

Summary
The innominate artery is a predilection site for atherosclerotic lesion formation in hyperlipidemic mice. The lesions at this site in chow-fed apo E−/− mice progress from fatty streaks through stages that include atheroma with large necrotic areas, fibro-fatty nodules containing chondrocyte-like cells and highly calcified, acellular plaques. The advanced lesions in the innominate arteries of the apo E−/− mice exhibit a reproducible frequency of intra-plaque hemorrhage that occurs primarily as a result of fissures through lateral fatty streaks that form adjacent to or on top of the established plaques. However, this plaque disruption is not equivalent to plaque rupture in human lesions where there is rupture of well formed fibrous caps. The plaque disruption in the lesions of the chow-fed apo E−/− mice also do not lead to formation of occlusive thrombi, the predominant marker of plaque rupture in humans. Thus, although the lesions in the innominate arteries of hyperlipidemic mice progress to very advanced stages of the disease, they are not, in our opinion a model in which to study the mechanisms of plaque rupture in humans. The advanced lesions in the innominate arteries of the apo E−/− mice may however be adequate models for studying vascular fibrosis and calcification.
Introduction
Focus on the innominate artery
As part of a study of the effects of 17-beta estradiol on progression of atherosclerosis in older male apolipoprotein E deficient mice (apo E−/−), we mapped the distribution of atherosclerotic lesions in whole mounts of the arterial tree in a large number of animals [1]. In the untreated chow-fed control mice between the ages of 24 and 60 weeks of age, we found that the most advanced lesions were located in the innominate artery. The innominate artery is a small segment of artery at the base of the right carotid artery situated between the ascending aortic arch and the branching of the right subclavian artery. It is also called the brachiocephalic trunk. To our knowledge, no other sites in mice develop as advanced disease as the innominate artery. This may be because the innominate artery is subjected to very irregular patterns of blood flow, being situated between the larger aortic arch and the branching of the right subclavian artery. This irregular blood flow likely reduces the mural shear stress and possibly accounts for the robust development of lesions. However, recent imaging approaches suggest that reduced shear stress may not play as significant a role in lesion formation in mice as it does in humans [2].
Over the past decade, we have focused on defining the pathology of advanced lesion development in the innominate arteries of primarily chow-fed apo E−/− mice [3–5]. Thus, in the first part of this discussion we describe our observations concerning the natural history of advanced lesion development at this site and speculate about the mechanisms that are associated with advanced plaque formation and progression. However, there are additional mouse models that also develop advanced disease in the innominate artery and these mice may have greater utility depending on the questions posed. Therefore, we also describe these additional models and discuss known differences to the chow-fed apo E−/− mice. Finally, we discuss whether any hyperlipidemic mice adequately model the advanced clinically relevant stages of lesions in humans with a particular focus on plaque rupture and thrombosis and make some recommendations for the use of terminology more appropriate to advanced atherosclerosis in mice.
Natural history of advanced plaque progression in the innominate artery
Figure 1 shows data compiled from a number of previous [1, 4–10] and ongoing studies showing the rate of increase of the average cross sectional lesion area in the innominate arteries of chow-fed apo E−/− mice between the ages of 24 and 104 weeks of age. Each time point represents the average area of lesions from at least 15 mice. The data shows that there is a fairly rapid rate of increase in lesion area as the lesions convert from foam cell rich fatty streaks (figure 2) to atheroma with large necrotic cores (figure 3) and then to fibro-fatty nodules (figure 4). This process occurs in mice between 24–50 weeks of age. Following formation of the fibro-fatty nodules, the rate of progression in lesion area slows down appreciably (figure 1). In mice from a year of age onward, there is a very slow progressive conversion of the fibrotic nodules to highly calcified plaques that eventually become devoid of cells (figure 5). The formation of the fibrotic nodules and the subsequent deposition of hydroxyapatite appears to involve chondrocyte-like cells [5].
Figure 1.

Figure 2. Fatty Streak.

This figure shows a fatty streak in the innominate artery consisting of macrophage-derived foam cells from a 20 week old chow-fed apo E−/− mouse. Movat’s pentachrome stain. 100X final maginifcation.
Figure 3. Large Atheromatous Plaque.
