth were observed in clones expressing low versus high telomerase activity (Fig. S2). In addition, immunoblotting showed no differences between high and low telomerase-expressing clones in their low levels of residual p16 (Fig. 4D). The range of telomerase activities observed in the p16-exposed cells may be due to differences in viral integration sites and hence differences in the robustness of the p16 induction. Nevertheless, these results clearly demonstrate that the hTERT-inhibitory function of p16 is irreversible and does not require maintenance of p16 expression.
To explore the possibility that p16-mediated suppression of hTERT expression might be attributable to changes in diffusible transcription factors, such as E2Fs, which are down-stream of pRb-family proteins, and have been previously shown to down regulate telomerase (Crowe et al. 2001; Won et al. 2002), we analyzed hTERT promoter activity. We chose high (0-2, 0-4) telomerase expressing clones from the untreated MCF7 population and low (26-1, 26-5) telomerase-expressing clones from a population exposed to DOX for 26 h. We transiently transfected the cells with plasmids containing different segments of the human hTERT promoter linked to a luciferase reporter gene. No significant differences were observed in hTERT promoter activities between the high and low telomerase-expressing clones (p > 0.2, Wilcoxon test for all reporter plasmids) (Fig. 4E). In addition, cells arrested by prolonged (48 hr) p16 exposure exhibited only minor (< 2-fold) while significant decreases in hTERT promoter activities (Fig. S3). Thus, in this system, repression of telomerase activity following p16 induction is not due to loss or gain of diffusible transcriptional activators/repressors of the proximal hTERT promoter, suggesting instead that chromatin modifications at the endogenous locus may alter its accessibility.
The hTERT gene and 5’ flanking sequences are located in a CpG island. However, attempts to correlate the methylation status of these regions with hTERT expression have yielded ambiguous results (Devereux et al. 1999). We examined two regions of the hTERT CpG island previously shown to undergo differential methylation and to contain transcriptional regulatory elements (Renaud et al. 2007). One region contains the core promoter that is necessary for hTERT expression; the other is the exonic region where the CTCF repressor binds. We analyzed the DNA methylation status of these regions in p16-exposed and control MCF7 clones, but found no obvious correlations between DNA methylation and telomerase activity (Fig. S4).
p16 induction results in increased methylation of lysine 27 on histone H3 associated with hTERT sequences
To determine whether p16 suppresses telomerase expression by modifying chromatin composition, we assayed the relative levels of specifically modified histones bound to the hTERT gene before and after p16 exposure. MCF7 cells were harvested immediately following 48 h exposure to DOX, or were treated with DOX for 24 h and allowed to recover and proliferate for 10-14 d before being harvested. As expected, transient p16 induction resulted in stable suppression of hTERT mRNA levels (Fig. 5A). We then performed chromatin immunoprecipitation (ChIP) with lysates from untreated or DOX-treated cells, as well as cells allowed to recover after DOX treatment, using specific antibodies against histone H3 with trimethylated lysine 27 (H3K27), or lysine 9 (H3K9); histone modifications generally associated with gene silencing (Berger 2007). Multiple sequences in both the hTERT promoter and protein-coding regions were examined (Fig 5B). Following p16 induction for 48 h, there were significant increases in the levels of hTERT sequences bound by H3K27 (Fig. 5C), indicating that H3 histones in this chromatin region underwent widespread K27 methylation. In cells exposed to p16 for just 24 h and allowed to recover proliferative potential, the magnitude and extent of H3K27 methylation were more limited, but still significantly greater than in unexposed cells (Fig. 5D). Similar results were obtained in MDA-MB-231 cells (Fig. S5). In contrast, little change occurred in the levels of hTERT sequences bound by H3K9 in DOX treated samples (Fig. S6). MDA-MB-231 cells allowed to recover, however, exhibited significant increases in hTERT sequences bound by H3K9, suggesting that H3K9 methylation may play a role in maintaining the long-term epigenetic effects of p16 exposure.
Fig. 5.

