

Abstract
Key studies leading to the discovery and definition of the role of endogenous fatty acid amide signaling molecules are summarized.
Introduction
Work conducted over the last twenty years has provided compelling evidence that the fatty acid amides serve as a new and additional class of endogenous signaling molecules. Herein, we review these studies and key elements of the work resulting in their discovery, biosynthesis, degradation, and fundamental endogenous role. These may be grouped largely into two classes, the fatty acid ethanolamides, of which anandamide is the prototypical member, and the fatty acid primary amides, of which oleamide is the most explored member. Because of their overlapping endogenous and signaling roles, a brief introduction to key structurally related fatty acid esters and ethers is also provided.
Fatty Acid Ethanolamides
Although isolated, identified and physiologically characterized as early as the mid 1900’s,1 the notion that the fatty acid ethanolamides serve as key fundamental signaling molecules gained substance with the discovery that anandamide represents an endogenous ligand for the newly identified cannabinoid receptors.
Anandamide
Isolation/Identification
Shortly following the identification and characterization of the cannabinoid receptors in 1988 (CB1) and 1993 (CB2),2 Mechoulam and coworkers isolated, identified, and characterized anandamide as an endogenous agonist of the receptors in 1992 (Figure 1).3 Although received with some skepticism at the time given the simplicity of the structure and the lack of precedent for this class of signaling molecules, anandamide, also known as N-arachidonoyl ethanolamide (AEA), is now widely accepted as an endogenous cannabinoid neurotransmitter. Its name is derived from the Indian Sanskrit word, ananda, which means ‘bringer of eternal bliss and tranquillity’. Anandamide is part of a large family of signaling lipids, the N-acylethanolamines (NAEs). It was the first endogenous ligand identified in a screen for ligands for the cannabinoid receptor shortly after their identification2 and was isolated from porcine brain extracts.3 This endocannabinoid inhibited the specific binding of a radiolabeled cannabinoid probe [3H]HU-243 to synaptosomal membranes in a manner typical of competitive ligands and produced a concentration-dependent inhibition of the electrically evoked twitch response of the mouse vas deferens, a characteristic effect of psychotropic cannabinoids.
Figure 1.
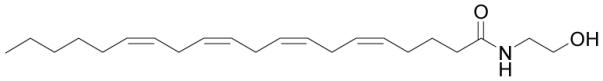
Structure of anandamide.
Biosynthesis/Metabolism
Despite over 20 years of study, the biosynthesis of anandamide and other NAEs is not yet fully characterized. It is generally accepted that N-acyl phosphatidylethanolamines (NAPEs) are the precursors for NAEs, but the precise enzymatic steps leading to release of NAEs from NAPEs are unclear. Several postulated routes for their synthesis are reported and discussed in the literature.4 The original model for the biosynthesis of NAEs follows the sequential action of (1) a calcium-dependent transacylase (CDTA) that transfers the sn-1 acyl chain of phospholipids onto the primary amine of phosphatidylethanolamine (PE) to generate N-acyl phosphatidyethanolamines (NAPEs), and (2) a D-type phospholipase that hydrolyzes NAPEs to produce NAEs (Scheme 1).5 Initial studies indicated that this two-step pathway might also contribute to the biosynthesis of anandamide. Anandamide, along with its NAE congeners and their respective NAPE precursors, is produced by neurons in a calcium-dependent manner,6 and a brain CDTA activity is capable of producing the anandamide precursor N-arachidonoyl PE in vitro.7 Subsequent characterization of an NAPE-selective phospholipase D (NAPE-PLD) revealed that this enzyme can convert N-arachidonoyl PE to anandamide in vitro.8 It was assumed that most, if not all NAEs, were biosynthesized in a common enzymatic pathway. However, investigations into the regulated production of NAEs suggested the existence of other biosynthetic pathways for all members of this lipid class. The generation and characterization of mice lacking the NAPE-PLD gene provided convincing evidence to support the existence of multiple biosynthetic pathways for NAEs in the nervous system.9 For the NAPE-PLD-independent pathways, two intermediates have been reported to date. The first is a glycerophospho-NAE intermediate, where the sn-1 or sn-2 O-acyl chains of NAPEs or both are first hydrolyzed to generate lyso-NAPEs and glycerophospho(GP)-NAEs, respectively,10 followed by cleavage of the phosphodiester bond of these intermediates to generate NAEs.11 The second involves the phospholipase C-dependent conversion of NAPEs to phospho-NAEs, followed by phosphatase-mediated hydrolysis of these intermediates to generate NAEs.12 Anandamide is not stored in cells but is formed when needed, then released from neurons on depolarization and rapidly inactivated. Its principle, if not exclusive, mechanism of inactivation at its sites of action is enzymatic hydrolysis by the membrane-bound serine hydrolase fatty acid amide hydrolase (FAAH).13,14
Scheme 1.

The metabolism of anandamide by human liver and kidney microsomes and the formation of epoxyeicosatrienoic ethanolamides and hydroxyeicosatetraenoic acid ethanolamides have also been studied and reported.15
Function
Anandamide binds to the central CB1 and peripheral CB2 cannabinoid receptors through which it is thought to exhibit its analgesic and anti-inflammatory effects.16 It has been reported to bind with higher affinity to CB1 (Ki = 89 nM)17 than to CB2 (Ki = 371 nM)18 in CHO cells in a radioligand binding assay using [3H]CP55940 and was also measured in other cell lines. Anandamide has been shown to behave as an agonist with greater efficacy at CB1 than CB2.19 Conflicting reports of its potency have been reported. Since anandamide is such an effective substrate for FAAH,13b such measurements in cell-based assays need to be carried out in the presence of a potent and selective FAAH inhibitor20 to insure accurate concentrations are maintained in the binding or functional assays. It also has effects, particularly in the vasculature, that cannot be explained by actions at either the CB1 or CB2 receptors. These effects may be mediated by novel G protein-coupled receptors, but genome searching has not yet revealed a strong candidate. Several approaches have suggested that an orphan receptor, GPR55, is a target for anandamide, but the pharmacology of this receptor is such that it cannot yet be categorically classified as a cannabinoid receptor.21
Anandamide is also reported to be an endogenous ligand for the vanilloid receptor (TRPV1) that is involved in the transduction of acute and inflammatory pain signals, activating the receptor in a PKC-dependent (protein kinase C-dependent) manner, leading first to the perception of pain and then desensitization providing what may be a second site of action for its analgesic effects.22
Anandamide modulates distinct and diverse physiological processes, including nociception,23 anxiety,24 inflammation,25 appetite,26 learning and memory.27 A study also reported that anandamide inhibits breast cancer cell proliferation.28
Palmitoyl ethanolamide
Isolation/Identification
Palmitoyl ethanolamide, or N-(2-hydroxyethyl)hexadecanamide (PEA), was first identified more than 50 years ago (Figure 2). It was isolated from egg yolk, hexane-extracted peanut meal, and soybean lecithin.29 This endogenous compound is present in the rat brain, liver and skeletal muscle.1
Figure 2.
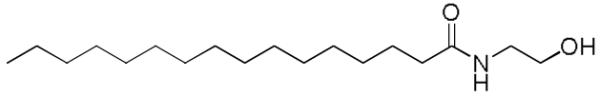
Structure of palmitoyl ethanolamide.
Biosynthesis/Metabolism
PEA and anandamide are synthesized from different precursors through the action of the same enzyme, the N-arachidonoyl-phosphatidyl-ethanolamine-selective phospholipase D30 and are hydrolyzed by the same amidase enzymes.
In addition to FAAH,13 an additional enzyme has been purified and characterized that exhibits a higher catalytic efficiency for palmitoyl ethanolamide than with anandamide.31 This enzyme is a lysosomal hydrolase with optimal activity at acidic pH and a different tissue distribution than FAAH. Esters, retroesters and retroamides of 16:0 palmitic acid were reported as selective inhibitors of the palmitoyl ethanolamide amidase.32
Function
Palmitoyl ethanolamide was shown to reduce allergic reactions and inflammation33,34 in animals along with influenza symptoms in humans.35 It was found to inhibit peripheral inflammation36,37 and mast-cell degranulation,38 as well as to exert neuroprotective39 and anti-nociceptive effects in rats and mice.40,23a These actions are accompanied by changes in nitric oxide production,41 neurotrophil influx,42 and expression of proinflammatory proteins such as inducible nitric oxide synthase and cyclooxygenase-2.43 The nuclear receptor peroxisome proliferator-activated receptor-α (PPAR-α) was indentified as the molecular target responsible for the anti-inflammatory properties of PEA.44 It was also reported that PEA has an anti-inflammatory effect on human adipocytes and could be a potentially interesting candidate molecule in the prevention of obesity-associated insulin resistance.45 Its anticonvulsant activity in mice has been reported, however, its precise mechanism of action remains to be elucidated.39
Analogues of palmitoyl ethanolamide, varying the fatty acid chain length from caproyl to stearoyl and the nature of the amide substituent, were evaluated for affinity to cannabinoid receptors and, like PEA itself, were reported to be inactive.46
An enhancement of the hypotensive effects of intrathecally (i.t.) injected endocannabinoids in the spinal cord by palmitoyl ethanolamide has been reported. The facilitative action of palmitoyl ethanolamide affects the vanilloid TRPV1 as well as the cannabinoid CB1 receptor-mediated effects of endocannabinoids on blood pressure control.47,48
Levels of palmitoyl ethanolamide along with other endogenous neuroprotective substrates were measured in different brain areas of R2/6 mice, a transgenic model of Huntington’s disease, versus wild-type animals.49 These studies suggest that drugs inhibiting endocannabinoid degradation might be useful to treat this disease.