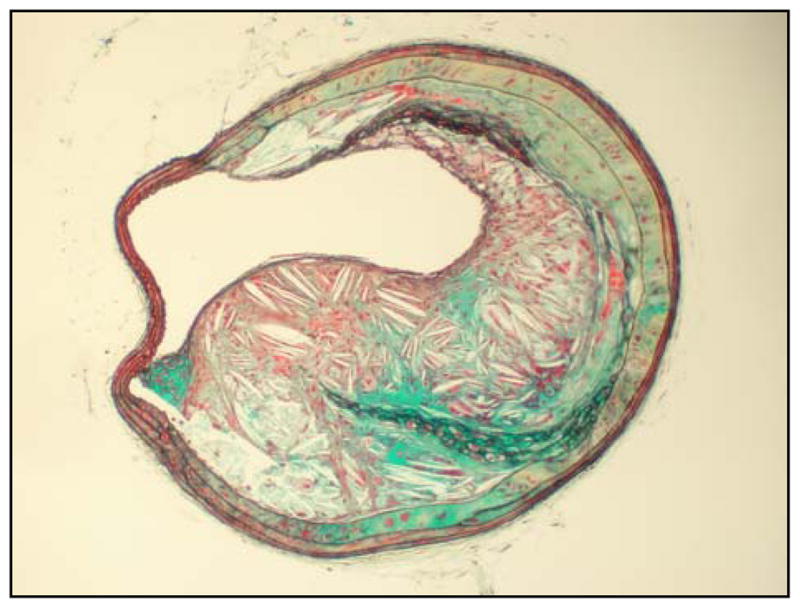
This micrograph shows a large plaque consisting of necrotic debris and extracellular cholesterol clefts in the innominate artery of a 36 week old chow-fed apo E−/− mouse. This necrotic plaque is covered by a very thin cap. This figure also demonstrates thickening of the media caused by invasion of the medial layer by plaque components. Movat’s pentachrome stain. 100× final magnification.
Figure 4. Fibro-fatty Nodule.

This figure shows an example of a fibrotic nodule containing many chondrocyte-like cells in the innominate artery of a 40 week old chow-fed apo E−/− mouse. Movat’s pentachrome stain. 100× final magnification.
Figure 5. Calcified, Acellular Plaque.

This micrograph shows an example of a highly calcified plaque that is practically devoid of cells. This plaque was from a 2 year old chow-fed apo E−/− mouse. Movat’s pentachrome stain. 100× final magnification.
The conversion of the fatty streaks consisting of intact macrophage-derived foam cells to an atheroma likely involves necrotic and apoptotic death of the foam cells mediated by oxidized LDL, free cholesterol loading and potentially a deficiency of p53 [11–14]. One analysis of the spatial distribution of TUNEL positive cells in the lesions of the apo E−/− and LDLR−/− mice showed that most positive cells are located within the lateral margins and along the edges of the necrotic core [14]. In the advanced lesions in the innominate artery of the chow-fed apo E−/− mice there are frequently aggregates of macrophage derived foam cells that form fatty streaks along the lateral margins of the established plaques (also referred to as lateral xanthomas) (figure 6). In our experience, it is these lateral fatty streaks that contain the highest frequency of TUNEL positive cells. These lateral fatty streaks become disrupted leading to intra-plaque hemorrhage (figure 7). The lateral fatty streaks will with time become secondary lesions that layer on top of the established plaques [4]. Thus, formation of the lateral fatty streaks appears to be one way that the overall advanced plaques progress. However, verification of this hypothesis awaits studies where formation of the lateral fatty streaks can specifically be blocked, perhaps by conditional knockout of adhesion molecules or chemokines expressed by cells within the advanced plaques [15].
Figure 6. Lateral Fatty Streak.

This micrograph shows an example of a new fatty streak forming on the lateral margin of a much larger established plaque in the innominate artery of a 40 week old apo E−/− mouse. Movat’s pentachrome stain. 400× final magnification.
Figure 7. Intra-plaque Hemorrhage.

This micrograph shows an example of pooled red blood cells indicative of intra-plaque hemorrhage in a lateral fatty streak in the innominate artery of a 50 week old chow-fed apo E-.- mouse. Movat’s pentachrome stain. 600× final magnification.
With the exception of formation of the necrotic core, it is currently unclear whether cell death plays a role in the progression of advanced lesions in mice. The combined data from a number of published studies have been very confusing in clarifying whether cell death contributes to lesion formation and progression. For example, knockout of the pro-apoptotic protein p53 in apoE*3-Leiden transgenic mice or apoE−/− mice reduced cell death but caused an increase in lesion area [16–18]. Similarly, transplantation of bone marrow from apoE−/− x ACAT-1−/− mice into apoE−/− mice increased cell death within the lesions, but also increased lesion area [19]. Conversely, reduction in cell death due to transplant of BAX−/− cells also led to an increase in lesion area in fat-fed LDLR−/− mice [20].
