

Abstract
Maintaining the chemical identity of DNA depends on ribonucleotide exclusion by DNA polymerases. However, ribonucleotide exclusion during DNA synthesis in vitro is imperfect. To determine if ribonucleotides are incorporated during DNA replication in vivo, we substituted leucine or glycine for an active site methionine in yeast DNA polymerase ε (Pol ε). Compared to wild type Pol ε, ribonucleotide incorporation in vitro was 3-fold lower for M644L and 11-fold higher for M644G Pol ε. This hierarchy was re-capitulated in vivo in yeast strains lacking RNase H2. Moreover, the pol2-M644G rnh201Δ strain progressed more slowly through S-phase, had elevated dNTP pools and generated 2–5 base pair deletions in repetitive sequences at a high rate and gene orientation-dependent manner. The data indicate that ribonucleotides are incorporated during replication in vivo, that they are removed by RNase H2-dependent repair, and that defective repair results in replicative stress and genome instability via DNA strand misalignment.
The integrity of DNA-based genomes is partly due to the ability of DNA polymerases to prevent incorporation of ribonucleotides 1, which contain a reactive 2′ hydroxyl on the ribose ring that would sensitize the DNA backbone to cleavage. The extent to which DNA polymerases exclude ribonucleoside triphosphates (rNTPs) during DNA synthesis depends on the identity of the polymerase and the base examined, with selectivity for insertion of dNTPs over rNTPs varying from 10-fold to greater than 106-fold (see 2 and references therein). The amount of ribonucleoside monophosphates (rNMPs) incorporated into DNA also depends on the rNTP:dNTP ratio, with rNTP pools in cells significantly exceeding dNTP pools 2–4. For these reasons, it may be that DNA polymerases incorporate rNMPs into DNA during replication in vivo. As an initial test of this possibility, we recently examined the ability of major S. cerevisiae replicative DNA polymerases, Pols α, δ and ε, to incorporate rNMPs during DNA synthesis in vitro. The results demonstrate that all three polymerases do indeed stably incorporate rNMPs into DNA 2. One major goal of the present study is to determine if this is also the case in vivo. Here we focus on Pol ε, which has been implicated in replicating the leading strand template 5,6, and which incorporates rNMPs into DNA somewhat more frequently than Pol δ 2. We have also shown that synthesis by Pol ε is partially impeded by a single rNMP in a DNA template 2. This suggests that unrepaired rNMPs in DNA might have biological consequences in vivo. Thus, anther goal of the present study is to examine what these consequences might be.
One well studied mechanism by which rNMPs are initially introduced into DNA is the RNA primase-dependent synthesis of ~10 nucleotide RNA primers that initiate Okazaki fragments every few hundred base pairs during lagging strand replication. Although these rNMPs are eventually removed during Okazaki fragment maturation 7,8, if any were to remain in DNA, they could confound interpretations regarding rNMPs incorporated by DNA polymerases. In order to focus here on rNMPs incorporated by DNA polymerases rather than by RNA primase, we sought to identify DNA polymerases that have altered ability to incorporate rNMPs during DNA synthesis. To accomplish this, we took advantage of studies showing that specific amino acids at the active sites of DNA polymerases act as a “steric gate” to prevent rNMP insertion (reviewed in 1, and see Fig. 4 in 9). In B family DNA polymerases, of which the yeast replicative polymerases are members, the steric gate is a conserved tyrosine whose replacement with other amino acids greatly reduces selectivity against rNMP insertion 10–12. In yeast, replacing this tyrosine in Pol ε with alanine results in formation of micro-colonies 13, making it difficult to study biological effects. Therefore, we turned our attention to the immediately adjacent hydrophobic amino acid that might also be important for rNTP exclusion, based on studies with A family polymerases 14,15. To test whether amino acid replacements for a conserved methionine (Met644) adjacent to the steric gate tyrosine (Tyr645) in yeast Pol ε might also have altered dNTP selectivity yet retain sufficient polymerase activity to promote growth of yeast cells, we chose to investigate two different replacements for Met644. One was glycine, because purified M644G Pol ε 5 retains high polymerase activity, and because a pol2-M644G yeast strain, unlike a pol2-Y645A strain 13, grows at a rate similar to a wild type (POL2) strain. A second replacement was with leucine (M644L). This choice was based on the idea that leucine might reduce rather than increase rNMP incorporation because Pol δ has a leucine at this position, and its dNTP selectivity is somewhat higher than that of wild type Pol ε 2.
In the present study, we have demonstrated that both variants of Pol ε do indeed have altered ability to incorporate ribonucleotides into DNA. We then studied the consequences of these variant polymerase alleles in yeast strains with and without RNH201, the gene encoding the catalytic subunit of RNase H2, whose substrate specificity in vitro has implicated it in removing single ribonucleotides from DNA (see 16–18 and review by 19). The properties of the double mutant yeast strains indicate that rNMPs are incorporated by Pol ε in vivo and that removing these rNMPs from DNA by an RNase H2-dependent repair process is important for maintaining high replication efficiency and genome stability.
RESULTS
Ribonucleotide insertion by Pol ε derivatives
As an initial test of the ability of wild type, M644L and M644G Pol ε to exclude rNTPs, we used 3′ exonuclease-deficient, 152 kDa N-terminal polymerization domains of the catalytic subunit that were previously described 5,20. We measured extension of DNA primer-templates in reactions containing a single correctly-paired dNTP or rNTP present at the concentrations previously determined to be present in yeast 2. All three enzymes inserted each of the four dNTPs and rNTPs, allowing band intensities to then be used to calculate the degree to which the three polymerases prefer to insert dNTPs as compared to rNTPs. The results show that the selectivity against rNMP insertion is higher for M644L Pol ε than for wild type Pol ε, which is in turn higher than that of M644G Pol ε (Fig. 1a). As a result, the difference in selectivity between M644L and M644G Pol ε is 38-fold for dC/rC, 53-fold for dT/rU, 100-fold for dA/rA and 170-fold for dG/rG.