p16 expression causes increased histone H3-K27 methylation of hTERT promoter and protein coding sequences. (A) hTERT mRNA levels in MCF7-tetR-tetp16 cells. Mean values and standard deviations are shown (n=3). U, untreated cells; T, cells treated with doxycycline for 48 hours; R, cells treated with doxycyline for 24 hours when 10-15% confluent, allowed to recover and harvested when sub-confluent. T and R values significantly different from U (p < 0.05, Student’s t-test) are denoted with a *. (B) Map of the hTERT promoter and protein coding regions with PCR amplification targets indicated by letters A through P. (C & D) Chromatin immunoprecipitations were performed with antibodies that specifically recognize trimethylated H3K27. The relative amounts of hTERT sequences bound to the immunoprecipitated material were determined by quantitative PCR and normalized to the amounts of β-actin sequences bound. Mean values and standard deviations are shown (n=3). T and R values significantly different from U (p < 0.05, Student’s t-test) are denoted with a *.
To determine whether p16 triggers epigenetic alterations throughout the genome, we used ChIP-enriched DNA to probe the binding of ENCODE regions (Birney et al. 2007) to modified histones prior to, during, or after induction of p16 expression in MCF7 cells. A significant reduction was observed in the number of sequences bound by H3K27 following p16 induction (Fig. S7), in agreement with previous reports (Bracken et al. 2007). Interestingly, the reduction in regions bound by H3K27 persisted after removal of DOX. Taken together with the concurrent increases we have observed in H3K27 bound by hTERT promoter sequences, these results suggest that H3K27 methylation is not simply reduced, but is stably redistributed following p16 induction.
Finally, to determine if the local increase in H3K27 bound to the hTERT promoter in p16-exposed cells is accompanied by an increase in EZH2, a histone methylase commonly associated with H3K27 methylation, we performed similar ChIP experiments using an EZH2-specific antibody (Fig. S8). Levels of EZH2 associated with the hTERT locus after p16 exposure were generally decreased rather than increased, suggesting that the local increase in H3K27 bound to the hTERT gene in p16-exposed cells is mediated by an alternative histone methylase.
Discussion
We propose that p16-mediated suppression of telomerase is a novel tumor-suppressing mechanism that is likely to be important in epithelial tissues that retain significant telomerase activity in adulthood. In agreement with this hypothesis, p16 suppression resulted in increased telomerase activity and significantly extended proliferative potential in cultured HMEC.
Our finding that transient induction of p16 has irreversible consequences may also explain its recently demonstrated role in decreasing the self-renewal potential of adult stem cells (Janzen et al. 2006; Krishnamurthy et al. 2006; Molofsky et al. 2006). Because limited p16 exposure can suppress telomerase without irreversibly affecting reentry into the cell cycle, it is possible that p16 plays a key role in the differentiation of telomerase-positive stem or progenitor cells into telomerase-negative cells that are capable of limited proliferation, and therefore are less susceptible to malignant transformation. In agreement with this model, embryonic stem (ES) cells contain constitutively hyperphosphorylated pRb and p107 proteins (Galderisi et al. 2006), and an inactive p16 pathway, which only begins to function at the onset of differentiation (White et al. 2005). In vivo, p16 induction occurs during mammary involution in rodents (Gadd et al. 2001). Such induction may suppress expression of genes involved in self-renewal in epithelial cells that remain after involution, thereby limiting their proliferation and protecting them from oncogenic transformation. This may explain why full term pregnancy is associated with reduced risk of breast cancer in humans (Kelsey et al. 1993).
p16-mediated heterochromatin formation during cellular senescence may involve activation of partial or aberrant differentiation programs. In fact, on the basis of morphological and functional features, it was argued over a decade ago that p16 induction at senescence is due to initiation of differentiation in senescent cells (Stein et al. 1999). Conversely, pRb-mediated transcriptional repression observed during senescence is also likely to be involved in terminal differentiation. For example, pRb-dependent histone H3K27 methylation of cell cycle genes has recently been reported in irreversibly arrested myotubes, but not reversibly quiescent murine embryo fibroblasts (Blais et al. 2007).