The formation of plaques with large necrotic cores is followed by conversion to highly fibrotic nodules consisting of the remnants of the necrotic core (extracellular cholesterol clefts), enmeshed in collagen and proteoglycans (figure 4). These nodules are highly cellular and we have recently shown that many of the cells within the nodules express markers of chondrocytes and osteoblasts [5]. For example, much of the collagen that is deposited in these nodules is type II collagen that is normally found in cartilage. This conversion of a plaque with a large necrotic zone to a stable fibrotic nodule may be a type of wound healing response that is accompanied by conversion of resident smooth muscle cells to chondrocyte-like cells or to recruitment of mesenchymal stem cells that mature into chondrocytes [21–24]. It is currently unknown what factors or structural properties within the plaques stimulate this phenotypic switch of the resident smooth muscle cells. In vitro studies suggest that elevated levels of phosphate, lipoproteins, hormones, and cytokines can induce smooth muscle cells to convert to calcifying cells [21, 23, 25–27].
Both the atheroma stage and the formation of the fibrotic nodules are accompanied by the invasion of the medial layer by plaque leading to a thickening of the media (figures 3 and 4). Occasionally, this process has led to aneurysms. However, in general it appears to be another way that the advanced plaques enlarge and progress.
We and others have reported that the advanced lesions in the innominate arteries of the apo E−/− mice exhibit a reproducible frequency of intra-plaque hemorrhage [4, 28, 29]. In the chow-fed apo E−/− mice, the hemorrhage occurs at all stages from the atheroma to the most advanced calcified lesions (figure 10). At the atheroma and fibrotic nodule stages, the hemorrhage is the result of disruptions of the lateral fatty streaks and the frequency corresponds with the stages at which there are the highest frequency and largest area of lateral fatty streaks (figure 11). These disruptions frequently involve small fissures through the aggregates of foam cells leading to pooling of red blood cells within the lateral fatty streaks (figure 8). Occasionally, the fissures can be larger leading to hemorrhage into the necrotic zones. However, as discussed later, we never see disruptions that are equivalent in magnitude to plaque ruptures through the fibrous cap of a human plaque nor do we see thrombi associated with the disruption of the mouse lesions. This is in part due to the failure of the advanced mouse lesions to form a thick fibrous cap. In most cases, the fibrous cap of the lesions in the innominate arteries are very thin and consist of a few layers of smooth muscle cells or layers of laminated elastin (figure 3).
Figure 10. Frequency of Intra-plaque Hemorrhage at Different Stages of Advanced Plaque Progression in the Innominate Arteries of Older Apo E−/− Mice.

This figure shows data compiled from a number of previous studies. Each data point represents the average frequency of intra-plaque hemorrhage in the advanced lesions in the innominate arteries of a minimum of 12–15, male, chow-fed apo E−/− mice at 20, 30, 36, 40, 54, and 60 weeks of age. Intra-plaque hemorrhage is defined as the presence of red blood cells observed in any of the serial sections of the advanced lesions in the innominate arteries.
Figure 11. Lateral Fatty Streak Area at Different Stages of Advanced Plaque Progression in the Innominate Arteries of Older Apo E−/− Mice.

This figure shows data compiled from a number of previous studies. Each data point represents the average cross sectional area of lateral fatty streaks measured in serial sections of advanced lesions in the innominate arteries of a minimum of 12–15, male, chow-fed apo E−/− mice at 20, 30, 36, 40, 60, 80 and 104 weeks of age.
Figure 8. Fissure in a Lateral Fatty Streak.

This micrograph shows a small fissure in a lateral fatty streak in the innominatge artery of a 50 week old chow-fed apo E−/− mouse. This image was from the same mouse as in figure 7 and the section was 50 microns proximal to that shown in figure 7. This suggests that the fissure may be the source of the intra-plaque hemorrhage. Movat’s pentachrome stain. 600× final magnification.
It is currently unclear what causes the disruption of the lateral fatty streaks. However, Gough et al have recently shown that over-expression of an auto-catalytically active MMP-9 by macrophages leads to a significant increase in the frequency of intra-plaque hemorrhage in the chow-fed apo E−/− mice [30]. Thus activation and/or death of the foam cells within the lateral fatty streaks may lead to the secretion of active MMPs with formation of fissures and intra-plaque hemorrhage. We have also occasionally observed intra-plaque hemorrhage in very old apo E−/− mice that have highly calcified plaques in the innominate arteries. In these very advanced lesions, the calcification often occupies more than half of the entire volume of the plaque and there are virtually no cells (figure 4). Thus, breakdown of this type of plaque may be due to the brittleness of the calcium.