Figure 1. rNMP incorporation and bypass by Pol ε derivatives.

(a) Discrimination against rNMP insertion, determined as described in reference 2. (b) SDS-PAGE analysis of 3 μg of purified Pol ε derivatives, visualized with Coomassie Blue. (c) Stable rNMP incorporation into DNA, determined as described in reference 2. The marker lanes (M) depict products generated by Pol α prior to gel purification. The percentages of alkali sensitive product and the percentages of rNMP incorporation per nucleotide synthesized are shown below each lane. (d) Frequency of rNMP incorporation by M644L (blue bars), wild type (green bars), and M644G Pol ε (red bars) at each of 22 template positions. (e) Phosphorimage of rGMP bypass products for reactions incubated for 20 min. The template is shown on the right; X denotes dG or rG. The asterisks denote the position of full-length products. NE, no enzyme. (f) Insertion, extension, and relative bypass probabilities for M644L, wild type and M644G Pol ε. Values are percentages and error bars are standard deviations for three time points, calculated as described in reference 32.
Stable incorporation of rNMPs by Pol ε derivatives
The above assay monitors the initial insertion reaction but not the subsequent extension reactions required to stably incorporate rNMPs into fully duplex DNA. In order to monitor stable incorporation of rNMPs into DNA in vitro, for comparisons to the studies in vivo described below, we expressed and purified each Pol ε derivative as an exonuclease-proficient, four-subunit holoenzyme (Fig. 1b). When assayed for DNA polymerase specific activity, M644L and M644G Pol ε were 62% and 63% as active, respectively, as wild type Pol ε. We then examined the ability of each of the three polymerases to stably incorporate rNMPs into DNA. Polymerization reactions were performed to extend a 40-mer primer hybridized to a 70-mer template, in reactions containing all four dNTPs and all four rNTPs at physiological concentrations. Full-length reaction products were isolated and subjected to alkaline hydrolysis under conditions previously shown to completely hydrolyze the DNA backbone at positions where a rNMP is present 2. For wild type Pol ε (Fig. 1c), 2.1% of the full-length products were alkali sensitive. Among the 22 different template positions quantified in this experiment, this corresponds to an average of one rNMP incorporated per 1000 dNMPs, with site-to-site variations observed that depend on the template base and the sequence context. These results for wild type Pol ε (Fig. 1c) are similar to those in our initial study 2. In a parallel reaction, M644L Pol ε had difficulty fully extending the primer in the presence of physiological rNTP and dNTP pools, requiring reactions to be scaled up to isolate equivalent amounts of full-length product. Full-length products generated by M644L Pol ε were more resistant to alkaline treatment, with 0.63% of the products hydrolyzed (Fig. 1c). This indicates that M644L Pol ε stably incorporates 3-fold fewer rNMPs than wild type Pol ε. In contrast, M644G Pol ε generated full-length products as readily as the wild type polymerase, and a remarkable 23% of these were hydrolyzed by alkaline treatment (Fig. 1c), i.e., one rNMP was incorporated for every 91 dNMPs. This is an 11-fold increase compared to wild type Pol ε and a 37-fold increase compared to M644L Pol ε. Quantification of site-specific band intensities revealed that rNMP incorporation by each of the three Pol ε derivatives varied widely along the template (Fig. 1d). Site-to-site variations exceeded 100-fold even from this survey of only 22 positions. The yeast nuclear genome contains 12 million base pairs, leading one to wonder if certain sequence contexts in the genome might be especially prone, or refractory, to rNMP incorporation.
Bypass of a single rNMP in a DNA template
We previously demonstrated that a rGMP in a DNA template partially impedes DNA synthesis by wild type Pol ε, with incorporation being problematic for insertion opposite the rGMP and for insertions opposite each of the next four template positions 2. When rGMP bypass by the Pol ε variants was examined (Fig. 1e), M644G Pol ε bypass parameters (Fig. 1f, gray bars) were similar to those of wild type Pol ε (black bars). For both enzymes, the efficiency of insertion of a dNMP opposite the rGMP was more efficient than was subsequent extension of the resulting primer that contained a dNMP paired with the rGMP in the template (Fig. 1f). In contrast, M644L Pol ε was less efficient in bypassing rGMP (Fig. 1e and Fig. 1f, white bars), and when attempting to copy either the control (fully DNA) template or the rGMP-containing template, the 3′ exonuclease activity of M644L Pol ε degraded the primer strand to a greater extent than the other two enzymes (Fig. 1e). This strongly suggests that the M644L replacement affects the partitioning of a primer terminus between the polymerase and 3′ exonuclease active sites, a property that can be further explored in the future. Increased DNA degradation by M644L Pol ε during attempts at primer extension may explain the lower yield of full-length products in the stable rNMP incorporation experiment described above.
Detecting rNMPs in genomic DNA of rnh201Δ strains
To determine if rNMPs incorporated into DNA by a DNA polymerase can be detected in vivo, we constructed six yeast strains. Three encode either M644L, wild type or M644G Pol ε in strains that are wild type for RNase H2, and three encode either M644L, wild type or M644G Polε in strains in which the gene encoding the catalytic subunit of RNase H2 was deleted (rnh201Δ). RNase H2 (formerly known as RNase H35 in Saccharomyces cerevisiae) can incise the DNA backbone on the 5′-side of a single ribonucleotide in duplex DNA 16–18. This property, and the fact that yeast RNase H2 prefers a single ribose over a stretch of riboses 16, led to the suggestion that it participates in removing single rNMPs from duplex DNA. Thus, the rationale here is to determine if rNMPs can be detected in yeast nuclear genomic DNA, in amounts that correlate with the rNMP incorporation properties of M644L, wild type and M644G Pol ε, and in amounts that correlate with the capacity for RNase H2-dependent repair of rNMPs in DNA. If so, the additional objective was to determine if unrepaired rNMPs that are incorporated into genomic DNA by a DNA polymerase exhibit phenotypes consistent with replicative stress.