The H3K27 chromatin mark has been associated with the histone methyltransferase activity of PRC2 polycomb repressor complexes (reviewed in (Kanno et al. 2008)). These developmentally regulated multimeric transcriptional repressor complexes are structurally and functionally diverse in mammalian cells. In general, trimethylation of H3K27 by PRC2 complexes is thought to cause the recruitment of PRC1 complexes, resulting in stable repression of target genes. ES cell differentiation is associated with widespread changes in the binding of PRC complexes (Boyer et al. 2006; Lee et al. 2006), as well as reduced hTERT expression, at least partly via epigenetic mechanisms (Wang et al. 2007).
Since trimethylated H3K27 is redistributed in breast cancer cells exposed to p16, and is ordinarily bound by PRC1 (Cao et al. 2002), it is likely that PRC complexes are also redistributed following p16 induction. In agreement with our findings, p16 induction has been found to increase binding of HPC2 (Cbx5) - a component of PRC1, to the cyclin A promoter in U2-OS cells (Dahiya et al. 2001). H3K27 is methylated by PRC2 component EZH2 (Kuzmichev et al. 2002). p16-mediated activation of pRb leads to reduction of EZH2 (Bracken et al. 2003), resulting in global reduction of H3K27. Interestingly, although PRC2 is responsible for H3K27 methylation in growing ES cells, two core components of the canonical PRC2 complex, Suz12 and EZH2, were not detected at the targets of pRb silencing in differentiated myotubes, raising the possibility that multiple distinct complexes may possess H3K27 histone methylase activity involved in targeting pRb-regulated genes (Blais et al. 2007). Similarly, we determined that the local increase in H3K27 bound to the hTERT gene in p16-exposed cells was not accompanied by increased EZH2, suggesting the involvement of an alternative histone methylase. Further determination of the cellular components that mediate p16-initiated silencing of genes such as hTERT may be useful in the design of strategies to compensate for the loss of the tumor suppressor, and stop the self-renewal of cancer-initiating cells.
Experimental procedures
Cell culture
HMEC were obtained from histologically normal reduction mammoplasty or prophylactic mastectomy tissues (UCSF Comprehensive Cancer Center Tissue Core) and grown in MEGM medium (Lonza, Walkersville, MD, USA). MDA-MB-231 and MCF7 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM with 10% fetal bovine serum (Tet System Approved FBS, Clontech, Mountain View, CA, USA).
Plasmids
Inducible expression of p16 was accomplished by inserting a p16 cDNA (Beausejour et al. 2003) in front of the DOX-inducible promoter of the pLenti CMV/TO Puro DEST (670-1) vector (Campeau et al. 2009). A retrovirus encoding pRB, p107, and p130 shRNAs (Chicas et al.) was generated in the LMP vector (Thermo Scientific, Huntsville, AL, USA).
Immunoblotting
Total protein lysates were collected and processed for immunoblotting by standard methods. Prominent Ponceau-stained protein bands were used as loading controls. Antibodies used included those against p16 (Ab-1; NeoMarkers, Fremont, CA, USA), total-pRb (05-377, Upstate Biotech., Lake Placid, NY, USA), phospho-pRb Ser 807/811 (9308, Cell Signaling, Beverly MA, USA), phospho-pRb Ser 795 (9301, Cell Signaling), p107 (sc-318, Santa Cruz Biotechnology, Santa Cruz, CA, USA), and p130 (sc-317, Santa Cruz Biotechnology).
Telomerase
Telomerase activity was measured using a commercial assay (Allied Biotech, Ijamsville, MD, USA) according to the manufacturer’s instructions. Relative levels of hTERT mRNA were quantitated using a real-time PCR assay (Hines et al. 2005).
Reporter Assays
pGL3-3396, pGL3-996 and pGL3-179 plasmids containing 3396, 996 or 179 base pair fragments upstream of the hTERT gene (Oh et al. 1999) in pGL3 (Promega, Madison, WI, USA) were provided by Dr. Tae Kook Kim (Korea Advanced Institute for Science and Technology). The plasmids were transfected into MCF7 clones using FuGene 6 (Roche, Indianapolis, IN, USA), and luciferase activities were determined 48 hours after transfection using the Dual Luciferase Assay (Promega).