Other Mouse Models of Advanced Disease
We have focused our work entirely on chow-fed apo E−/− mice. However, Jackson and colleagues have done extensive studies of the development of advanced lesions in the innominate arteries of apo E−/− mice on a mixed genetic background and fed a diet enriched with lard and cholesterol [28, 31–36]. The addition of this diet significantly elevates the plasma lipid levels as compared to the chow-fed apo E−/− mice and induces a very rapid formation of lesions at this site. Like the more slowly developing lesions in the chow-fed mice, these authors report a high frequency of intra-plaque hemorrhage associated with plaque disruption. Unlike the lesions in the chow-fed mice however, the high fat feeding leads to a significant increase in mortality of the mice within a few months and to formation of thrombi that may or may not account for the death of these mice. While we don’t believe that either the chow-fed or fat-fed apo E−/− mice develop plaque disruptions equivalent to what is observed with human advanced plaques, (and don’t see any thrombosis in the chow-fed mice), Jackson and colleagues have suggested that the fat fed mice are a model for studying events leading to plaque rupture in humans. We direct readers to two recent reviews from both Jackson et al and from us that provide a good discussion of our differences in point of view regarding plaque rupture in mice [31, 32, 37, 38].
The low-density lipoprotein deficient mouse (LDL-R−/−) is also a very popular model for studying atherosclerosis. However, unlike the apo E−/− mice, the LDL-R−/− mice require a high fat diet to induce measurable lesion formation. Nevertheless, the LDL-R−/− mice do form lesions in the innominate arteries that are remarkably similar in size and composition to those we’ve described in the chow-fed apo E−/− mice (figure 9) [39]. This is especially true when the high-fat diet is coupled with diabetes in the LDL-R−/− mice [40–43]. Another recent model of advanced atherosclerosis is the SR-B1−/− x apo E−/− mice double knockout mice [44–46]. These mice have extremely elevated plasma lipids and have been reported to develop occlusive lesions in the coronary arteries leading to myocardial infarctions. To date, the nature of the lesions in the innominate arteries of these mice has not been reported. Welch et al have also recently reported that apo E−/− mice that are simultaneously deficient in Npc-1 have high rates of thrombosis and medial degradation associated with lesions in the aortic sinus [47]. However, again the nature of the lesions in the innominate arteries of these mice has not yet been reported. Additional models that may prove useful for the study of advanced lesions in the innominate arteries are the LDL-R−/− x apo E−/−, apobec−/− x apo E−/− and the human apo B100 transgenic x LDL-R−/− or apo E−/− mice [48–53].
Figure 9. Advanced Lesion in the Innominate Artery of a Fat-fed LDL-R−/− Mouse.

This micrograph shows an advanced lesion in the innominate artery of an LDL-R−/− mouse. The mouse was fed a high fat and cholesterol containing diet for 8 months starting at 8 weeks of age. The size and composition of the plaque is remarkably similar to lesions found in the chow-fed apo E−/− mice. Movat’s pentachrome stain. 100× final magnification.
Defining stable versus unstable: lack of true plaque rupture in mice and recommendations for use of appropriate terminology
As noted, in our experience with the older chow-fed apo E−/− mice, the only marker of plaque disruption that we’ve observed is intra-plaque hemorrhage (figure 7). This hemorrhage is most frequently the result of fissures within the lateral fatty streaks and do not involve rupture of a fibrous cap (figure 8). Because of this, we recommend that the terms “disruption” and “fissure” be used rather than “rupture” to describe the cause of the intra-plaque hemorrhage. Again, we direct the reader to the recent reviews and accompanying letters to the editor that list recommended terminology for mouse lesions and address differences in opinion on whether these disruptions can be considered as models of plaque rupture in human plaques [31, 32, 37, 38]. In our opinion, mouse and human lesions are not equivalent and disrupted mouse lesions likely can not provide strong evidence for the mechanisms underlying plaque rupture in humans.