DNA was isolated from the six yeast strains, and aliquots of each sample were treated with either 0.3 M KCl or 0.3 M KOH, the latter having been demonstrated to completely hydrolyze DNA molecules containing a single rNMP 2. When analyzed in a neutral agarose gel, DNA samples treated with KCl (Fig. 2a, left panel) were high molecular weight and indistinguishable from untreated samples also analyzed in the same manner (not shown). When equivalent amounts of these DNA preparations were treated with KOH and subjected to electrophoresis in an alkaline agarose gel (Fig. 2a, right panel), the genomic DNA samples isolated from the three rnh201Δ strains were observed to be more sensitive to alkaline hydrolysis than were DNA samples isolated from the three corresponding RNH201 strains. Similar results were obtained in a pol2-M644G strain deleted for the RNH202 gene, which encodes one of the two non-catalytic subunits of yeast RNase H2. As expected, when assayed as previously described 16, no RNase H2 activity was detected in extracts prepared from the rnh201Δ strains, while RNase H2 activity was present in extracts of the RNH201 strains (Fig. 2b).
Figure 2. rNMPs in genomic DNA of Pol ε strains ± RNase H2.
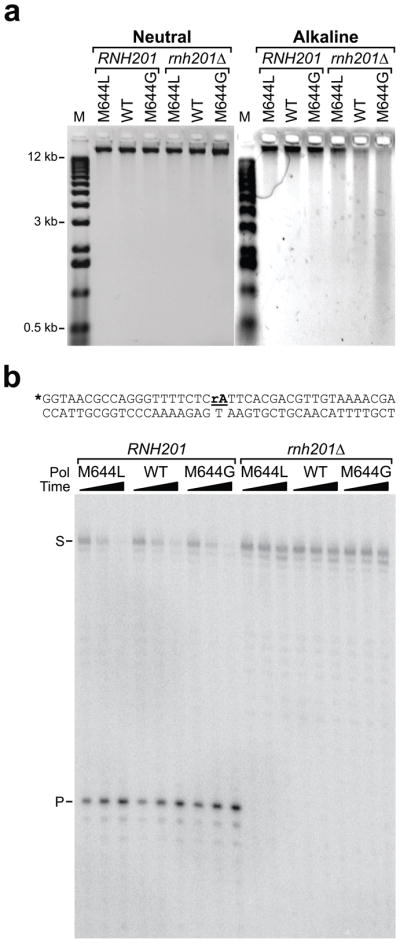
(a) Alkali sensitivity of yeast genomic DNA. The marker lanes (M) are DNA fragments whose lengths in kilobase pairs are 12, 11, 10, 9, 8.1, 7.1, 6.1, 5.1, 4.1, 3.1, 2.0, 1.6, 1.0 and 0.52. (b) Cleavage of a double-stranded substrate containing a single ribonucleotide by yeast extracts. The position of the single ribonucleotide (rA) in the double-stranded oligonucleotide substrate is underlined. The 5′-end-labeled strand is indicated by and asterisk, S is substrate and P is cleavage product.
As illustrated by the relative intensities of the high molecular weight bands (Fig. 2a, right panel), DNA from the pol2-M644G rnh201Δ strain was more sensitive to alkaline hydrolysis than was DNA from the POL2 rnh201Δ strain, which was in turn more sensitive than was DNA from the pol2-M644L rnh201Δ strain. This correlates with the relative propensities of M644L, wild type and M644G Pol ε to incorporate rNMPs during DNA synthesis in vitro (Fig. 1c,d). Moreover, alkaline hydrolysis generated the highest proportion of the shortest DNA fragments with genomic DNA from the pol2-M644G rnh201Δ strain (Fig. 2a, right panel), demonstrating that genomic DNA from this strain contains more unrepaired rNMPs than the POL2 rnh201Δ and pol2-M644L rnh201Δ strains. The fact that small fragments were much less abundant in the equivalent strains that encode wild type RNase H2, even including the pol2-M644G strain, demonstrates that rNMPs incorporated by DNA polymerases were efficiently repaired by an RNase H2-dependent process.
Altered phenotypes due to unrepaired rNMPs in DNA
To examine possible consequences of unrepaired rNMPs incorporated into DNA in vivo, we compared several properties of the six yeast strains. All six strains grew at similar rates in rich (YPDA) medium (Fig. 3a,b) and colonies were of similar size on YPDA plates (Fig. 3a). When progression through the cell cycle in an asynchronously growing cell population was examined (Fig. 3c), pol2-M644L cells slightly accumulated in S phase. This accumulation may be related to the slightly lower catalytic activity of M644L Pol ε compared to wild type Pol ε and/or to altered partitioning between polymerase and exonuclease activity (Fig. 1e). Notably, the slight accumulation of cells in S phase was not increased further by inactivation of RNase H2. The situation differed somewhat for the pol2-M644G strain, where cells also accumulated in S phase, but the effect was slightly exacerbated in the rnh201Δ strain (Fig. 3c). In addition, dNTP pools were elevated in the pol2-M644G rnh201Δ strain in comparison to the other five strains (Fig. 3d). The accumulation of cells in S phase and the increase in dNTP pools are both characteristics of replication stress 21.
Figure 3. Characteristics of Pol ε strains ± RNase H2.

(a) Growth of serial dilutions of overnight cultures plated on YPDA agar plates. (b) Growth in liquid YPDA medium, represented as the mean ± SD. (c) Flow cytometry histograms obtained as described in reference 36. (d) Analysis of dNTP levels, determined as described in reference 2. Two independent isolates were analyzed for each genotype.