DNA methylation analyses
Genomic bisulfite sequencing of the promoter and first exon of hTERT was performed as previously described (Renaud et al. 2007).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation was performed as previously described (Kim et al. 2005). Briefly, cells were cross-linked with 1% methanol-free formaldehyde for 10 min. at room temperature, followed by quenching with 0.125M glycine for 5 min. Following washes, cells were resuspended in lysis buffer at concentrations of 1 × 107 cells per 0.5 ml. Chromosomal DNA was disrupted using an Ultrasonic Liquid Processor (MISONIX, Inc., Farmingdale, NY, USA). The sizes of DNA fragments obtained were determined by agarose gel electrophoresis with a 200-1000 base pair range considered acceptable. Immunoprecipitation was performed overnight using antibodies obtained from commercial sources: (H3K27me3, Upstate 07-449; H3K9me3, Abcam (Cambridge, MA, USA) ab8898; EZH2, Abcam ab3748) followed by 1 hr incubation with pre-blocked Protein-A or -G conjugated Dynabeads (Invitrogen, Carlsbad, CA, USA). Following washes, reversal of cross-linking and proteinase K treatment, enriched DNAs were purified using QIAquick PCR purification kits (Qiagen, Valencia, CA, USA). Each experiment included IgG and beads-only negative controls. The relative amounts of targeted sequences bound to the immunoprecipitated material were determined by quantitative PCR and normalized to the amounts of β-actin sequences bound.
hTERT and β-actin promoter PCR
Primers for hTERT promoter and coding region amplification are described (Table S1). Primers for β-actin were F: aaatgctgcactgtgcggcgaa, R: tgctcgcgggcggacgcggtctcgg. Quantitative PCR was performed using a commercial kit (Finnzymes, Woburn, MA, USA).
pRb family qRT-PCR
Primers for pRb, p107, p130, and stably expressed reference gene, TBP (TATA box binding protein; NM_003194) transcript quantification are described (Table S2). Quantitative RT-PCR was performed using a commercial kit (SYBR GreenER kit, Invitrogen, CA, USA)
ChIP/CHIP
Amplification, labeling, and hybridization were performed as previously described (Kim et al. 2005). Briefly, blunt ends were created on ChIP-enriched DNA with T4 DNA polymerase (New England Biolabs, Beverly, MA, USA). The DNA was then ligated to linkers (oJW102, 5-GCGGTGACCCGGGAGATCTGAATTC-3; oJW103, 5-GAATTCAGATC-3) and subjected to ligation-mediated PCR. Amplified DNA was labeled with fluorescent Cy3 and Cy5 dCTP (95040-166, VWR, Brisbane, CA, USA) using a BioPrime Array CGH labeling system (18095012, Invitrogen, Carlsbad, CA, USA). Equal amounts of Cy3 (Input) and Cy5 (ChIP) labeled DNA were hybridized to custom ENCODE arrays. Hybridization and washing of the arrays were performed using of a TECAN HS 4800 Pro Hybridization Station (Tecan, San Jose, CA, USA). Array data was normalized using median normalization following background correction. Enrichment was calculated relative to input genomic DNA extracted from sonicated chromatin. The average enrichment ratio for each array probe was calculated for triplicate experiments and deemed significant for those elements with P<0.001. GEO accession numbers GSM359288 – 359314.
Supplementary Material
Acknowledgments
We thank Dr. Tae Kook Kim for providing reagents, and Dr. Alfred Au and the UCSF Comprehensive Cancer Center Tissue Core for reduction mammaplasty samples.