Oleoyl ethanolamide
Isolation/Biosynthesis
Oleoyl ethanolamide (OEA) is a natural analog of the endogenous cannabinoid anandamide (Figure 3). It is produced, like anandamide, in cells in a stimulus-dependent manner and is rapidly eliminated by enzymatic hydrolysis,13 suggesting a function in cellular signaling.6 OEA, along with AEA and PEA, has been shown to be present in human seminal plasma, mid-cycle oviductal fluid, follicular fluid, amniotic fluid, milk, and fluids from malignant ovarian cysts.50
Figure 3.
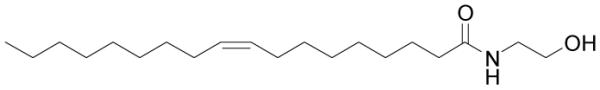
Structure of oleoyl ethanolamide.
Function
Oleoyl ethanolamide mainly modulates feeding and energy homeostasis and is thought to act by binding to peroxisome proliferator-activated receptor-α (PPAR-α).26,51 It is reported to not bind to or activate cannabinoid receptors. OEA was found to excite sensory neurons and produce visceral increased sensitivity to pain via activation of the TRPV1 receptor.52,53,54 However, a recent study described this agent as an antinociceptive substance in two models of visceral and inflammatory pain in both mouse and rat.55
It has also been reported that the appetite suppressant activity of OEA may be derived from its action as an efficacious agonist of GPR119,56 a highly expressed receptor in pancreatic islets and in the colon57 that has a distant similarity to biogenic amine and cannabinoid receptors (~ 40% identity in the transmembrane regions). However, weight loss mediated by OEA is not seen in mice lacking PPAR-α, but remains fully intact in mice lacking GPR119.58
Long chain saturated and unsaturated alkyl sulfonamide and propyl sulfonamide derivatives, analogs of oleoyl ethanolamide, have been evaluated in vivo and in vitro as PPAR-α activators. Additionally, the anorexic effects of the compounds have been studied in vivo in food-deprived rats. Among the active compounds, N-octadecyl-N’-propylsulfonamide has been identified as a potent hypolipidemic compound, a potent feeding suppressant, and a concentration dependent activator of PPAR-α.59
Steaoryl ethanolamide
Identification
Stearoyl ethanolamide (SEA) is a fully saturated C18 N-acyl ethanolamide (Figure 4). It has been shown to accompany anandamide in many tissues including rat central neurons,6 brain,60 and testis,61 mouse neuroblastoma,62 murine basophiles and macrophages.63 Palmitoyl and stearoyl ethanolamides have been found to be the two most abundant N-acyl ethanolamides in most tissues.
Figure 4.
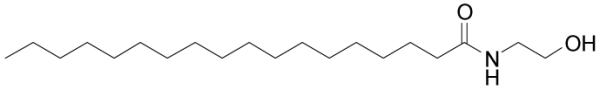
Structure of stearoyl ethanolamide.
Function
The endogenous role of stearoyl ethanolamide has yet to be fully elucidated. It does not bind cannabinoid receptors, however it can affect cell signaling and elicit biological effects potentially through targets other than cannabinoid receptors. Although these pathways are not yet understood, stearoyl ethanolamide has been shown to enhance AP-1 transcriptional activity mediated by the extracellular-signed-regulated protein kinase (ERK) mitogen-activated protein kinase (MAP kinase) pathway. In 2001, steaoryl ethanolamide was shown to stimulate AP-1 activity in mouse epidermal JB6 P+ cells through the ERK MAP kinase pathway.64
It is known that high levels of saturated versus unsaturated ethanolamides accumulate in injured tissue.65 By employing a murine model of passive IgE-induced cutaneous anaphylaxis, stearoyl ethanolamide was shown to possess anti-inflammatory properties in vivo. The results demonstrated that an acute systemic administration of stearoyl ethanolamide markedly conteracts the edema in the pinna (ipsilateral ear) of adult mice caused by cutaneous anaphylaxis.
Linoleoyl ethanolamide
Identification/Biosynthesis
As with other members of this class, linoleoyl ethanolamide (Figure 5) was detected in porcine brain and murine peritoneal macrophages.66 In addition, linoleoyl ethanolamide also has been isolated from mouse J774 macrophages and N18 neuroblastoma cells62 as well as RBL-2H3 leukocytes.67 The biosynthesis has not been studied in detail, but is presumed to be analogous to that of more frequently studied N-acyl ethanolamides, including anandamide.
Figure 5.
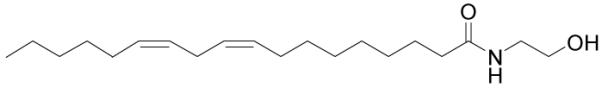
Structure of linoleoyl ethanolamide.
Function
Linoleoyl ethanolamide is approximately 4-fold less potent than anandamide at causing catalepsy in mice and it does not prolong sleep time.68 Hanus and coworkers reported that it binds to cannabinoid receptors and inhibits the electrically evoked twitch response of mouse isolated vas deferense similar to anandamide and other N-acyl ethanolamides.69 However, linoleoyl ethanolamide has been found to only weakly bind CB1 and CB2 receptors, inhibiting the binding of [3H]CP-55,940 with Ki values of 10 μM and 25 μM, respectively.70 In addition, linoleoyl ethanolamide may be involved in regulation of food intake by selective prolongation of feeding latency and post-meal interval. It appears to be formed locally in the intestine, where it activates PPAR-α.71
Fatty Acid Primary Amides
Less appreciated, but equally important, the endogenous fatty acid primary amides72 emerged as candidate signaling molecules with the discovery,73 disclosure,74 and surprisingly structural selectivity75 that oleamide displays in exerting a fundamental role in regulating sleep. In this work and key to the field, its rapid enzymatic inactivation by hydrolysis led to the detection,74 characterization,13 and study of fatty acid amide hydrolase (FAAH), which regulates the activity of fatty acid primary amides at their sites of action. An especially attractive feature of this class of fatty acid amide signaling molecules76 is the fact that they are capped as a primary amide analogous to the widely recognized peptide signaling molecules suggesting conserved strategies for their biosynthesis, precursor storage, and release. Often overlooked in the screening for endogenous ligands and because of their rapid degradation (hydrolysis) by fatty acid amide hydrolase (FAAH),13b it is likely that the most important fundamental endogenous role of many members of this class remain to be defined.
Oleamide
Identification
Fatty acid primary amides form a group of endogenous lipid messengers of growing interest. In 1995, groups at Scripps isolated a novel lipid in the cerebrospinal fluid of sleep-deprived cats.73 It was shortly thereafter identified as oleamide, the primary amide of oleic acid.74,75 Oleamide or cis-9,10-octadecenamide has since attracted wide interest being the first fatty acid primary amide to be identified as a signaling molecule (Figure 6). In addition to serving as a chemical messenger signaling sleep,74,77 it exhibits cannabinoid-like activity,78 and has been shown to have direct agonist action at CB1 cannabinoid receptors.78,79 Oleamide has also been observed to interact directly with voltage-gated Na+ channels and allosterically with GABAA and several 5-hydroxytryptamine (5-HT) receptor subtypes.
Figure 6.
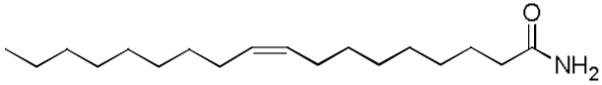
Structure of oleamide.
Biosynthesis
One of the key unanswered questions is how endogenous oleamide is produced. Currently there are several proposed pathways for its biosynthesis that have some experimental support. Glutamine can serve as an ammonia source for many amination reactions in vivo. A modest glutamine-dependent biosynthesis of oleamide from oleic acid was observed in rat brain microsomes61 and similar observations in mouse neuroblastoma cells have been reported.80 A second pathway has been suggested in which oleamide can be endogenously derived from its glycine adduct. This biosynthetic route entails the production of the amide of oleic acid with glycine or the N-terminal glycine of a peptide by an unidentified enzyme, followed by the oxidative cleavage of this acyl glycine by peptidylglycine α-amidating monooxygenase (PAM). PAM is a well-characterized enzyme involved in the production of C-terminally amidated neuropeptides.81 Recent in vitro studies have demonstrated that PAM efficiently generates oleamide from its corresponding glycine adduct.82,83 Merkler and coworkers have also shown that the N18TG2 cell line can synthesize oleamide from oleic acid,84 thereby demonstrating that these cells contain the necessary catalytic activities for generating oleamide.
Function
Most prominent among its effects is the ability of oleamide to induce natural physiological sleep. Unlike typical sleep aids that act as CNS depressants, oleamide induces sleep indistinguishable from physiological sleep without the side effects of such sedatives or hypnotics. A key feature to emerge from these studies was the observation that removal of the cis double bond, its conversion to a trans double bond, or even its movement along the fatty acid chain by a single carbon reduced or abolished the sleep-inducing effects of the compound.74
The characteristic tetrad of effects evoked by cannabinoid receptor agonists in vivo is hypothermia, hypoactivity, analgesia and catalepsy. Observations concerning the direct interaction of oleamide with cannabinoid receptors are conflicting. However, oleamide produces a dose-dependent hypothermia and a decrease in locomotor activity in both mice and rats.85,86 It induces catalepsy in mice, but not in rats87 and it produces antinoception in both species.88 Studies using the selective CB1 antagonist SR141716A have also produced conflicting results. SR141716A was shown to reverse the effects of oleamide on sleep,89 locomotor activity and antinoception, yet failed to reverse locomotion.90 In both studies, SR141716A failed to reverse oleamide-induced hypothermia. An unusual, and likely irrelevant, “entourage effect”91,16d of oleamide was proposed to account for its biological properties suggesting that they arise instead from the actions (concentration) of anandamide that are potentiated by competitive hydrolysis of oleamide by the enzyme fatty acid amide hydrolase (FAAH). In addition, Cheer78 and more recently Leggett79 demonstrated that oleamide does bind the CB1 receptor. Using radiolabeled ligand binding studies, it was shown that oleamide inhibits agonist [3H]CP55940 binding to CB1 receptors. Oleamide also acts as an agonist at CB1 as shown by an increase in [35S]GTPγS binding in rat brain slices in a pattern that mimicks that of the cannabinoid receptor agonist HU-210 and this receptor stimulation was blocked by the CB1 antagonist SR141716A. These studies indicate that oleamide is an endogenous cannabinoid receptor full agonist with selectivity for CB1 over CB2. Characteristic of the challenges in interpreting the results of such reports is the rapid hydrolysis and inactivation of oleamide by fatty acid amide hydrolase (FAAH).13,92 Studies enlisting cell-based binding or functional assays should be carried out in the presence of a FAAH inhibitor in order to ensure maintenance of accurate concentrations of oleamide and such variations may account for the distinctions observed in many of the conflicting reports.