Mutator effect of unrepaired rNMPs in DNA
We previously suggested that rNMPs in DNA could be mutagenic 2. To test this, we measured spontaneous mutation rates in the six yeast strains using three different reporter genes. One measures the rate at which a single base insertion in the LYS2 gene reverts to lysine prototrophy via indels that restore the correct reading frame. In the strain encoding wild type Polε, deletion of RNH201 elevated the Lys+ reversion rate by 2.1-fold (Table 1, line 2). When a second, independently isolated strain was examined (Table 1, line 8), a 1.4-fold increase was observed. Both results are consistent with other studies 22,23 demonstrating that deletion of RNH201 increases spontaneous mutagenesis in strains with wild type DNA polymerases. When we examined the pol2-M644L mutant strain, no increase in reversion to Lys+ was observed upon deletion of RNH201. However, when the pol2-M644G mutant strain was examined, deletion of RNH201 increased the Lys+ reversion rate by 25-fold and 18-fold (Table 1, lines 6 and 12). These results were extended to measurements of mutation rates at the CAN1 locus, where a variety of mutations result in resistance to canavanine. At this locus, deletion of RNH201 in both the POL2 and pol2-M644L strains increased spontaneous mutation rates by about 2-fold. However, the rnh201Δ-dependent increases in mutation rates in the pol2-M644G strains were much greater (31-fold and 25-fold; Table 1, lines 6 and 12).
Table 1.
Mutation rates in single and double mutant yeast strains.
| Genotype | Lys+ | CANr | 5-FOAr | |||
|---|---|---|---|---|---|---|
| Rate (× 10−8) | a Relative | Rate (× 10−8) | a Relative | Rate (× 10−8) | a Relative | |
| URA3 in Orientation 1 | ||||||
| Wild Type | 1.4 (1.2 – 1.8) | 1.0 | 20 (19 – 29) | 1.0 | 3.4 (2.5 – 7.4) | 1.0 |
| POL2 rnh201Δ | 3.0 (2.3 – 3.9) | 2.1 | 31 (27 – 35) | 1.6 | 3.6 (2.2 – 5.6) | 1.1 |
| M644L RNH201 | 0.94 (0.74 – 1.5) | 0.7 | 17 (11 – 31) | 0.9 | 3.1 (2.6 – 5.0) | 0.9 |
| M644L rnh201Δ | 1.3 (0.77 – 2.0) | 0.9 | 31 (27 – 64) | 1.6 | 5.9 (4.5 – 9.4) | 1.7 |
| M644G RNH201 | 1.1 (0.88 – 2.1) | 0.8 | 93 (89 – 170) | 4.7 | 23 (18 – 53) | 6.8 |
| M644G rnh201Δ | 35 (30 – 40) | 25 | 620 (450 – 1100) | 31 | 57 (51 – 440) | 17 |
| URA3 in Orientation 2 | ||||||
| Wild Type | 2.0 (1.5 – 2.5) | 1.0 | 21 (18 – 32) | 1.0 | 4.8 (3.8 – 7.8) | 1.0 |
| POL2 rnh201Δ | 2.7 (2.4 – 4.2) | 1.4 | 38 (34 – 51) | 1.8 | 7.5 (6.5 – 13) | 1.6 |
| M644L RNH201 | 0.85 (0.62 – 1.9) | 0.4 | 19 (17 – 25) | 0.9 | 5.2 (2.4 – 16) | 1.1 |
| M644L rnh201Δ | 1.0 (0.79 – 1.4) | 0.5 | 35 (29 – 51) | 1.7 | 5.1 (4.0 – 7.2) | 1.1 |
| M644G RNH201 | 1.0 (0.86 – 1.5) | 0.5 | 130 (65 – 360) | 6.2 | 8.5 (5.7 – 19) | 1.8 |
| M644G rnh201Δ | 36 (33 – 130) | 18 | 530 (420 – 670) | 25 | 110 (83 – 200) | 23 |
Rates are relative to the wild type strain. Measurements were performed as described in Methods. Values in parentheses are 95% confidence limits, calculated as described in reference 37.
Specificity of mutations due to unrepaired rNMPs in DNA
To explore the nature and the mechanism of mutagenesis resulting from unrepaired rNMPs incorporated by Pol ε, we measured mutation rates and mutational specificity at the URA3 locus, where a wide variety of mutations result in resistance to 5-fluoro-orotic acid. Measurements were performed with URA3 present immediately adjacent to ARS306 on chromosome III 2, in either orientation 1 (Table 1, lines 1–6) or in the opposite orientation (orientation 2) (Table 1, lines 7–12). In both orientations, mutation rates were substantially higher in the pol2-M644G rnh201Δ strain than in the other strains (Table 1, lines 6 and 12), again indicating that the majority of the mutagenesis depends on both M644G Pol ε and loss of RNase H2. Sequence analysis of independent ura3 mutants from the pol2-M644G rnh201Δ strains revealed mutation spectra dominated by deletions of 2–5 base pairs (depicted as colored lines in Fig. 4). In comparison to pol2-M644G single mutant strains (from data in 5), the overall increase in the rate of these deletions in the pol2-M644G rnh201Δ strain was 220-fold in orientation 1 and 440-fold in orientation 2. This indicates that RNase H2 strongly suppresses these deletions. Although the 2–5 base deletions were scattered throughout the URA3 coding sequence, all occurred in sequences that were either perfect repeats (Fig. 4, solid boxes) or imperfect repeats (dashed boxes). Several hotspots for deletions are orientation-specific. For example, deletion of CA from a CACA repeat at positions 216–219 occurred 91 times in orientation 2 (rate of 72 × 10−8) but only once in orientation 1 (rate of 0.45 × 10−8), corresponding to a rate difference of 160-fold. Not all repeat sequences were equally unstable. In orientation 2, 91 deletions occurred at a CACA repeat at positions 216–219, but no deletions were observed in either orientation at a CACA repeat at positions 362–365 (green box in Fig. 4). Also notable is the appearance of a hotspot for G-C to A-T substitutions at position 768 in orientation 1 (Fig. 4). This substitution was only observed once in the pol2-M644G mutant spectrum, i.e., it is rnh201Δ-dependent by about 80-fold. Serving as an “internal control” is the orientation 1-specific hotspot for A-T to T-A substitutions at position 686. The rate of this substitution is independent of the status of RNase H2.