Funding This work was supported by a Flight Attendant Medical Research Institute Young Clinical Investigator Award No. 032122 (AVB), a Komen for the Cure Research Grant No. BCTR0707231 (AVB), National Institutes of Health grants AG09909 and AG17242 (JC) and the Office of Energy Research, Office of Health and Biological Research, US Department of Energy under Contract No. DE-AC03-76SF00098 (PY, JC). C.H. was supported by a Komen for the Cure Postdoctoral Fellowship No. 0707408
Footnotes
Author contributions E.B. and W.J.L. performed protein expression analyses. S.M. and Y.Z. performed cell culture experiments and telomerase activity assays. J.B. performed DNA methylation analyses. E.C. and F.R. constructed the vectors used. A.B. performed virus production. M.v.S., L.L. and B.R. performed ChIP and ChIP/CHIP experiments. C.H., R.M., and J.C. were involved in the study design. A.V.B. and P.Y. designed the study, analyzed the data, and wrote the manuscript.
References
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. Embo J. 2003;22:4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belair CD, Yeager TR, Lopez PM, Reznikoff CA. Telomerase activity: a biomarker of cell proliferation, not malignant transformation. Proc Natl Acad Sci U S A. 1997;94:13677–13682. doi: 10.1073/pnas.94.25.13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais A, van Oevelen CJ, Margueron R, Acosta-Alvear D, Dynlacht BD. Retinoblastoma tumor suppressor protein-dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. J Cell Biol. 2007;179:1399–1412. doi: 10.1083/jcb.200705051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Monch K, Minucci S, Porse BT, Marine JC, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One. 2009;4:e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, Lowe SW. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 17:376–387. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe DL, Nguyen DC, Tsang KJ, Kyo S. E2F-1 represses transcription of the human telomerase reverse transcriptase gene. Nucleic Acids Res. 2001;29:2789–2794. doi: 10.1093/nar/29.13.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiya A, Wong S, Gonzalo S, Gavin M, Dean DC. Linking the Rb and polycomb pathways. Mol Cell. 2001;8:557–569. doi: 10.1016/s1097-2765(01)00346-x. [DOI] [PubMed] [Google Scholar]
- Dai CY, Enders GH. p16 INK4a can initiate an autonomous senescence program. Oncogene. 2000;19:1613–1622. doi: 10.1038/sj.onc.1203438. [DOI] [PubMed] [Google Scholar]
- Devereux TR, Horikawa I, Anna CH, Annab LA, Afshari CA, Barrett JC. DNA methylation analysis of the promoter region of the human telomerase reverse transcriptase (hTERT) gene. Cancer Res. 1999;59:6087–6090. [PubMed] [Google Scholar]
- Duan J, Chen Z, Liu P, Zhang Z, Tong T. Wild-type p16INK4a suppresses cell growth, telomerase activity and DNA repair in human breast cancer MCF-7 cells. Int J Oncol. 2004;24:1597–1605. [PubMed] [Google Scholar]
- Gadd M, Pisc C, Branda J, Ionescu-Tiba V, Nikolic Z, Yang C, Wang T, Shackleford GM, Cardiff RD, Schmidt EV. Regulation of cyclin D1 and p16(INK4A) is critical for growth arrest during mammary involution. Cancer Res. 2001;61:8811–8819. [PubMed] [Google Scholar]
- Galderisi U, Cipollaro M, Giordano A. The retinoblastoma gene is involved in multiple aspects of stem cell biology. Oncogene. 2006;25:5250–5256. doi: 10.1038/sj.onc.1209736. [DOI] [PubMed] [Google Scholar]
- Hines WC, Fajardo AM, Joste NE, Bisoffi M, Griffith JK. Quantitative and spatial measurements of telomerase reverse transcriptase expression within normal and malignant human breast tissues. Mol Cancer Res. 2005;3:503–509. doi: 10.1158/1541-7786.MCR-05-0031. [DOI] [PubMed] [Google Scholar]