In humans, the predominant marker of plaque rupture is occlusive thrombi. We have not to date, seen any thrombi associated with the fissures that lead to intra-plaque hemorrhage in the innominate arteries of the older apo E−/− mice. It is unclear why thrombi do not form at these sites. It may be that a high fat diet is required and that something about the massive hyperlipidemia that results makes the mice hypercoagulable. It may also be due to the fissures providing insufficient exposure of pro-thrombotic factors to the blood or perhaps extremely rapid and efficient thrombolysis. Nevertheless, unlike in humans, thrombosis can not be used a marker of plaque disruption in the mouse.
Many authors refer to “stable” versus “unstable” plaques in mouse lesions based on the size of the necrotic core, thinness of the fibrous cap and the relative content of macrophages and connective tissue. Again, we reject use of the terms “stable” and “unstable” in describing mouse lesions. First, in our experience, the advanced lesions that form in the innominate arteries of the apo E−/− mice do not usually have well formed fibrous caps. Second, the fissures through the lateral fatty streaks that lead to pooling of red blood cells, most often do not reach the necrotic core and thus, the size of the necrotic core is not relevant. Third, although the fissures occur through the lateral fatty streaks consisting primarily of macrophage-derived foam cells, in our previous studies the overall content of macrophages was not associated with an increased frequency of intra-plaque hemorrhage. Finally, the amount of connective tissue in the plaque also does not dictate whether there will be a reduced frequency of fissure through the lateral fatty streak. In fact, we have seen intra-plaque hemorrhage occurring in the fibrotic nodules or the most advanced, acellular, calcified lesions as long as lateral fatty streaks continue to layer on top of or adjacent to the plaque.
Cited References
- 1.Rosenfeld ME, Kauser K, Martin-McNulty B, Polinsky P, Schwartz SM, Rubanyi GM. Estrogen inhibits the initiation of fatty streaks throughout the vasculature but does not inhibit intra-plaque hemorrhage and the progression of established lesions in apolipoprotein E deficient mice. Atherosclerosis. 2002;164:251–259. doi: 10.1016/s0021-9150(02)00178-8. [DOI] [PubMed] [Google Scholar]
- 2.Feintuch A, Ruengsakulrach P, Lin A, Zhang J, Zhou YQ, Bishop J, Davidson L, Courtman D, Foster FS, Steinman DA, Henkelman RM, Ethier CR. Hemodynamics in the mouse aortic arch as assessed by MRI, ultrasound, and numerical modeling. Am J Physiol Heart Circ Physiol. 2007;292:H884–892. doi: 10.1152/ajpheart.00796.2006. [DOI] [PubMed] [Google Scholar]
- 3.Seo HS, Lombardi DM, Polinsky P, Powell-Braxton L, Bunting S, Schwartz SM, Rosenfeld ME. Peripheral vascular stenosis in apolipoprotein E-deficient mice. Potential roles of lipid deposition, medial atrophy, and adventitial inflammation. Arterioscler Thromb Vasc Biol. 1997;17:3593–3601. doi: 10.1161/01.atv.17.12.3593. [DOI] [PubMed] [Google Scholar]
- 4.Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587–2592. doi: 10.1161/01.atv.20.12.2587. [DOI] [PubMed] [Google Scholar]
- 5.Rattazzi M, Bennett BJ, Bea F, Kirk EA, Ricks JL, Speer M, Schwartz SM, Giachelli CM, Rosenfeld ME. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: potential role of chondrocyte-like cells. Arterioscler Thromb Vasc Biol. 2005;25:1420–1425. doi: 10.1161/01.ATV.0000166600.58468.1b. [DOI] [PubMed] [Google Scholar]
- 6.Bea F, Blessing E, Bennett B, Levitz M, Wallace EP, Rosenfeld ME. Simvastatin promotes atherosclerotic plaque stability in apoE-deficient mice independently of lipid lowering. Arterioscler Thromb Vasc Biol. 2002;22:1832–1837. doi: 10.1161/01.atv.0000036081.01231.16. [DOI] [PubMed] [Google Scholar]