Figure 4. Mutational spectra in pol2-M644G rnh201Δ strains.

The coding strand of the 804 base pair URA3 open reading frame is shown. The sequence changes observed in independent ura3 mutants are depicted in red above the coding sequence for URA3 orientation 1, and in blue below the coding sequence for URA3 orientation 2. Letters indicate single base substitutions, closed triangles indicate single base additions, open triangles indicate single base deletions, and short lines above or below the coding sequence indicate 2–5 base deletions. Solid boxes enclose perfect direct repeat sequences, and dashed boxes enclose imperfect direct repeat sequences. The solid green box highlights a CACA repeat at which no deletions were observed.
DISCUSSION
Having previously found that yeast replicative DNA polymerases stably incorporate rNMPs into DNA during synthesis in vitro 2, the goal here was to determine whether rNMPs are incorporated by a DNA polymerase in vivo, and if so, with what consequences. To address these questions, we identified Pol ε derivatives with robust replicative capacity but altered propensity to incorporate rNMPs into DNA. Given its location, it is no surprise that replacing Met644 with other amino acids alters discrimination against rNTPs. What is novel and important is that the two replacements studied here have opposite effects, with leucine decreasing and glycine increasing rNMP incorporation. This allows two important interpretations regarding whether a DNA polymerase incorporates rNMPs into DNA in vivo. The first derives from the fact that the greatest abundance of alkali-sensitive sites in genomic DNA occurs in the yeast strain encoding M644G Pol ε and lacking RNase H2, which correlates with the rNMP incorporation data. This strongly implies that M644G Pol ε does incorporate rNMPs into DNA in vivo. The second interpretation is that even wild type Pol ε incorporates rNMPs into DNA. This is based on the observation that the alkali sensitivity of genomic DNA from the single rnh201Δ mutant strain encoding wild type Pol ε is greater than the alkali sensitivity of the genomic DNA from the pol2-M644L rnh201Δ double mutant strain (Fig. 2a).
DNA fragments resulting from treatment with alkali span a broad range of lengths between ~500 and 10,000 nucleotides (Fig. 2a), being shortest in the pol2-M644G rnh201Δ double mutant strain. Thus a large number of rNMPs have accumulated in the genome. They appear to have accumulated non-randomly, because (i) the fragment distribution is very broad (Fig. 2a), (ii) there is wide site-to-site variation in rNMP incorporation observed in vitro (Fig. 1c,d), (iii) some genomic DNA from the pol2-M644G rnh201Δ double mutant strain remains resistant to alkali even though the average product of hydrolysis by alkali is short (Fig. 2a), and (iv) Pol ε may primarily replicate the leading strand template 5,6, such that the rNMPs may be primarily incorporated into one DNA strand. In the future, it will be interesting to examine the distribution of rNMPs across the yeast genome in rnh201Δ strains.
A recent review of eukaryotic RNases H 19 posed two outstanding questions about RNases H - what are their in vivo substrates and in which processes are they involved? RNase H2 is comprised of a catalytic subunit plus two accessory subunits 24. The gene encoding the second subunit (RNH202) contains a C-terminal PCNA interacting motif (a PIP box), and this subunit interacts with PCNA 19. The genes encoding all three subunits interact genetically with other yeast genes, including FEN1 (see 19 and references therein), and two studies 22,25 led to the suggestion that yeast RNase H2 cooperates with FEN1 to remove RNA primers during Okazaki fragment maturation. However, a variety of ribonucleotide-containing substrates can be cleaved by yeast RNase H2 and its human homolog (reviewed in 19), including duplex DNA containing a single rNMP 16–18. This led to a second, non-exclusive hypothesis that RNase H2 cooperates with FEN1 to remove rNMPs from DNA that have been incorporated by DNA polymerases. The rNMP incorporation data (Fig. 1c,d) and the alkaline hydrolysis data (Fig. 2a) strongly support the latter hypothesis, i.e., that one substrate for yeast RNase H2 is a rNMP incorporated into DNA by Pol ε. This repair pathway appears to be efficient because the genomic DNA from the pol2-M644G rnh201Δ strain is much more sensitive to alkaline hydrolysis than is genomic DNA from the pol2-M644G strain with intact RNase H2 (Fig. 2a). The possible involvement of FEN1 and PCNA suggests that rNMP repair may share common features with long patch BER. There may be additional pathways for rNMP removal, as is the case for many common DNA lesions.
DNA synthesis by M644G Pol ε is slightly inhibited by rGMP in a DNA template (Fig. 1e,f) and unrepaired rNMPs are present in the nuclear genome in pol2-M644G rnh201Δ cells (Fig. 2a). Despite these facts, the pol2-M644G rnh201Δ strain has a normal colony size and growth rate (Fig. 3a,b), indicating that rNMPs incorporated into DNA by M644G Pol ε are tolerated reasonably well. Nonetheless, they are not completely innocuous because the pol2-M644G rnh201Δ double mutant strain progresses more slowly through S-phase and has elevated dNTP pools, both characteristics of replicative stress 21. A more striking consequence is mutagenesis. Our data in strains with wild type DNA polymerases (Table 1) are consistent with previous reports 22,23 that rnh201Δ strains have elevated mutation rates, with the appearance of a four base deletion being especially prevalent. In those reports, the increased mutagenesis was suggested to result from aberrant processing of the 5′ ends of Okazaki fragments. Our data demonstrate a non-exclusive mechanism, i.e., that mutagenesis results from processing of unrepaired rNMPs incorporated by a DNA polymerase. To our knowledge, the mutagenic specificity observed in the pol2-M644G rnh201Δ strain (Fig. 4) is unique among DNA polymerase-dependent mutational spectra. The high mutation rates for 2–5 base pair deletions (Table 2) depend on both M644G Pol ε and on loss of RNase H2. These facts and the observation that the 2–5 base pair deletions are in repetitive sequence motifs (Fig. 4), suggest that following M644G Pol ε incorporation of rNMPs during DNA replication, subsequent processing of unrepaired rNMPs results in DNA strand misalignments containing unpaired nucleotides stabilized by adjacent correct base pairs. Such misalignments could arise in subsequent rounds of replication, or possibly during processing of nicked or gapped intermediates generated when RNase H2 is defective.