- Itahana K, Zou Y, Itahana Y, Martinez JL, Beausejour C, Jacobs JJ, Van Lohuizen M, Band V, Campisi J, Dimri GP. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003;23:389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Kanno R, Janakiraman H, Kanno M. Epigenetic regulator polycomb group protein complexes control cell fate and cancer. Cancer Sci. 2008;99:1077–1084. doi: 10.1111/j.1349-7006.2008.00797.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kelsey JL, Gammon MD, John EM. Reproductive factors and breast cancer. Epidemiol Rev. 1993;15:36–47. doi: 10.1093/oxfordjournals.epirev.a036115. [DOI] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurelli R, Zambruno G, Guerra L, Abbruzzese C, Dimri G, Gellini M, Bondanza S, Dellambra E. Inactivation of p16INK4a (inhibitor of cyclin-dependent kinase 4A) immortalizes primary human keratinocytes by maintaining cells in the stem cell compartment. Faseb J. 2006;20:1516–1518. doi: 10.1096/fj.05-4480fje. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006;126:503–514. doi: 10.1016/j.cell.2006.05.052. [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Oh S, Song YH, Kim UJ, Yim J, Kim TK. In vivo and in vitro analyses of Myc for differential promoter activities of the human telomerase (hTERT) gene in normal and tumor cells. Biochem Biophys Res Commun. 1999;263:361–365. doi: 10.1006/bbrc.1999.1366. [DOI] [PubMed] [Google Scholar]
- Pavletich NP. Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol. 1999;287:821–828. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- Renaud S, Loukinov D, Abdullaev Z, Guilleret I, Bosman FT, Lobanenkov V, Benhattar J. Dual role of DNA methylation inside and outside of CTCF-binding regions in the transcriptional regulation of the telomerase hTERT gene. Nucleic Acids Res. 2007;35:1245–1256. doi: 10.1093/nar/gkl1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Nakagawa K, Hamada K, Hirose S, Harada H, Kohno S, Nagato S, Ohnishi T. Introduction of p16INK4a inhibits telomerase activity through transcriptional suppression of human telomerase reverse transcriptase expression in human gliomas. Int J Oncol. 2004;24:1213–1220. [PubMed] [Google Scholar]
- Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Song LB, Zeng MS, Liao WT, Zhang L, Mo HY, Liu WL, Shao JY, Wu QL, Li MZ, Xia YF, et al. Bmi-1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Cancer Res. 2006;66:6225–6232. doi: 10.1158/0008-5472.CAN-06-0094. [DOI] [PubMed] [Google Scholar]
- Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999;19:2109–2117. doi: 10.1128/mcb.19.3.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty TD, Crawford YG, Holst CR, Fordyce CA, Zhang J, McDermott K, Kozakiewicz K, Gauthier ML. Genetic and epigenetic changes in mammary epithelial cells may mimic early events in carcinogenesis. J Mammary Gland Biol Neoplasia. 2004;9:263–274. doi: 10.1023/B:JOMG.0000048773.95897.5f. [DOI] [PubMed] [Google Scholar]
- Wang S, Hu C, Zhu J. Transcriptional silencing of a novel hTERT reporter locus during in vitro differentiation of mouse embryonic stem cells. Mol Biol Cell. 2007;18:669–677. doi: 10.1091/mbc.E06-09-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J, Stead E, Faast R, Conn S, Cartwright P, Dalton S. Developmental activation of the Rb-E2F pathway and establishment of cell cycle-regulated cyclin-dependent kinase activity during embryonic stem cell differentiation. Mol Biol Cell. 2005;16:2018–2027. doi: 10.1091/mbc.E04-12-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J, Yim J, Kim TK. Opposing regulatory roles of E2F in human telomerase reverse transcriptase (hTERT) gene expression in human tumor and normal somatic cells. Faseb J. 2002;16:1943–1945. doi: 10.1096/fj.02-0311fje. [DOI] [PubMed] [Google Scholar]
- Yaswen P, Stampfer MR. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. Int J Biochem Cell Biol. 2002 doi: 10.1016/s1357-2725(02)00047-x. [DOI] [PubMed] [Google Scholar]
- Zhu L, Xie E, Chang LS. Differential roles of two tandem E2F sites in repression of the human p107 promoter by retinoblastoma and p107 proteins. Mol Cell Biol. 1995;15:3552–3562. doi: 10.1128/mcb.15.7.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.