It is also likely that not all sites of action and perhaps not even the major site of action of oleamide have yet been identified. Consequently, oleamide’s endogenous site of action remains unclear, but it has been shown to modulate both serotonergic and GABAergic receptor types in vitro, two neurotransmitter systems typically associated with the control of sleep-wake processes in vivo. Basile and coworkers quantified the changes in oleamide levels in the CSF of sleep-deprived rats, demonstrating a 3- to 4-fold increase in the compound’s concentration upon sleep deprivation for 6 or more hours.88 It has been shown that the GABAA receptor antagonist bicuculline reverses oleamide-induced hypothermia and analgesia and elimination of the β subunit of the GABAA receptor prevents oleamide-induced sleep.93 Oleamide’s endogenous and temporal associations are consistent with those required of cannabinoid, serotonergic, GABAnergic, or ion channel neurotransmission which may be involved in sleep induction.94
It has also been shown that inhibitory synaptic currents in rat GABAA receptors are sensitive to modulation by oleamide. Oleamide reversibly induces GABAA currents and depresses the frequency of spontaneous excitatory and inhibitory synaptic activity in cultured networks.95 Synthetic depressant drugs are recognized as allosteric modulators of ion channel targets like the GABAA receptor and voltage-gated Na+ channels. Oleamide has been found to be a nonselective modulator of inhibitory ionotropic receptors and has been shown to act indirectly at an allosteric site on the GABAA receptor in a fashion analogous to benzodiazepine binding.96
Studies have indicated that oleamide affects multiple neuropathway systems. Oleamide has been shown to modulate the signaling of several 5-hydroxytryptamine (5-HT) receptor subtypes, including 5-HT1A, 5-HT2A/C, and 5-HT7.97 Previous studies by Huidroboro-Toro and Harris98 indicate that oleamide potentiates 5-HT2A/C-mediated chlorine currents in frog oocyte systems. In this system, the chlorine currents elicited by 5-HT2A/C result from a signaling cascade involving phosphoinositide hydrolysis and inosital trisphosphate stimulation. Thomas and coworkers99 measured the effect of oleamide directly on phosphoinositide hydrolysis and demonstrated that oleamide substantially increases 5-HT-induced hydrolysis in P11 cells. Functional studies by Thomas and coworkers100 and Hedlund101 indicate that oleamide acts at an allosteric site on the 5-HT7 receptor to influence G protein signaling regulating cyclic AMP formation. Oleamide has demonstrated a 50% increase in cyclic AMP production in HeLa cells expressing the 5-HT7 receptor. In addition, oleamide induced a concentration dependent increase in cyclic AMP formation that could not be inhibited by clozapine suggesting that it acts at a site distinct from the primary 5-HT binding site. Oleamide has also been shown to activate 5-HT7 neurons in mouse thalamus and hypothalamus.
Oleamide has also been reported to interact with gap junctions, and has been utilized as a tool to inhibit their function. It has been reported that oleamide blocks dye transfer between rat glial cells in culture102 and blocks junctions formed by cells expressing Cx32 (β1 connexin), but does not block Ca2+-wave propagation between glial cells.103,104 Other compounds traditionally used as inhititors of gap junction communication, like heptanol, block not only gap junction communication, but also intracellular Ca2+ signaling. Thus, oleamide might have selective effects on the permeability of gap junctions, an effect that can be exploited. In view of the importance of gap junctions in the cardiovascular system, the heart, endothelial cells, and vascular muscle, this aspect of its biology is of particular relevance.
Additional fatty acid primary amides
The primary amides of oleic (18:19), palmitic (16:0), palmitoleic (16:19), elaidic (18:19-trans), and linoleic (18:29,12) were identified in human plasma before physiological roles were established (Figure 7).105 Linoleamide was found to induce sleep and increase cytosolic Ca2+ levels in MDCK tubular cells.106 Arachidonamide has been reported to affect gap junction communication.102 Erucamide (22:113) has also been identified as the major angiogenic component in bovine mesenterial fluids stimulating new blood vessel formation and was reported to act as a modulator of water balance.107 More recently, additional fatty acid amides including stearamide, palmitamide, and myristamide were isolated from human tear gland secretions.108 However, the actions of these signaling molecules remain to be elucidated. The fact that arachidonamide, the primary amide of arachidonic acid, is the best substrate for FAAH being hydrolyzed and inactivated faster than oleamide (3-fold) or anandamide (2-fold),13b suggests that it represents a key signaling molecule in this class. Because its physiological role is yet to be defined, arachidonamide represents a prime candidate to examine in existing or new targets for the fatty acid amides and should be done so in the presence of FAAH inhibitors to block its rapid inactivation.
Figure 7.
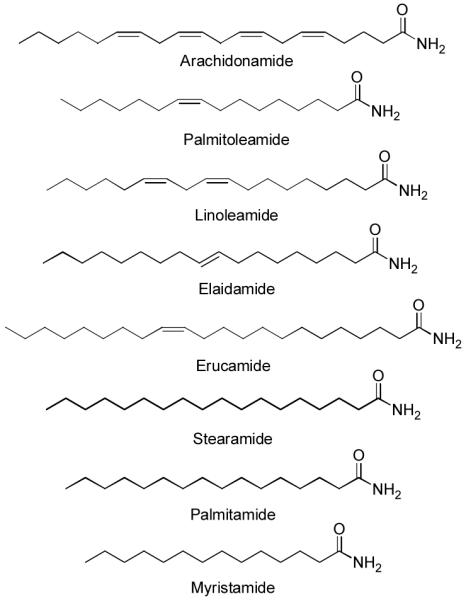
Additional endogenous fatty acid primary amides.
Glycine amidesa
An intriging series of N-acyl glycinamides bearing fatty acid acyl groups have been identified as endogenous fatty acid amides that are attracking increasing attention. At present, it is not yet clear whether they serve as chemical signaling molecules in their own right, or whether they are simply biosynthetic precursors to the active fatty acid primary amides.
N-Arachidonoylglycine
Isolation/Identification
N-Arachidonoylglycine (NAGly) has been isolated from cell cultures treated with anandamide (AEA),109 from extracts of mammalian brain,110 and has been synthesized as an analog of anandamide for structure-activity testing (Figure 8).111 Its biosynthesis is poorly understood to date, and two primary biosynthetic pathways have been proposed. One suggests that NAGly is formed by an enzymatically regulated conjugation of arachidonic acid and glycine. The other suggests that NAGly is an oxidative metabolite of AEA through the action of an alcohol dehydrogenase. In vivo and in vitro assays measuring metabolites with LC/MS/MS support the hypothesis that NAGly is a metabolite product of AEA by both oxidative metabolism of AEA and through the conjugation of glycine to arachidonic acid that is released during AEA hydrolysis by FAAH.112 It is notable that endogenous levels of NAGly are greater than those of anandamide in the CNS.
Figure 8.
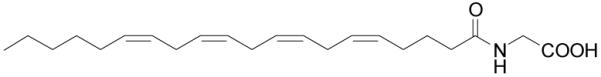
Structure of N-arachidonoylglycine.
Function
NAGly is a very poor ligand for the CB1 and CB2 receptors but has shown pain-relieving and anti-inflammatory effects in rodents.113 Other signaling effects of NAGly have been indentified, including activation of the orphan G protein coupled receptors GPR18114 and GPR92.115 An inhibitory interaction with the glycine transporter GLYT2a116 and inhibition of AEA hydrolysis by FAAH have also been reported.117 It is also a substrate of cyclooxygenase 2, producing an Gly amino acid conjugate of prostaglandins.118 These observations have been interpreted to suggest that effects of NAGly may be derived from an increase in the concentration of AEA, or from modulating the ratio of prostaglandins from the pro-inflammatory PGE2 towards the inflammation-resolving J prostaglandins.119
N-Palmitoylglycine
Isolation/Identification
N-palmitoylglycine (PalGly) is produced in high levels after cellular stimulation (KCl-induced depolarization of F-11 cells) in rat skin and spinal cord (Figure 9). It is present in 100-fold greater amounts in skin and 3-fold greater amounts in brain compared to anandamide.120
Figure 9.
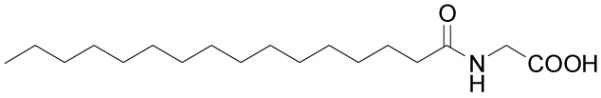
Structure of N-palmitoylglycine.
Function
PalGly was up-regulated in FAAH knock-out mice suggesting a pathway for enzymatic regulation. It potently inhibits heat-evoked firing of nociceptive neurons in rat dorsal horn, and induced transient calcium influx in native adult dorsalroot ganglion (DRG) cells and a DRG-like cell line (F-11). It also contributed to the production of NO through calcium-sensitive nitric-oxide synthase enzymes present in F-11 cells and this activity was inhibited by the nitric-oxide synthase inhibitor 7-nitroindazole.120
N-Oleoylglycine
Identification/Biosynthesis
N-Oleoyglycine (OlGly) was first isolated from rat brain matrix and later detected in rat skin, lung, liver, kidney, heart, testes and spinal cord (Figure 10).121 The N-acyl glycines are produced in vivo from the fatty acyl-CoA thioesters and glycine by acyl-CoA:glycine N-acyltransferase (ACGNAT). Mueller and Driscoll demonstrated that cytochrome c catalyzes the formation of oleoylglycine from oleoyl-CoA, glycine and hydrogen peroxide.122 Oleoylglycine has been proposed to be an important intermediate in the PAM-mediated biosynthesis of oleamide from oleic acid. In experiments with N18TG2 cells, Merkler detected oleoylglycine by mass spectroscopy as an intermediate in this biosynthetic pathway.123
Figure 10.