Also of interest is the hotspot for G to A transitions at base pair 768 (Fig. 4). Like the 2–5 base pair deletions, these mutations also depend on pol2-M644G and rnh201Δ. Base substitutions were not anticipated, at least to the extent that rNMPs retain normal base pairing potential. Surprisingly, among 12 possible base substitutions, only G to A substitutions were enhanced, and only at this one position among many G-C base pairs in the URA3 coding sequence where G to A substitutions are known to result in resistance to 5-FOA (unpublished database, available upon request). The observation that an unrepaired rNMP in DNA can promote formation of a base-base mismatch may be mechanistically linked to the deletions at imperfect repeat sequences (Fig. 4). For example, formation of a mismatch during rNMP bypass involving an imperfect repeat of the appropriate sequence could allow primer relocation to create a misaligned primer-template in which the terminal base pairs are now matched and contain deoxynucleotides in both strands at the primer terminus, thus facilitating extension. In support of this idea are previous studies with normal templates indicating that misinsertion followed by primer relocation can give rise to single base deletions and larger deletions between direct repeats 26–28.
This study highlights a source of genome instability that has been understudied and warrants further investigation. For example, one wonders if mismatches generated by processing of rNMPs in DNA are subject to mismatch repair (MMR), either by the MutSα pathway (e.g., for G to A at 768) or by the MutSβ pathway (e.g., for 2–5 base pair deletions). Experiments are underway to test this possibility. The fact that unrepaired rNMPs incorporated into DNA by M644G Pol ε are highly mutagenic also provides a highly sensitized genetic background to study alleles of three-subunit RNase H2, such as those lacking the PIP box or that act as surrogates for missense mutations in human RNase H2 subunits associated with Aicardi-Goutières syndrome 29.
METHODS
Polymerase specific activity
Measurements were performed as described 30, in reactions containing 56 μM dTTP and 80 μM dATP, dGTP and dCTP, incubated at 30°C for 4 minutes.
Discrimination against rNMP insertion
Selectivity for dNTPs was determined as described 2, using 3′-exonuclease-deficient, 152 kDa N-terminal polymerization domains of the Pol ε catalytic subunit 5,20.
Stable incorporation of rNMPs into DNA
Four-subunit M644L, wild type and M644G Pol ε was purified as described 31. Their ability to incorporate rNMPs into DNA were assessed as described 2. Reactions (40 μL) contained 4.0 pmol (100 nM) of 70-mer template annealed to a 5′-[γ-32P]-labeled 40-mer DNA primer, and 10 nM wild type, M644L or M644G Pol ε. The nucleotide concentrations used were: dATP, 16 μM; dCTP, 14 μM; dGTP, 12 μM; dTTP, 30 μM; rATP, 3000 μM; rCTP, 500 μM; rGTP, 700 μM; and rUTP, 1700 μM 2. To obtain sufficient full-length products with M644L Pol ε, the reaction was scaled up 8-fold. All three polymerases generated multiple chain lengths surrounding the position for full-length chains, with M644L Polε generating a slightly higher proportion of shorter products, perhaps due to altered partitioning as discussed in Results.
Bypass of a rGMP in a DNA template
The efficiency with which the 4-subunit Pol ε derivatives bypass a rGMP was determined as described 2. Reactions (30μL) contained 2.0 pmol of 65-mer template annealed to a 5′-[γ-32P]-labeled 40-mer DNA primer, and 20, 1.7 or 5.0 fmol, respectively, of M644L, wild type or M644G Pol ε. Bypass parameters were quantified as described 32.
Construction of yeast strains
Strains were isogenic derivatives of strain Δ|(−2)|-7B-YUNI300 (MATa CAN1 his7-2 leu2-Δ::kanMX ura3-Δ trp1-289 ade2-1 lys2-ΔGG2899-2900) 33. The pol2-M644G mutant strain has been described 5. The pol2-M644L mutation was introduced into a haploid Δ|(−2)|-7B-YUNI300 strain via integration-excision using plasmid p173 encoding a portion of POL2 34 containing the Met644Leu mutation. Primers used to introduce the Met644Leu mutation into p173 via site-directed mutagenesis were 5′-CATGTAGATGTCGCCTCTTTATACCCAAACATCATGAC-3′ and 5′-GTCATGATGTTTGGGTATAAAGAGGCGACATCTACATG-3′. rnh201Δ variants of POL2, pol2-M644L and pol2-M644G strains were generated by deletion-replacement of RNH201 via transformation with a PCR product containing the hygromycin-resistance cassette (HYG-R) flanked by 60 nucleotides of sequence homologous to intergenic regions upstream and downstream of the RNH201 ORF. Transformants arose primarily by homologous recombination replacing RNH201 with HYG-R, and deletion of RNH201 and replacement by HYG-R was verified by PCR analysis. The URA3 reporter was then introduced in either orientation 1 or orientation 2 at position AGP1 33 by transformation of a PCR product containing URA3 and its endogenous promoter flanked by sequence targeting the reporter to AGP1, as previously described 6.