- 7.Bea F, Blessing E, Bennett BJ, Kuo CC, Campbell LA, Kreuzer J, Rosenfeld ME. Chronic inhibition of cyclooxygenase-2 does not alter plaque composition in a mouse model of advanced unstable atherosclerosis. Cardiovasc Res. 2003;60:198–204. doi: 10.1016/s0008-6363(03)00464-4. [DOI] [PubMed] [Google Scholar]
- 8.Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, Schwartz SM, Giachelli CM, Rosenfeld ME. Osteoprotegerin Inactivation Accelerates Advanced Atherosclerotic Lesion Progression and Calcification in Older ApoE−/− Mice. Arterioscler Thromb Vasc Biol. 2006 doi: 10.1161/01.ATV.0000236428.91125.e6. [DOI] [PubMed] [Google Scholar]
- 9.Blessing E, Bea F, Kuo CC, Campbell LA, Chesebro B, Rosenfeld ME. Lesion progression and plaque composition are not altered in older apoE−/− mice lacking tumor necrosis factor-alpha receptor p55. Atherosclerosis. 2004;176:227–232. doi: 10.1016/j.atherosclerosis.2004.05.033. [DOI] [PubMed] [Google Scholar]
- 10.Blessing E, Campbell LA, Rosenfeld ME, Chesebro B, Kuo CC. A 6 week course of azithromycin treatment has no beneficial effect on atherosclerotic lesion development in apolipoprotein E-deficient mice chronically infected with Chlamydia pneumoniae. J Antimicrob Chemother. 2005;55:1037–1040. doi: 10.1093/jac/dki128. [DOI] [PubMed] [Google Scholar]
- 11.Yao PM, Tabas I. Free cholesterol loading of macrophages induces apoptosis involving the fas pathway. J Biol Chem. 2000;275:23807–23813. doi: 10.1074/jbc.M002087200. [DOI] [PubMed] [Google Scholar]
- 12.Tabas I. p53 and atherosclerosis. Circ Res. 2001;88:747–749. doi: 10.1161/hh0801.090536. [DOI] [PubMed] [Google Scholar]
- 13.Lutgens E, Daemen M, Kockx M, Doevendans P, Hofker M, Havekes L, Wellens H, de Muinck ED. Atherosclerosis in APOE*3-Leiden transgenic mice: from proliferative to atheromatous stage. Circulation. 1999;99:276–283. doi: 10.1161/01.cir.99.2.276. [DOI] [PubMed] [Google Scholar]
- 14.Harada K, Chen Z, Ishibashi S, Osuga J, Yagyu H, Ohashi K, Yahagi N, Shionoiri F, Sun L, Yazaki Y, Yamada N. Apoptotic cell death in atherosclerotic plaques of hyperlipidemic knockout mice. Atherosclerosis. 1997;135:235–239. doi: 10.1016/s0021-9150(97)00167-6. [DOI] [PubMed] [Google Scholar]
- 15.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guevara NV, Kim HS, Antonova EI, Chan L. The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat Med. 1999;5:335–339. doi: 10.1038/6585. [DOI] [PubMed] [Google Scholar]
- 17.Mercer J, Figg N, Stoneman V, Braganza D, Bennett MR. Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ Res. 2005;96:667–674. doi: 10.1161/01.RES.0000161069.15577.ca. [DOI] [PubMed] [Google Scholar]
- 18.van Vlijmen BJ, Gerritsen G, Franken AL, Boesten LS, Kockx MM, Gijbels MJ, Vierboom MP, van Eck M, van De Water B, van Berkel TJ, Havekes LM. Macrophage p53 deficiency leads to enhanced atherosclerosis in APOE*3-Leiden transgenic mice. Circ Res. 2001;88:780–786. doi: 10.1161/hh0801.089261. [DOI] [PubMed] [Google Scholar]
- 19.Su YR, Dove DE, Major AS, Hasty AH, Boone B, Linton MF, Fazio S. Reduced ABCA1-mediated cholesterol efflux and accelerated atherosclerosis in apolipoprotein E-deficient mice lacking macrophage-derived ACAT1. Circulation. 2005;111:2373–2381. doi: 10.1161/01.CIR.0000164236.19860.13. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. 2005;25:174–179. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 22.Tintut Y, Alfonso Z, Saini T, Radcliff K, Watson K, Bostrom K, Demer LL. Multilineage potential of cells from the artery wall. Circulation. 2003;108:2505–2510. doi: 10.1161/01.CIR.0000096485.64373.C5. [DOI] [PubMed] [Google Scholar]
- 23.Watson KE, Bostrom K, Ravindranath R, Lam T, Norton B, Demer LL. TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J Clin Invest. 1994;93:2106–2113. doi: 10.1172/JCI117205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abedin M, Tintut Y, Demer LL. Mesenchymal stem cells and the artery wall. Circ Res. 2004;95:671–676. doi: 10.1161/01.RES.0000143421.27684.12. [DOI] [PubMed] [Google Scholar]