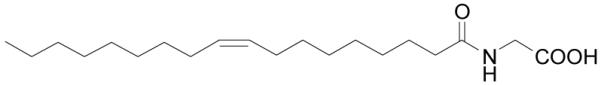
Structure of N-oleoylglycine.
Function
Chatuervedi suggested that oleoylglycine possesses biological activity that is independent of its conversion to oleamide. Oleoylglycine was found to be equipotent with oleamide in decreasing locomotion and body temperature.124 However, the full extent of it’s actions have yet to be elucidated.
Other N-acyl glycines
Isolation
Along with oleoylglycine, stearoyl (StrGly), linoleoyl (LinGly) and docosahexaenoyl glycine (DocGly) were also detected in rat brain, skin, liver, kidney, spinal cord, heart and testes (Figure 11). Levels in the skin, lungs, and spinal cord were highest in stearoyl, oleoyl, and docosahexaenoyl glycine while levels of linoleoyl glycine in the spinal cord were lower than all the other N-acyl glycines measured.121
Figure 11.
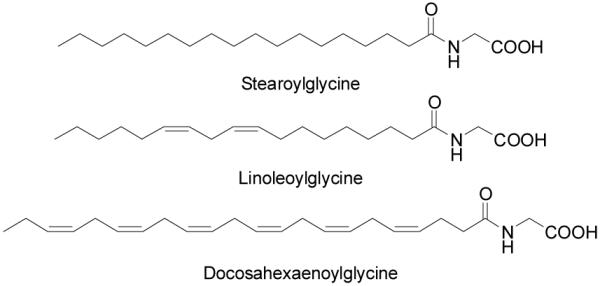
Additional N-acyl glycines.
Function
Burstein demonstrated that docosahexaenoyl and linoleoyl glycine suppress proliferation of the murine macrophage cell line, RAW264.7, whereas oleoylglycine had no effect.125 Many of these acyl glycines have yet to be carefully studied.
N-Acyl taurines
Recent efforts using highly sensitive MS techniques and comparative global metabolomic profiling of FAAH knockout and wild type mice led to the identification of a new class of endogenous fatty acid amides in the CNS.126
Identification/Biosynthesis
In 2004, Cravatt and coworkers discovered the presence and a 10-fold increase of long chain (≥C20) saturated N-acyl taurines (NATs) in the central nervous system of FAAH knockout mice.126,127 N-Acyl taurines isolated in the central nervous system were highly enriched in long chain saturated and monounsaturated N-acyl chains while those found in the kidney and liver were enriched in polyunsaturated N-acyl chains.128
The identity of the enzyme responsible for NAT biosynthesis remains to be elucidated. However, high levels of an activity capable of biosynthesizing NATs from fatty acyl CoA and taurine were detected in the liver and kidney.129 The bile acid-CoA:amino acid N-acyltransferase (BAT) enzyme responsible for bile salt production is also enriched in the liver.130 This enzyme could potentially catalyze the formation of NATs. Consistent with this premise, human BAT has been shown to form N-acyl glycines when incubated with fatty acyl CoA substrates in vitro.131
Function
N-Arachidonyl taurine (Figure 12), in particular, was found to activate multiple members of the transient receptor potential (TRP) family of calcium channels, including TRPV1 and TRPV4,132 both of which are expressed in the kidney. These channels have been proposed to play a role in the regulation of blood pressure and osmotic sensation. It has been noted that elevations in endogenous levels of NATs following acute or chronic inactivation of FAAH, suggests that NATs could form a major lipid signaling system, similar to N-acyl ethanolamides.132
Figure 12.
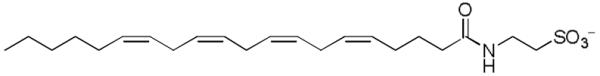
Structure of an N-acyl taurine.
Key Structurally Related Fatty Acid Derived Signaling Molecules
2-Arachidonylglycerol
Isolation/Identification
2-Arachidonylglycerol (2-AG) was isolated in 1995 from canine gut133 and rat brain (Figure 13).134 It represents a second cannabinoid receptor ligand class and possesses an ester versus amide. It was the first putative endogenous cannabinoid receptor agonist isolated from peripheral tissue. Unlike anandamide, 2-AG is present at relatively high levels in the central nervous system (100-fold higher than anandamide) and it is the most abundant molecular species of monoacylglycerol found in mouse and rat brain. It has also been found in low amounts in the liver, spleen, lung and kidney.135
Figure 13.
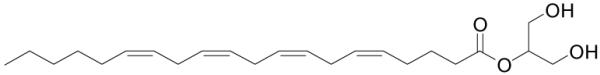
Structure of arachidonylglycerol.
Function
The formation of 2-AG is calcium-dependent and is mediated by the activities of phospholipase C (PLC) and diacylglycerol lipase (DAGL).134 The hydrolysis of 2-arachidonylglycerol to arachidonic acid and glycerol in the mouse brain, is mainly attributed to monoacylglycerol lipase, MAGL (~85%) with the remaining 15% mostly catalyzed by two uncharacterised enzymes alpha/beta-hydrolase domains 6 and 12 (ABHD6 and ABHD12).136 FAAH was identified as the next largest contributor to 2-AG hydrolysis accounting for ~1% of total membrane activity.
2-Arachidonylglycerol binds both CB1 and CB2 and acts as a full agonist.137 Despite substantial degradation, 2-AG has been shown to be a potent full efficacy agonist mediating CB1 receptor-dependent G-protein activation in rat cerebellar membranes. It has been reported that 2-AG was more potent than AEA in stimulating [35S]GTPγS binding to rat cerebellar membranes.138
A series of conformationally constrained analogs at the glycerol moiety of 2-AG incorporating its key of pharmacophore features into a six-membered carbocyclic ring system were synthesized and were tested for their affinity for CB1 and CB2 receptors. All the compounds had affinity for the cannabinoid receptors comparable to 2-AG.139
2-Arachidonoyl glyceryl ether
Isolation/Identification
Chemically, noladin ether (NE), or 2-arachidonoyl glyceryl ether (2-AGE), is the 2-glyceryl ether of arachidonyl alcohol and structurally resembles 2-AG (Figure 14). It was initially extracted from porcine140 and rat141 brain in moderate concentrations. The presence of noladin in body tissue is disputed. Although a Japanese group could not detect it in the brains of mice, hamsters, guinea-pig or pigs,142 two other groups successfully detected it in animal tissues.141,143
Figure 14.
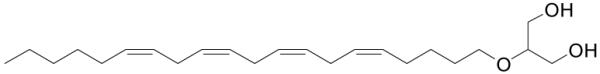
Structure of 2-arachidonoyl glyceryl ether.
Function
Noladin ether binds as a full agonist to CB1140 and is a weaker binder to CB2.144 It shows agonist behavior on both receptors and is a partial agonist for the TRPV1 receptor.145 Upon binding CB2 receptors, it inhibits adenylate cyclase, stimulates ERK-MAPK, and regulates calcium transients.144 It lowers intraocular pressure,146 increases the uptake of GABA in the globus pallidus146 and is neuroprotective by binding and activating PPAR-α.147 In comparison to 2-AG, it is metabolically more stable resulting in a longer duration of action.148
The synthesis of dimethylheptyl (DMP) analogs, with a different tail length of 2-AG and 2-AGE have been reported and showed a distinct decrease of potency towards CB1 receptors.149 Another series of mono- and diphosphate esters of NE have been reported and showed an enhancement in water-solubility compared to NE.150 Two regioisomers and 13 analogs of noladin ether were synthesized and tested for their interaction with CB1 receptors in rat brain membrane. The results showed that a C-20 tetra-unsaturated moiety is necessary for high affinity.151
Virodhamine (O-arachidonoyl ethanolamine)
Identification
Virodhamine was discovered in 2002 by Porter and coworkers during the development of a bioanalytical method to measure anandamide levels in tissue. A second peak with the same mass as anandamide but with a different retention time was detected and isolated and found to be arachidonic acid and ethanolamine joined by an ester linkage, in contrast to the amide linkage in anandamide (Figure 15). Virodhamine has been isolated from rat brain and human hippocampus in levels similar to anandamide. In the peripheral tissues, levels of virodhamine isolated were 2- to 9-fold higher than anandamide.152
Figure 15.
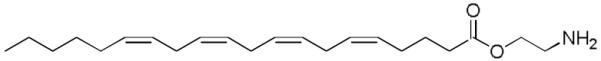
Structure of virodhamine.
Biosynthesis
It is not yet defined how virodhamine is produced, stored, or degraded. Virodhamine could be generated from a fatty acid ethanolamine and arachadonic acid by a transphosphotidylation reaction catalyzed by phospholipase D. It is also possible that virodhamine could be produced from anandamide by an enzymatically catalyzed rearrangement of the ethanolamine portion of the molecule from an amide linkage to an ester.153 Neither mechanism has been examined. It is also possible that it serves as an additional precursor of anandamide.