Detection of alkali-sensitive sites in genomic DNA
Yeast strains were cultured in 50 ml of YPDA to 1 × 107 cells per ml. Genomic DNA was isolated using a yeast DNA purification kit from Epicentre Biotechnologies. Either KOH or KCl was added to 10 μg of genomic DNA to a final concentration of 0.3 M in 40 μl volumes and incubated at 55°C for 2 hours. Following treatment, 6X alkaline loading buffer (300 mM KOH, 6 mM EDTA, 18% Ficoll (Type 400), 0.15% bromocresol green, 0.25% xylene cyanol) was added to KOH-treated samples. Neutral loading buffer (30% glycerol in TE buffer, 0.25% bromophenol blue, 0.25% xylene cyanol) was added to KCl-treated samples. Electrophoresis of alkaline-treated samples was performed using a 50 mM NaOH, 1 mM EDTA 1% agarose alkaline gel with 50 mM NaOH, 1 mM EDTA electrophoresis buffer 35. Electrophoresis of KCl-treated samples was performed using a 1% agarose gel and TBE buffer. Electrophoresis of the samples was at 1 V/cm for 18 hours. Alkaline gels were neutralized by soaking in 1M Tris HCl pH 8.0, 1.5 M NaCl for 1 hour, and then stained with 0.5 μg/ml ethidium bromide.
RNase H2 cleavage assay
Preparation of DNA containing a ribonucleotide was performed by 5′ end-labeling 1 μg of a 41-mer oligonucleotide containing rAMP (5′-GGTAACGCCAGGGTTTTCTCrATTCACGACGTTGTAAAACGA) with [γ-32P] ATP by T4 polynucleotide kinase. The labeled oligonucleotide was annealed to 1.5 μg of a complementary oligonucleotide (5-TCGTTTTACAACGTCGTGAATGAGAAAACCCTGGCGTTACC). Un-annealed oligonucleotide was removed by binding to Benzoylated Naphthoylated DEAE-cellulose after adjusting NaCl concentration to 1 M. Unincorporated label and BND-cellulose were removed by G-25 sephadex spin column. RNase H2 activity in yeast cell free extracts was monitored following conditions used by Rydberg and Game 16. 0.15 μg of extract was incubated with 100 fmoles of substrate in a 10 μl reaction at 30°C in buffer containing 10 mM Tris pH 7.5, 35 mM KCl, 1 mM DTT, 4 mM MgCl2 and 5% Glycerol. Incubation times varied from 2.5 minutes to 60 minutes. Reactions were terminated with an equal volume of 10 mM EDTA, 80% Formamide, 0.25% Xylene Cyanol FF and 0.25% Bromophenol Blue. Samples were heated to 75° C for 5 minutes prior to separation by electrophoresis using a 12% polyacrylamide 8 M urea gel, and the gel was dried and analyzed after exposure to a phosphor screen.
Growth characteristics of yeast strains
Strains were grown in YPDA media (1% yeast extract, 2% bacto-peptone, 250 mg/l adenine, 2% dextrose, 2% agar for plates). For growth rate analysis, (i) overnight cultures grown in liquid YPDA media were diluted 10-fold and spotted on a YPDA agar plate, and (ii) two independent colonies for each strain were grown overnight in liquid YPDA, re-inoculated into fresh YPDA at OD600 = 0.1, and the OD600 was measured every hour. Flow cytometry was performed as described 36.
Measurement of dNTP and rNTP pools
Measurements of dNTP and rNTP levels in extracts prepared from logarithmically, asynchronously growing yeast cells were performed by HPLC analysis, as described 2.
Spontaneous mutation rates and sequence analysis
Spontaneous mutation rates were measured by fluctuation analysis as described previously 37. For each ura3 mutant that was sequenced, an independent colony was patched to YPDA and then replica plated to media containing 5-fluoro-orotic acid (5-FOA). Genomic DNA from a single 5-FOA resistant colony from each patched colony was isolated and the ura3 gene was PCR-amplified and sequenced.
Acknowledgments
We thank Katarzyna Bebenek and Jessica Williams for thoughtful comments on the manuscript, and the NIEHS Molecular Genetics Core for technical support in DNA sequence analysis of ura3 mutants. This work was supported in part by Project Z01 ES065070 (to T.A.K.) from the Division of Intramural Research of the NIH, NIEHS, in part by the Swedish Foundation for Strategic Research, the Swedish Research Council and the Swedish Cancer Society (to A.C.), and in part by the Swedish Research Council, the Swedish Cancer Society, Smärtafonden and the fund for Basic Science-oriented Biotechnology, Medical Faculty of Umeå University (to E.J.).
Footnotes
AUTHOR CONTRIBUTIONS
SANM, DK, ABC, BEW and DLW performed the experiments and analyzed the data; EBL contributed reagents; EJ, AC and TAK designed the experiments and the analyzed data; TAK wrote the manuscript; all authors edited the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Joyce CM. Choosing the right sugar: how polymerases select a nucleotide substrate. Proc Natl Acad Sci U S A. 1997;94:1619–22. doi: 10.1073/pnas.94.5.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nick McElhinny SA, et al. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Natl Acad Sci U S A. 2010;107:4949–4954. doi: 10.1073/pnas.0914857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kornberg A, Baker T. DNA replication. Freeman; New York: 1992. [Google Scholar]
- 4.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 5.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–30. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–44. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossi ML, Purohit V, Brandt PD, Bambara RA. Lagging strand replication proteins in genome stability and DNA repair. Chem Rev. 2006;106:453–73. doi: 10.1021/cr040497l. [DOI] [PubMed] [Google Scholar]
- 8.Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–5. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLucia AM, Grindley ND, Joyce CM. An error-prone family Y DNA polymerase (DinB homolog from Sulfolobus solfataricus) uses a ‘steric gate’ residue for discrimination against ribonucleotides. Nucleic Acids Res. 2003;31:4129–37. doi: 10.1093/nar/gkg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonnin A, Lazaro JM, Blanco L, Salas M. A single tyrosine prevents insertion of ribonucleotides in the eukaryotic-type phi29 DNA polymerase. J Mol Biol. 1999;290:241–51. doi: 10.1006/jmbi.1999.2900. [DOI] [PubMed] [Google Scholar]