- 25.Parhami F, Basseri B, Hwang J, Tintut Y, Demer LL. High-density lipoprotein regulates calcification of vascular cells. Circ Res. 2002;91:570–576. doi: 10.1161/01.res.0000036607.05037.da. [DOI] [PubMed] [Google Scholar]
- 26.Shioi A, Katagi M, Okuno Y, Mori K, Jono S, Koyama H, Nishizawa Y. Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: roles of tumor necrosis factor-alpha and oncostatin M derived from macrophages. Circ Res. 2002;91:9–16. doi: 10.1161/01.res.0000026421.61398.f2. [DOI] [PubMed] [Google Scholar]
- 27.Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation. 2000;102:2636–2642. doi: 10.1161/01.cir.102.21.2636. [DOI] [PubMed] [Google Scholar]
- 28.Johnson JL, Jackson CL. Atherosclerotic plaque rupture in the apolipoprotein E knockout mouse. Atherosclerosis. 2001;154:399–406. doi: 10.1016/s0021-9150(00)00515-3. [DOI] [PubMed] [Google Scholar]
- 29.Rosenfeld ME, Carson KG, Johnson JL, Williams H, Jackson CL, Schwartz SM. Animal models of spontaneous plaque rupture: the holy grail of experimental atherosclerosis research. Curr Atheroscler Rep. 2002;4:238–242. doi: 10.1007/s11883-002-0025-3. [DOI] [PubMed] [Google Scholar]
- 30.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson CL. Defining and defending murine models of plaque rupture. Arterioscler Thromb Vasc Biol. 2007;27:973–977. doi: 10.1161/01.ATV.0000261545.53586.f0. [DOI] [PubMed] [Google Scholar]
- 32.Jackson CL, Bennett MR, Biessen EA, Johnson JL, Krams R. Assessment of unstable atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2007;27:714–720. doi: 10.1161/01.ATV.0000261873.86623.e1. [DOI] [PubMed] [Google Scholar]
- 33.Johnson JL, Fritsche-Danielson R, Behrendt M, Westin-Eriksson A, Wennbo H, Herslof M, Elebring M, George SJ, McPheat WL, Jackson CL. Effect of broad-spectrum matrix metalloproteinase inhibition on atherosclerotic plaque stability. Cardiovasc Res. 2006;71:586–595. doi: 10.1016/j.cardiores.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 34.Johnson JL, Baker AH, Oka K, Chan L, Newby AC, Jackson CL, George SJ. Suppression of atherosclerotic plaque progression and instability by tissue inhibitor of metalloproteinase-2: involvement of macrophage migration and apoptosis. Circulation. 2006;113:2435–2444. doi: 10.1161/CIRCULATIONAHA.106.613281. [DOI] [PubMed] [Google Scholar]
- 35.Rodgers KJ, Watkins DJ, Miller AL, Chan PY, Karanam S, Brissette WH, Long CJ, Jackson CL. Destabilizing role of cathepsin S in murine atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2006;26:851–856. doi: 10.1161/01.ATV.0000203526.75772.4b. [DOI] [PubMed] [Google Scholar]
- 36.Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci U S A. 2005;102:15575–15580. doi: 10.1073/pnas.0506201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falk E, Schwartz SM, Galis ZS, Rosenfeld ME. Putative murine models of plaque rupture. Arterioscler Thromb Vasc Biol. 2007;27:969–972. doi: 10.1161/01.ATV.0000261572.33474.e0. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz SM, Galis ZS, Rosenfeld ME, Falk E. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:705–713. doi: 10.1161/01.ATV.0000261709.34878.20. [DOI] [PubMed] [Google Scholar]
- 39.MacDougall ED, Kramer F, Polinsky P, Barnhart S, Askari B, Johansson F, Varon R, Rosenfeld ME, Oka K, Chan L, Schwartz SM, Bornfeldt KE. Aggressive very low-density lipoprotein (VLDL) and LDL lowering by gene transfer of the VLDL receptor combined with a low-fat diet regimen induces regression and reduces macrophage content in advanced atherosclerotic lesions in LDL receptor-deficient mice. Am J Pathol. 2006;168:2064–2073. doi: 10.2353/ajpath.2006.051009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamharzi N, Renard CB, Kramer F, Pennathur S, Heinecke JW, Chait A, Bornfeldt KE. Hyperlipidemia in concert with hyperglycemia stimulates the proliferation of macrophages in atherosclerotic lesions: potential role of glucose-oxidized LDL. Diabetes. 2004;53:3217–3225. doi: 10.2337/diabetes.53.12.3217. [DOI] [PubMed] [Google Scholar]