Virodhamine’s ability to block anandamide transport, which is largely mediated by intracellular FAAH hydrolysis, suggests that it may be degraded in a manner similar to anandamide. Since FAAH has been shown to have both amidase and esterase activity, it could be responsible for virodhamine’s hydrolysis in vivo.154
Function
Virodhamine produced dose dependent hypothermia in mice. In a [35S]GTPγS functional binding assay, EC50 values for virodhamine matched those reported for anandamide and WIN 55,212-2, both of which behave as agonists at CB1 and CB2 receptors, but it was found to be less efficacious than both anandamide and WIN 55,212-2. At CB2, virodhamine acted as a full agonist. However it acted as a partial agonist at CB1 with a maximal efficacy of 61% compared with anandamide.62 Virodhamine has been shown to relax both human and rat mesenteric arteries through endothelial cannabinoid receptors.155 Virodhamine has also been reported to have activity at the orphan receptor, GRP55.156
N-Acyl dopamine
Identification/Biosynthesis
It was suggested that certain N-acyl dopamines may exist in mammalian tissues and serve as TRPV1 ligands.157 N-Arachidonyl dopamine was first synthesized prior to its endogenous detection in 2002 in rat and bovine nervous tissues (Figure 16).158 More recently, several other N-acyl dopamines including, palmitoyl, stearoyl, and oleoyl dopamine have been detected.159 These compounds share a structural similarity with the potent TRPV1 agonist, capsaicin.
Figure 16.
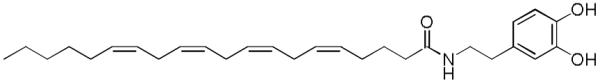
Structure of arachidonyl dopamine.
A proposed biosynthesis of arachidonyl dopamine proceeds via condensation of arachidonic acid with tyrosine and the subsequent conversion of N-arachidonoyltyrosine to N-arachidonyl dopamine by tyrosine hydroxylase and l-aromatic amino-acid decarboxylase.158
Function
To date, only arachidonyl dopamine was found to have any significant biological activity. It enhances calcium influx in cultured dorsal root ganglion neurons and TRPV1-transfected human embryonic kidney (HEK) cells.160 Patch-clamp studies of cultured dorsal root ganglion neurons showed that arachidonyl dopamine elicited reversible responses, which were blocked by both the CB1 antagonist SR141617 and the TRPV1 antagonist, capsazepine. In behavioral experiments using non-anesthesized rats, arachidonyl dopamine caused thermal hyperalgesia.129 Whether arachidonyl dopamine or related N-acyl derivatives constitute true signaling molecules or reflect metabolic artifacts is yet to be established.
Conclusions
Work conducted over the past 20 years has provided compelling evidence that fatty acid amides serve as a new and additional class of endogenous signaling molecules. To date, these fall largely into the two classes of ethanolamides and the primary amides. In spite of their simple structures and their apparent lack of distinguishing features, they exhibit surprisingly selective physiological activity that has been directly related to their selective activation (agonist) of receptor mediated events. Because of their rapid inactivation largely by enzymatic hydrolysis, they are synthesized, released, and inactivated proximal to their sites of action providing a temporal and spatial pharmacological control of their effects. To date, these have impacted some of the most fundamental processes including pain perception (analgesia), inflammation, sleep, and feeding behavior suggesting they may be among the first and most fundamental of our present day classes of signaling molecules. Devoted efforts to characterizing their synthesis, storage, and release as well as continued efforts to identify their site(s) of action are sure to not only reveal new biology not yet appreciated, but to provide new approaches and targets for therapeutic intervention in some of our most fundamental clinical disorders.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Bachur NR, Masek K, Melmon KL, Udenfriend S. J. Biol. Chem. 1965;240:1019. [PubMed] [Google Scholar]
- 2.(a) Devane WA, Dysarz FA, Johnson MR, Melvin LS, Howlett AC. Mol. Pharmacol. 1988;34:605. [PubMed] [Google Scholar]; (b) Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Nature. 1990;346:561. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]; (c) Munro S, Thomas KL, Abu-Shaar M. Nature. 1993;365:61. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 3.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 4.Okamoto Y, Wang J, Morishita J, Ueda N. Chem. Biodiv. 2007;4:1842. doi: 10.1002/cbdv.200790155. [DOI] [PubMed] [Google Scholar]
- 5.(a) Natarajan V, Reddy PV, Schmid PC, Schmid HH. Biochim. Biophys. Acta. 1982;712:342. doi: 10.1016/0005-2760(82)90352-6. [DOI] [PubMed] [Google Scholar]; (b) Schmid PC, Reddy PV, Natarajan V, Schmid HH. J. Biol.Chem. 1983;258:9302. [PubMed] [Google Scholar]; (c) Ahn K, McKinney MK, Cravatt BF. Chem. Rev. 2008;108:1687. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz J-C, Piomelli D. Nature. 1994;372:686. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 7.Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. J. Neurosci. 1996;16:3934. doi: 10.1523/JNEUROSCI.16-12-03934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cadas H, Di Tomaso E, Piomelli D. J. Neurosci. 1997;17:1226. doi: 10.1523/JNEUROSCI.17-04-01226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leung D, Saghatelian A, Simon GM, Cravatt BF. Biochemistry. 2006;45:4720. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Natarajan V, Schmid PC, Reddy PV, Schmid HH. J. Neurochem. 1984;42:1613. doi: 10.1111/j.1471-4159.1984.tb12750.x. [DOI] [PubMed] [Google Scholar]
- 11.(a) Simon GM, Cravatt BF. J. Biol. Chem. 2006;281:26465. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]; (b) Simon GM, Cravatt BF. J. Biol. Chem. 2008;281:9341. doi: 10.1074/jbc.M707807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim HY, Kunos G. Proc. Natl. Acad. Sci. U.S.A. 2006;103:13345. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu J, Wang L, Harvey-White J, Huang BX, Kim HY, Luquet S, Palmiter RD, Krystal G, Rai R, Mahadevan A, Razdan RK, Kunos G. Neuropharmacology. 2008;54:1. doi: 10.1016/j.neuropharm.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]; (b) Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF. Bioorg. Med. Chem. Lett. 2000;10:2613. doi: 10.1016/s0960-894x(00)00528-x. [DOI] [PubMed] [Google Scholar]; (c) Fowler CJ, Jonsson KD, Tiger G. Biochem. Pharmacol. 2001;62:517. doi: 10.1016/s0006-2952(01)00712-2. [DOI] [PubMed] [Google Scholar]; (d) Kaczocha M, Hermann A, Glaser ST, Bojesen IN, Deutsch DG. J. Biol. Chem. 2006;281:9066. doi: 10.1074/jbc.M509721200. [DOI] [PubMed] [Google Scholar]; (e) Day TA, Rakhshan F, Deutsch DG, Barker EL. Mol. Pharmacol. 2001;59:1369. doi: 10.1124/mol.59.6.1369. [DOI] [PubMed] [Google Scholar]
- 14.Giang DK, Cravatt BF. Proc. Natl. Acad. Sci. U.S.A. 1997;94:2238. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snider NT, Kornilov AM, Kent UM, Hollenberg PF. J. Pharmacol. Exp. Ther. 2007;321:590. doi: 10.1124/jpet.107.119321. [DOI] [PubMed] [Google Scholar]
- 16.(a) Axelrod J, Felder CC. Neurochem. Res. 1998;23:575. doi: 10.1023/a:1022418217479. [DOI] [PubMed] [Google Scholar]; (b) Di Marzo V, De Petrocellis L, Bisogno T, Melck D. Lipids. 1999;34:S319. doi: 10.1007/BF02562332. [DOI] [PubMed] [Google Scholar]; (c) Di Marzo V. Biochim. Biophys. Acta. 1998;1392:153. doi: 10.1016/s0005-2760(98)00042-3. [DOI] [PubMed] [Google Scholar]; (d) Mechoulam R, Fride E, Di Marzo V. Eur. J. Pharmacol. 1998;359:1. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]; (e) Di Marzo V, Bisogno T, De Petrocellis L, Melck D, Martin BR. Curr. Med. Chem. 1999;6:721. [PubMed] [Google Scholar]; (f) Martin BR, Mechoulam R, Razdan RK. Life Sci. 1999;65:573. doi: 10.1016/s0024-3205(99)00281-7. [DOI] [PubMed] [Google Scholar]
- 17.Adams IB, Ryan W, Singer M, Thomas BF, Compton DR, Razdan RK, Martin BR. J. Pharmacol. Exp. Ther. 1995;273:1172. [PubMed] [Google Scholar]
- 18.Showalter VM, Compton DR, Martin BR, Abood MR. J. Pharmacol. Exp. Ther. 1996;278:989. [PubMed] [Google Scholar]
- 19.Pertwee RG. Curr. Med. Chem. 1999;6:635. [PubMed] [Google Scholar]
- 20.Review: Seierstad M, Breitenbucher JG. J. Med. Chem. 2008;51:7327. doi: 10.1021/jm800311k. For one class of potent FAAH inhibitors, see: Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5044. doi: 10.1073/pnas.97.10.5044. Boger DL, Miyauchi H, Hedrick MP. Bioorg. Med. Chem. Lett. 2001;11:1517. doi: 10.1016/s0960-894x(01)00211-6. Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Aceredo O, Guimaraes CRW, Jorgensen WL, Cravatt BF. J. Med. Chem. 2005;48:1849. doi: 10.1021/jm049614v. Leung D, Du W, Hardouin C, Cheng H, Hwang I, Cravatt BF, Boger DL. Bioorg. Med. Chem. Lett. 2005;15:1423. doi: 10.1016/j.bmcl.2004.12.085. Romero FA, Hwang I, Boger DL. J. Am. Chem. Soc. 2006;128:14004. doi: 10.1021/ja064522b. Romero FA, Du W, Hwang I, Rayl TJ, Kimball FS, Leung D, Hoover HS, Apodaca RL, Breitenbucher JG, Cravatt BF, Boger DL. J. Med. Chem. 2007;50:1058. doi: 10.1021/jm0611509. Hardouin C, Kelso MJ, Romero FA, Rayl TJ, Leung D, Hwang I, Cravatt BF, Boger DL. J. Med. Chem. 2007;50:3359. doi: 10.1021/jm061414r. Kimball FS, Romero FA, Ezzili C, Garfunkle J, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. J. Med. Chem. 2008;51:937. doi: 10.1021/jm701210y. Garfunkle J, Ezzili C, Hochstatter DG, Rayl TJ, Hwang I, Boger DL. J. Med. Chem. 2008;51:4392. doi: 10.1021/jm800136b. DeMartino JK, Garfunkle J, Hochstatter DG, Cravatt BF, Boger DL. Bioorg. Med. Chem. Lett. 2008;18:5842. doi: 10.1016/j.bmcl.2008.06.084. Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. J. Am. Chem. Soc. 2009;131:10497. doi: 10.1021/ja902694n. Mileni M, Garfunkle J, Ezzili C, Kimball FS, Cravatt BF, Stevens RC, Boger DL. J. Med. Chem. 2010;53:230. doi: 10.1021/jm9012196.