- 11.Gardner AF, Jack WE. Determinants of nucleotide sugar recognition in an archaeon DNA polymerase. Nucleic Acids Res. 1999;27:2545–53. doi: 10.1093/nar/27.12.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang G, Franklin M, Li J, Lin TC, Konigsberg W. A conserved Tyr residue is required for sugar selectivity in a Pol alpha DNA polymerase. Biochemistry. 2002;41:10256–61. doi: 10.1021/bi0202171. [DOI] [PubMed] [Google Scholar]
- 13.Pavlov YI, Shcherbakova PV, Kunkel TA. In vivo consequences of putative active site mutations in yeast DNA polymerases alpha, epsilon, delta, and zeta. Genetics. 2001;159:47–64. doi: 10.1093/genetics/159.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Astatke M, Ng K, Grindley ND, Joyce CM. A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proceedings of the National Academy of Science. 1998;95:3402–3407. doi: 10.1073/pnas.95.7.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinkai A, Patel PH, Loeb LA. The conserved active site motif A of Escherichia coli DNA polymerase I is highly mutable. J Biol Chem. 2001;276:18836–42. doi: 10.1074/jbc.M011472200. [DOI] [PubMed] [Google Scholar]
- 16.Rydberg B, Game J. Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. Proc Natl Acad Sci U S A. 2002;99:16654–9. doi: 10.1073/pnas.262591699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eder PS, Walder JA. Ribonuclease H from K562 human erythroleukemia cells. Purification, characterization, and substrate specificity. J Biol Chem. 1991;266:6472–9. [PubMed] [Google Scholar]
- 18.Eder PS, Walder RY, Walder JA. Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie. 1993;75:123–6. doi: 10.1016/0300-9084(93)90033-o. [DOI] [PubMed] [Google Scholar]
- 19.Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Regulation of B family DNA polymerase fidelity by a conserved active site residue: characterization of M644W, M644L and M644F mutants of yeast DNA polymerase epsilon. Nucleic Acids Res. 2007;35:3076–86. doi: 10.1093/nar/gkm132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chabes A, et al. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112:391–401. doi: 10.1016/s0092-8674(03)00075-8. [DOI] [PubMed] [Google Scholar]
- 22.Qiu J, Qian Y, Frank P, Wintersberger U, Shen B. Saccharomyces cerevisiae RNase H(35) functions in RNA primer removal during lagging-strand DNA synthesis, most efficiently in cooperation with Rad27 nuclease. Mol Cell Biol. 1999;19:8361–71. doi: 10.1128/mcb.19.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen JZ, Qiu J, Shen B, Holmquist GP. Mutational spectrum analysis of RNase H(35) deficient Saccharomyces cerevisiae using fluorescence-based directed termination PCR. Nucleic Acids Res. 2000;28:3649–56. doi: 10.1093/nar/28.18.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeong HS, Backlund PS, Chen HC, Karavanov AA, Crouch RJ. RNase H2 of Saccharomyces cerevisiae is a complex of three proteins. Nucleic Acids Res. 2004;32:407–14. doi: 10.1093/nar/gkh209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frank P, Braunshofer-Reiter C, Karwan A, Grimm R, Wintersberger U. Purification of Saccharomyces cerevisiae RNase H(70) and identification of the corresponding gene. FEBS Lett. 1999;450:251–6. doi: 10.1016/s0014-5793(99)00512-8. [DOI] [PubMed] [Google Scholar]
- 26.Bebenek K, Kunkel TA. Frameshift errors initiated by nucleotide misincorporation. Proc Natl Acad Sci U S A. 1990;87:4946–50. doi: 10.1073/pnas.87.13.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunkel TA, Hamatake RK, Motto-Fox J, Fitzgerald MP, Sugino A. Fidelity of DNA polymerase I and the DNA polymerase I-DNA primase complex from Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:4447–58. doi: 10.1128/mcb.9.10.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai H, Yu H, McEntee K, Kunkel TA, Goodman MF. Purification and properties of wild-type and exonuclease-deficient DNA polymerase II from Escherichia coli. J Biol Chem. 1995;270:15327–35. doi: 10.1074/jbc.270.25.15327. [DOI] [PubMed] [Google Scholar]
- 29.Crow YJ, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–6. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 30.Hamatake RK, et al. Purification and characterization of DNA polymerase II from the yeast Saccharomyces cerevisiae. Identification of the catalytic core and a possible holoenzyme form of the enzyme. J Biol Chem. 1990;265:4072–83. [PubMed] [Google Scholar]
- 31.Chilkova O, Jonsson BH, Johansson E. The quaternary structure of DNA polymerase epsilon from Saccharomyces cerevisiae. J Biol Chem. 2003;278:14082–6. doi: 10.1074/jbc.M211818200. [DOI] [PubMed] [Google Scholar]
- 32.Kokoska RJ, McCulloch SD, Kunkel TA. The efficiency and specificity of apurinic/apyrimidinic site bypass by human DNA polymerase η and Sulfolobus solfataricus Dpo4. J Biol Chem. 2003;278:50537–45. doi: 10.1074/jbc.M308515200. [DOI] [PubMed] [Google Scholar]
- 33.Pavlov YI, et al. Correlation of somatic hypermutation specificity and A-T base pair substitution errors by DNA polymerase eta during copying of a mouse immunoglobulin kappa light chain transgene. Proc Natl Acad Sci U S A. 2002;99:9954–9. doi: 10.1073/pnas.152126799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirchner JM, Tran H, Resnick MA. A DNA polymerase epsilon mutant that specifically causes +1 frameshift mutations within homonucleotide runs in yeast. Genetics. 2000;155:1623–32. doi: 10.1093/genetics/155.4.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Journal. 2. Cold Spring Harbor Press; 1989. pp. 6.20–6.21. [Google Scholar]
- 36.Sabouri N, Viberg J, Goyal DK, Johansson E, Chabes A. Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res. 2008;36:5660–7. doi: 10.1093/nar/gkn555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shcherbakova PV, Kunkel TA. Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol Cell Biol. 1999;19:3177–83. doi: 10.1128/mcb.19.4.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]