- 41.Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, Chait A, Bornfeldt KE. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest. 2004;114:659–668. doi: 10.1172/JCI17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reaven P, Merat S, Casanada F, Sutphin M, Palinski W. Effect of streptozotocin-induced hyperglycemia on lipid profiles, formation of advanced glycation endproducts in lesions, and extent of atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 1997;17:2250–2256. doi: 10.1161/01.atv.17.10.2250. [DOI] [PubMed] [Google Scholar]
- 43.Schreyer SA, Vick C, Lystig TC, Mystkowski P, LeBoeuf RC. LDL receptor but not apolipoprotein E deficiency increases diet-induced obesity and diabetes in mice. Am J Physiol Endocrinol Metab. 2002;282:E207–214. doi: 10.1152/ajpendo.2002.282.1.E207. [DOI] [PubMed] [Google Scholar]
- 44.Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, Rosenberg RD, Schrenzel M, Krieger M. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res. 2002;90:270–276. doi: 10.1161/hh0302.104462. [DOI] [PubMed] [Google Scholar]
- 45.Braun A, Zhang S, Miettinen HE, Ebrahim S, Holm TM, Vasile E, Post MJ, Yoerger DM, Picard MH, Krieger JL, Andrews NC, Simons M, Krieger M. Probucol prevents early coronary heart disease and death in the high-density lipoprotein receptor SR-BI/apolipoprotein E double knockout mouse. Proc Natl Acad Sci U S A. 2003;100:7283–7288. doi: 10.1073/pnas.1237725100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang S, Picard MH, Vasile E, Zhu Y, Raffai RL, Weisgraber KH, Krieger M. Diet-induced occlusive coronary atherosclerosis, myocardial infarction, cardiac dysfunction, and premature death in scavenger receptor class B type I-deficient, hypomorphic apolipoprotein ER61 mice. Circulation. 2005;111:3457–3464. doi: 10.1161/CIRCULATIONAHA.104.523563. [DOI] [PubMed] [Google Scholar]
- 47.Welch CL, Sun Y, Arey BJ, Lemaitre V, Sharma N, Ishibashi M, Sayers S, Li R, Gorelik A, Pleskac N, Collins-Fletcher K, Yasuda Y, Bromme D, D’Armiento JM, Ogletree ML, Tall AR. Spontaneous atherothrombosis and medial degradation in Apoe−/−, Npc1−/− mice. Circulation. 2007;116:2444–2452. doi: 10.1161/CIRCULATIONAHA.107.701276. [DOI] [PubMed] [Google Scholar]
- 48.Strom A, Ahlqvist E, Franzen A, Heinegard D, Hultgardh-Nilsson A. Extracellular matrix components in atherosclerotic arteries of Apo E/LDL receptor deficient mice: an immunohistochemical study. Histol Histopathol. 2004;19:337–347. doi: 10.14670/HH-19.337. [DOI] [PubMed] [Google Scholar]
- 49.Osuga J, Yagyu H, Ohashi K, Harada K, Yazaki Y, Yamada N, Ishibashi S. Effects of apo E deficiency on plasma lipid levels in mice lacking APOBEC-1. Biochem Biophys Res Commun. 1997;236:375–378. doi: 10.1006/bbrc.1997.6951. [DOI] [PubMed] [Google Scholar]
- 50.Powell-Braxton L, Veniant M, Latvala RD, Hirano KI, Won WB, Ross J, Dybdal N, Zlot CH, Young SG, Davidson NO. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med. 1998;4:934–938. doi: 10.1038/nm0898-934. [DOI] [PubMed] [Google Scholar]
- 51.Veniant MM, Pierotti V, Newland D, Cham CM, Sanan DA, Walzem RL, Young SG. Susceptibility to atherosclerosis in mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. J Clin Invest. 1997;100:180–188. doi: 10.1172/JCI119511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Purcell-Huynh DA, Farese RV, Jr, Johnson DF, Flynn LM, Pierotti V, Newland DL, Linton MF, Sanan DA, Young SG. Transgenic mice expressing high levels of human apolipoprotein B develop severe atherosclerotic lesions in response to a high-fat diet. J Clin Invest. 1995;95:2246–2257. doi: 10.1172/JCI117915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Linton MF, Farese RV, Jr, Chiesa G, Grass DS, Chin P, Hammer RE, Hobbs HH, Young SG. Transgenic mice expressing high plasma concentrations of human apolipoprotein B100 and lipoprotein(a) J Clin Invest. 1993;92:3029–3037. doi: 10.1172/JCI116927. [DOI] [PMC free article] [PubMed] [Google Scholar]