- 21.(a) Brown AJ, Wise A. Patent WO0186305. 2001; (b) Drmota E, Greasley P, Groblewski T. Patent WO2004074844. 2004; (c) Brown AJ, Hiley CR. Vit. Hormones. 2009;81:111. doi: 10.1016/S0083-6729(09)81005-4. [DOI] [PubMed] [Google Scholar]
- 22.(a) Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Nature. 1999;400:452. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]; (b) Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, Chambers JK, Randall AD, Davis JB. Br. J. Pharmacol. 2000;129:227. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Smart D, Jerman JC. Trends Pharmacol. Sci. 2000;21:134. doi: 10.1016/s0165-6147(00)01459-0. [DOI] [PubMed] [Google Scholar]; (d) Olah Z, Karai L, Iadarola MJ. J. Biol. Chem. 2001;276:31163. doi: 10.1074/jbc.M101607200. [DOI] [PubMed] [Google Scholar]
- 23.(a) Calignano A, La Rana G, Giuffrida A, Piomelli D. Nature. 1998;394:277. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]; (b) Walker JM, Huang SM, Strangman NM, Tsou K, Sanudo-Pena MC. Proc. Natl. Acad. Sci. U.S.A. 1999;96:12198. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaetani S, Cuomo V, Piomelli D. Trends Mol. Med. 2004;9:474. doi: 10.1016/j.molmed.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 25.(a) Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10821. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Massa F, Marsicano G, Hermann H, Cannich A, Monory K, Cravatt BF, Ferri GL, Sibaev A, Storr M, Lutz B. J. Clin. Investig. 2004;113:1202. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lambert DM, Vandevoorde S, Jonsson KO, Fowler C. J. Curr. Med. Chem. 2002;9:663. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez de Fonseca F, Navarro M, Gomez R, Escuredo L, Nava F, Fu J, Murillo-Rodriguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. Nature. 2001;414:209. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- 27.(a) Lichtman AH, Varvel SA, Martin BR. Prostaglandins Leukot. Essent. Fatty Acids. 2002;66:269. doi: 10.1054/plef.2001.0351. [DOI] [PubMed] [Google Scholar]; (b) Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B. Nature. 2002;418:530. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 28.De Petrocellis L, Melck D, Palmisano A, Bisogno T, Laezza C, Bifulco M, Di Marzo V. Proc. Natl. Acad. Sci. U.S.A. 1998;95:8375. doi: 10.1073/pnas.95.14.8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meisinger MAP, Ormond RE, Ganley OH, Jacob TA, Kuehl FA., Jr. J. Am. Chem. Soc. 1957;79:5577. [Google Scholar]
- 30.Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. J. Biol. Chem. 2004;279:5298. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- 31.Ueda N, Yamanaka K, Yamamoto S. J. Biol. Chem. 2001;276:35552. doi: 10.1074/jbc.M106261200. [DOI] [PubMed] [Google Scholar]
- 32.Vandevoorde S, Tsuboi K, Ueda N, Jonsson KO, Fowler CJ, Lambert DM. J. Med. Chem. 2003;46:4373. doi: 10.1021/jm0340795. [DOI] [PubMed] [Google Scholar]
- 33.Perlik F, Raskova H, Ellis J. Acta Physiol. Acad. Sci. Hung. 1971;39:395. [PubMed] [Google Scholar]
- 34.Lambert DM, Vandervoorde S, Jonsson KO, Fowler CJ. Curr. Med. Chem. 2002;9:663. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- 35.Kahlich R, Klima J, Cihla F, Frankova V, Masek K, Rosicky M, Matousek F, Bruthans J. J. Hyg. Epidemiol. Microbiol. Immunol. 1979;23:11. [PubMed] [Google Scholar]
- 36.Mazzari S, Canella R, Petrelli L, Manolongo G, Leon A. Eur. J. Pharmacol. 1996;300:227. doi: 10.1016/0014-2999(96)00015-5. [DOI] [PubMed] [Google Scholar]
- 37.Berdyshev E, Boichot E, Corbel M, Germain N, Lagente V. Life Sci. 1998;63:PL125. doi: 10.1016/s0024-3205(98)00324-5. [DOI] [PubMed] [Google Scholar]
- 38.Aloe L, Leon A, Levi-Montalcini R. Agents Actions. 1993;39:C145. doi: 10.1007/BF01972748. [DOI] [PubMed] [Google Scholar]
- 39.Lambert DM, Vandevoorde S, Diependaele G, Govaerts SJ, Robert AR. Epilepsia. 2001;42:321. doi: 10.1046/j.1528-1157.2001.41499.x. [DOI] [PubMed] [Google Scholar]
- 40.Jaggar SI, Hasnie FS, Sellaturay S, Rice AS. Pain. 1998;76:189. doi: 10.1016/s0304-3959(98)00041-4. [DOI] [PubMed] [Google Scholar]
- 41.Ross RA, Brochie HC, Pertwee RG. Eur. J. Pharmacol. 2000;401:121. doi: 10.1016/s0014-2999(00)00437-4. [DOI] [PubMed] [Google Scholar]
- 42.Farquhar-Smith WP, Rice AS. Anesthesiology. 2003;99:1391. doi: 10.1097/00000542-200312000-00024. [DOI] [PubMed] [Google Scholar]
- 43.Costa B, Conti S, Giagnoni G, Colleoni M. Br. J. Pharmacol. 2002;137:413. doi: 10.1038/sj.bjp.0704900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. Mol. Pharmacol. 2005;67:15. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 45.Hoareau L, Buyse M, Festy F, Ravanan P, Gonthier MP, Matias I, Petrosino S, Tallet F, Lefebvre d’Hellencourt C, Cesari1 M, Di Marzo V, Roche R. Obesity. 2009;17:431. doi: 10.1038/oby.2008.591. [DOI] [PubMed] [Google Scholar]
- 46.Lambert DM, DiPaolo F, Sonveaux P, Kanyonyo M, Govaerts SJ, Hermans E, Bueb JL, Delzenne NM. Biochem. Biophys. Acta. 1999;1440:266. doi: 10.1016/s1388-1981(99)00132-8. [DOI] [PubMed] [Google Scholar]
- 47.Del Carmen García M, Adler-Graschinsky A, Celuch SM. Eur. J. Pharmacol. 2009;610:75. doi: 10.1016/j.ejphar.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 48.Ho WSV, Barrett DA, Randall MD. Br. J. Pharmacol. 2008;155:837. doi: 10.1038/bjp.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bisogno T, Martire A, Petrosino S, Popoli P, Di Marzo V. Neurochem. Intern. 2008;52:307. doi: 10.1016/j.neuint.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 50.Schuel H, Burkman LJ, Lippes J, Crickard K, Forester E, Piomelli D, Giuffrida A. Chem. Phys. Lipids. 2002;121:211. doi: 10.1016/s0009-3084(02)00158-5. [DOI] [PubMed] [Google Scholar]
- 51.Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez de Fonseca F. Nature. 2003;425:90. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 52.Ahern GP. J. Biol. Chem. 2003;278:30429. doi: 10.1074/jbc.M305051200. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Miyares RL, Ahern GP. J. Physiol. 2005;564:541. doi: 10.1113/jphysiol.2004.081844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lo Verme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. J. Pharmacol. Exp. Ther. 2006;319:1051. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- 55.Suardíaz M, Estivill-Torrús G, Goicoechea C, Bilbao A, Rodríguez de Fonseca F. Pain. 2007;133:99. doi: 10.1016/j.pain.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 56.Overton HA, Babbs AJ, Doel SM, Fyfe MCT, Gardner LS, Griffin G, Jackson HC, Proctor MJ, Rasamison CM, Tang-Christensen M, Widdowson PS, Williams GM, Reynet C. Cell Metab. 2006;3:167. doi: 10.1016/j.cmet.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 57.(a) Soga T, Ohishi T, Matsui T, Saito T, Matsumoto M, Takasaki J, Matsumoto S, Kamohara M, Hiyama H, Yoshida S, Momose K, Ueda Y, Matsushime H, Kobori M, Furuichi K. Biochem. Biophys. Res. Commun. 2005;326:744. doi: 10.1016/j.bbrc.2004.11.120. [DOI] [PubMed] [Google Scholar]; (b) Chu A-L, Jones RM, He H, Carroll C, Gutierrez V, Lucman A, Moloney M, Gao H, Mondala H, Bagnol D, Unett D, Liang Y, Demarest K, Semple G, Behan DP, Leonard J. Endocrinology. 2007;148:2601. doi: 10.1210/en.2006-1608. [DOI] [PubMed] [Google Scholar]; (c) Chu ZL, Carroll C, Alfonso J, Gutierrez V, He H, Lucman A, Pedraza M, Mondala H, Gao H, Bagnol D, Chen R, Jones RM, Behan DP, Leonard J. Endocrinology. 2008;149:2038. doi: 10.1210/en.2007-0966. [DOI] [PubMed] [Google Scholar]
- 58.Lan H, Vassileva G, Corona A, Liu L, Baker H, Golovko A. Diabetes: Molecular Genetics, Signalling Pathways and Integrated Physiology. 2007 Abstract 253. [Google Scholar]
- 59.Cano C, Pavon J, Serrano A, Goya P, Paez JA, Rodriguez de Fonseca F, Macias-Gonzalez M. J. Med. Chem. 2007;50:389. doi: 10.1021/jm0601102. [DOI] [PubMed] [Google Scholar]
- 60.Schmid PC, Krebsbach RJ, Perry SR, Dettmer TM, Maasson JL, Schmid HHO. FEBS Lett. 1995;375:117. doi: 10.1016/0014-5793(95)01194-j. [DOI] [PubMed] [Google Scholar]
- 61.Sugiura T, Kondo S, Kodaka T, Tonegawa T, Nakane S, Yamashita A, Ishima Y, Waku K. Biochem. Mol. Biol. Int. 1996;40:931. doi: 10.1080/15216549600201553. [DOI] [PubMed] [Google Scholar]
- 62.Di Marzo V, De Petrocellis L, Sepe N, Buono A. Biochem. J. 1996;316:977. doi: 10.1042/bj3160977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bigosno T, Maurelli S, Melck D. J. Biol. Chem. 1997;272:3315. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- 64.Berdyshev EV, Schmid PC, Krebsbach RJ, Hillard CJ, Huang C, Chen N, Dong Z, Schmid HO. Biochem. J. 2001;360:67. doi: 10.1042/0264-6021:3600067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carbonare MD, Giudice ED, Stecca A, Colavito D, Fabris M, D’Arrigo A, Bernardini D, Dam M, Leon A. J. Neuroendoc. 2008;20:26. doi: 10.1111/j.1365-2826.2008.01689.x. [DOI] [PubMed] [Google Scholar]
- 66.Hanus L, Gopher A, Almog S, Mechoulam R. J. Med. Chem. 1993;36:3032. doi: 10.1021/jm00072a026. [DOI] [PubMed] [Google Scholar]
- 67.Bisogno T, Venriglia M, Milone A, Mosca M, Cimino G, Di Marzo V. Biochim. Biophys. Acta. 1997;1345:338. doi: 10.1016/s0005-2760(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe K, Matsunaga T, Nakamura S, Kimura T, Ho IK, Yoshimura H, Yamamoto I. Biol. Pharm. Bull. 1999;22:366. doi: 10.1248/bpb.22.366. [DOI] [PubMed] [Google Scholar]
- 69.Pertwee R, Griffin G, Hanus L, Mechoulam R. Eur. J. Pharmacol. 1994;259:115. doi: 10.1016/0014-2999(94)90499-5. [DOI] [PubMed] [Google Scholar]
- 70.Lin S, Khanolkar AD, Fan P, Goutopoulos A, Qin C, Papahadjis D, Makriyannis A. J. Med. Chem. 1998;41:5353. doi: 10.1021/jm970257g. [DOI] [PubMed] [Google Scholar]
- 71.Hansen HS, Diep TA. Biochem. Pharmacol. 2009;78:553. doi: 10.1016/j.bcp.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 72.Boger DL, Henriksen SJ, Cravatt BF. Current Pharmaceutical Design. 1998;4:303. [PubMed] [Google Scholar]
- 73.Lerner RA, Suizdak G, Prospero-Garcia O, Hendriksen SJ, Boger DL, Cravatt BF. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9505. doi: 10.1073/pnas.91.20.9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cravatt BF, Prospero-Garcia O, Szuidak S, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. Science. 1995;268:1506. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- 75.Cravatt BF, Lerner RA, Boger DL. J. Am. Chem. Soc. 1996;118:580. [Google Scholar]
- 76.(a) Patterson JE, Ollmann IR, Cravatt BF, Boger DL, Wong CH, Lerner RA. J. Am. Chem. Soc. 1996;118:5938. [Google Scholar]; (b) Boger DL, Sato H, Lerner AE, Austin BJ, Patterson JE, Patricelli MP, Cravatt BF. Bioorg. Med. Chem. Lett. 1999;9:265. doi: 10.1016/s0960-894x(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 77.(a) Huitron-Resendiz S, Gombart L, Cravatt BF, Henriksen S. J. Exp. Neurol. 2001;172:235. doi: 10.1006/exnr.2001.7792. [DOI] [PubMed] [Google Scholar]; (b) Huitron-Resendiz, Sanchez-Alavez M, Will DN, Cravatt BF, Henriksen SJ. Sleep. 2004;27:857. doi: 10.1093/sleep/27.5.857. [DOI] [PubMed] [Google Scholar]
- 78.Cheer JF, Cadogan AK, Marsden CA, Fone KCF, Kendall DA. Neuropharmacology. 1999;38:533. doi: 10.1016/s0028-3908(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 79.Leggett JD, Aspley S, Beckett SRG, D’Antona AM, Kendall DA. Br. J. Pharmacol. 2004;141:253. doi: 10.1038/sj.bjp.0705607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bisogno T, Sepe N, De Petrocellis L, Mechoulam R, Di Marzo V. Biochem. Biophys. Res. Commun. 1997;239:473. doi: 10.1006/bbrc.1997.7431. [DOI] [PubMed] [Google Scholar]
- 81.Driscoll WJ, Mueller SA, Eipper BA, Mueller GP. Mol. Pharmacol. 1999;55:1067. doi: 10.1124/mol.55.6.1067. [DOI] [PubMed] [Google Scholar]
- 82.Wilcox BJ, Ritenour-Rogers KJ, Asser AS, Baumgart LE, Baumgart MA, Boger DL, DeBlessio JL, deLong MA, Glufke U, Henz ME, King L, III, Merkler KA, Patterson JE, Robleski JJ, Vederas JC, Merkler DJ. Biochemistry. 1999;38:3235. doi: 10.1021/bi982255j. [DOI] [PubMed] [Google Scholar]
- 83.Merkler DJ, Chew GH, Gee AJ, Merkler KA, Sorondo JPO, Johnson ME. Biochemistry. 2004;43:12667. doi: 10.1021/bi049529p. [DOI] [PubMed] [Google Scholar]
- 84.Ritenour-Rogers KJ, Driscoll WJ, Merkler KA, Merkler DJ, Mueller GP. Biochem. Biophys. Res. Commun. 2000;267:521. doi: 10.1006/bbrc.1999.1977. [DOI] [PubMed] [Google Scholar]
- 85.Huitron-Resendiz S, Gombart L, Cravatt BF, Henriksen SJ. Exp. Neurol. 2001;172:235. doi: 10.1006/exnr.2001.7792. [DOI] [PubMed] [Google Scholar]
- 86.Mechoulam R, Fride E, Hanus L, Sheskin T, Bisogno T, Di Marzo V, Bayewitch M, Vogel Z. Nature. 1997;389:25. doi: 10.1038/37891. [DOI] [PubMed] [Google Scholar]
- 87.Federova I, Hahsimoto A, Fecik RA, Hedrick MP, Hanus LO, Boger DL, Rice KC, Basile AS. J. Pharmacol. Exp. Ther. 2001;299:332. [PubMed] [Google Scholar]
- 88.Murillo-Rodriguez E. Progress in Neuro-Psycopharmacology and Biological Psychiatry. 2008;32:1420. doi: 10.1016/j.pnpbp.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 89.Basile AS, Hanus L, Mendelson WB. NeuroReport. 1999;10:947. doi: 10.1097/00001756-199904060-00010. [DOI] [PubMed] [Google Scholar]
- 90.Lichtman AH, Hawkins EG, Griffin G, Cravatt BF. J. Pharmacol. Exp. Ther. 2002;302:73. doi: 10.1124/jpet.302.1.73. [DOI] [PubMed] [Google Scholar]
- 91.Lambert DM, Di Marzo V. Curr. Med. Chem. 1999;6:757. [PubMed] [Google Scholar]
- 92.Laposky AD, Homanics GE, Basile A, Mendelson WB. NeuroRep. 2001;12:4134. doi: 10.1097/00001756-200112210-00056. [DOI] [PubMed] [Google Scholar]
- 93.Verdon B, Zheng J, Nicholson RA, Ganellin CR, Less G. Br. J. Pharmacol. 2000;129:283. doi: 10.1038/sj.bjp.0703051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Less G, Edwards MD, Hassoni AA, Ganellin CR, Galanakis D. Br. J. Pharmacol. 1998;124:873. doi: 10.1038/sj.bjp.0701918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Coyne L, Lees G, Nicholson RA, Zheng J, Neufield KD. Br. J. Pharmacol. 2002;135:1977. doi: 10.1038/sj.bjp.0704651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yost CS, Hampson AJ, Leonoudakis D, Koblin DD, Bornheim LM, Gray AT. Anesth. Analg. 1998;86:1294. doi: 10.1097/00000539-199806000-00031. [DOI] [PubMed] [Google Scholar]
- 97.Boger DL, Patterson JE, Jin Q. Proc. Natl. Acad. Sci. U.S.A. 1998;95:4102. doi: 10.1073/pnas.95.8.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huidobro-Toro JP, Harris RA. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8078. doi: 10.1073/pnas.93.15.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thomas EA, Carson MJ, Neal MJ, Sutcliffe JG. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14115. doi: 10.1073/pnas.94.25.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thomas EA, Cravatt BF, Sutcliffe JG. J. Neurochem. 1999;21:2370. doi: 10.1046/j.1471-4159.1999.0722370.x. [DOI] [PubMed] [Google Scholar]
- 101.Hedlund PB, Carson MJ, Sutcliffe JG, Thomas EA. Biochem. Pharmacol. 1999;58:1807. doi: 10.1016/s0006-2952(99)00274-9. [DOI] [PubMed] [Google Scholar]
- 102.Boger DL, Sato H, Lerner RA, Guan X, Gilula NB. Bioorg. Med. Chem. Lett. 1999;9:1151. doi: 10.1016/s0960-894x(99)00148-1. [DOI] [PubMed] [Google Scholar]
- 103.Guan X, Cravatt BF, Ehring GR, Hall JE, Boger DL, Lerner RA, Gilula NB. J. Cell Biol. 1997;139:1785. doi: 10.1083/jcb.139.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Boger DL, Patterson JE, Guan X, Cravatt BF, Lerner RA, Gilula NB. Proc. Natl. Acad. Sci. U.S.A. 1998;95:4810. doi: 10.1073/pnas.95.9.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Arafat ES, Trimble JW, Andersen RN, Dass C, Desiderio DM. Life Sci. 1989;45:1679. doi: 10.1016/0024-3205(89)90278-6. [DOI] [PubMed] [Google Scholar]
- 106.Huang JK, Jan CR. Life Sci. 2001;68:997. doi: 10.1016/s0024-3205(00)01002-x. [DOI] [PubMed] [Google Scholar]
- 107.(a) Wakamatsu K, Masaki T, Itoh F, Kondo K, Sudo K. Biochem. Biophys. Res. Commun. 1990;168:423. doi: 10.1016/0006-291x(90)92338-z. [DOI] [PubMed] [Google Scholar]; (b) Hamberger A, Stenhagen G. Neurochem. Res. 2003;28:177. doi: 10.1023/a:1022364830421. [DOI] [PubMed] [Google Scholar]
- 108.Nichols KK, Ham BM, Nichols JJ, Ziegler C, Green-Church KB. Invest. Ophtalmol. Vis. Sci. 2007;48:34. doi: 10.1167/iovs.06-0753. [DOI] [PubMed] [Google Scholar]
- 109.Burstein SH, Rossetti RG, Yagen B, Zurier RB. Prostaglandins Other Lipid Mediat. 2000;61:29. doi: 10.1016/s0090-6980(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 110.Huang SM, Bisogno T, Petros TJ, Chang SY, Zavitsanos PA, Zipkin RE, Sivakumar R, Coop A, Maeda DY, De Petrocellis L, Burstein S, Di Marzo V, Walker JM. J. Biol. Chem. 2001;276:42639. doi: 10.1074/jbc.M107351200. [DOI] [PubMed] [Google Scholar]
- 111.Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. J. Med. Chem. 1997;40:659. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- 112.Bradshaw HH, Rimmerman N, Shu-Jung Hu S, Benton VM, Stuart JM, Masuda K, Cravatt BF, O’Dell DK, Walker JM. BMC Biochemistry. 2009;10:1. doi: 10.1186/1471-2091-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.(1) Succar R, Mitchell VA, Vaughan CW. Mol. Pain. 2007;3:1. doi: 10.1186/1744-8069-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]; (2) Vuong LAQ, Mitchell VA, Vaughan CW. Neuropharmacol. 2008;54:189. doi: 10.1016/j.neuropharm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 114.(a) Kohno M, Hasegawa H, Inoue A, Muraoka M, Miyazaki T, Oka K, Yasukawa M. Biochem. Biophys. Res. Commun. 2006;347:827. doi: 10.1016/j.bbrc.2006.06.175. [DOI] [PubMed] [Google Scholar]; (b) McHugh D, Hu SS-J, Rimmerman N, Juknat A, Vogel Z, Walker JM, Bradshaw HB. BMC Neuroscience. 2010;11:44. doi: 10.1186/1471-2202-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oh DY, Yoon JM, Moon MJ, Hwang J-L, Choe H, Lee JY, Kim J, Kim S, Rhim H, O’Dell DK, Walker JM, Na HS, Lee MG, Kwon HB, Kim K, Seong JY. J. Biol. Chem. 2008;283:21054. doi: 10.1074/jbc.M708908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wiles AL, Pearlman R-J, Rosvall M, Aubrey KR, Vandenberg RJ. J. Neurochem. 2006;99:781. doi: 10.1111/j.1471-4159.2006.04107.x. [DOI] [PubMed] [Google Scholar]
- 117.Burstein SH, Huang SM, Petros TJ, Rossetti RG, Walker JM, Zurier RB. Biochem. Pharmacol. 2002;64:1147. doi: 10.1016/s0006-2952(02)01301-1. [DOI] [PubMed] [Google Scholar]
- 118.Prusakiewicz JJ, Kingsley PJ, Kozak KR, Marnett LJ. Biochem. Biophys. Res. Commun. 2002;296:612. doi: 10.1016/s0006-291x(02)00915-4. [DOI] [PubMed] [Google Scholar]
- 119.Burstein SH, Adams JK, Bradshaw HB, Fraioli C, Rossetti RG, Salmonson RA, Shaw JW, Walker JM, Zipkin RE, Zurier RB. Bioorg. Med. Chem. 2007;15:3345. doi: 10.1016/j.bmc.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rimmerman N, Bradshaw HB, Hughes HV, Chen JS-H, Hu SS-J, McHugh D, Vefring E, Jahnsen JA, Thompson EL, Masuda K, Cravatt BF, Burstein S, Vasko MR, Prieto AL, O’Dell DK, Walker M. Mol. Pharmacol. 2008;74:213. doi: 10.1124/mol.108.045997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bradshaw HB, Rimmerman N, Hu SS-J, Burstein S, Walker JM. Vit. Hormones. 2009;81:191. doi: 10.1016/S0083-6729(09)81008-X. [DOI] [PubMed] [Google Scholar]
- 122.Mueller GP, Driscoll WJ. J. Biol. Chem. 2007;282:22364. doi: 10.1074/jbc.M701801200. [DOI] [PubMed] [Google Scholar]
- 123.Merkler DJ, Chew GH, Gee AJ, Merkler KA, Sorondo J-P, Johnson ME. Biochemistry. 2004;43:12667. doi: 10.1021/bi049529p. [DOI] [PubMed] [Google Scholar]
- 124.Chaturvedi S, Driscoll WJ, Elliot BM, Faraday MM, Grunberg NE, Mueller GP. Prostaglandins Other Lipid Mediat. 2006;81:136. doi: 10.1016/j.prostaglandins.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Burstein SH, Adams JK, Bradshaw HB, Fraioli C, Rossetti RG, Salmonsen RA, Shaw JW, Walker JM, Zipkin RE, Zurier RB. Bioorg. Med. Chem. 2007;15:3345. doi: 10.1016/j.bmc.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siuzdak G, Cravatt BF. Biochemistry. 2004;43:14332. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- 127.Saghetelian A, Cravatt BF. Life Sci. 2005;77:1759. doi: 10.1016/j.lfs.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 128.Saghatelian A, McKinney MK, Bandell M, Smith CA, Patapoutian A, Cravatt BF. Biochemistry. 2006;45:9007. doi: 10.1021/bi0608008. [DOI] [PubMed] [Google Scholar]
- 129.Hay DW, Carey MC. Hepatology. 1990;12:6S. [PubMed] [Google Scholar]
- 130.Falany CN, Fortinberry H, Leitner EH, Barnes S. J. Lipid Res. 1997;38:1139. [PubMed] [Google Scholar]
- 131.O’Byrne J, Hunt MC, Rai DK, Saeki M, Alexson SE. J. Biol. Chem. 2003;278:34237. doi: 10.1074/jbc.M300987200. [DOI] [PubMed] [Google Scholar]
- 132.McKinney MK, Cravatt BF. Biochemistry. 2006;45:9016. doi: 10.1021/bi0608010. [DOI] [PubMed] [Google Scholar]
- 133.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR. Biochem. Pharmacol. 1995;50:83. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 134.Stella N, Schweitzer P, Piomelli D. Science. 1997;388:773. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 135.Kondo S, Kondo H, Nakane S, Kodaka T, Tokumara A, Waku K, Sugiura T, Waku K, Sugiura T. FEBS Lett. 1998;429:152. doi: 10.1016/s0014-5793(98)00581-x. [DOI] [PubMed] [Google Scholar]
- 136.Blankman JL, Simon GM, Cravatt BF. Chem. Biol. 2007;14:1347. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.(a) Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. Biochem. Biophys. Res. Commun. 1995;215:89. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]; (b) Sugiura T, Kodaka T, Nakane S, Miyashita T, Kondo S, Suhara Y, Takayama H, Waku K, Seki C, Baba N, Ishima Y. J. Biol. Chem. 1999;274:2794. doi: 10.1074/jbc.274.5.2794. [DOI] [PubMed] [Google Scholar]
- 138.Savinainen JR, Jarvinen T, Laine K, Laitinen JT. Br. J. Pharmacol. 2001;134:664. doi: 10.1038/sj.bjp.0704297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Vadivel SK, Vardarajan S, Duclos RI, Jr., Wood JT, Guo J, Makriyannis A. Bioorg. Med. Chem. Lett. 2007;17:5959. doi: 10.1016/j.bmcl.2007.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hanus L, Abu-Lafi S, Fride E, Breuer A, Vogel Z, Shalev DE, Kustanovich I, Mechoulam R. Proc. Natl. Acad. Sci. U.S.A. 2001;98:3662. doi: 10.1073/pnas.061029898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fezza F, Bisogno T, Minassi A, Appendino G, Mechoulam R, Di Marzo V. FEBS Lett. 2002;513:294. doi: 10.1016/s0014-5793(02)02341-4. [DOI] [PubMed] [Google Scholar]
- 142.Oka S, Tsuchie A, Tokumura A, Muramatsu M, Suhara Y, Takayama H, Waku K, Sugiura T. J. Neurochem. 2003;85:1374. doi: 10.1046/j.1471-4159.2003.01804.x. [DOI] [PubMed] [Google Scholar]
- 143.Richardson D, Ortori CA, Chapman V, Kendall DA, Barrett DA. Analytical Biochem. 2007;360:216. doi: 10.1016/j.ab.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 144.Shoemaker JL, Joseph BK, Ruckle MB, Mayeux PR, Prather PL. J. Pharmacol. Exp. Ther. 2005;314:868. doi: 10.1124/jpet.105.085282. [DOI] [PubMed] [Google Scholar]
- 145.Duncan M, Millns P, Smart D, Wright JE, Kendall DA, Ralevic V. Br. J. Pharmacol. 2004;142:509. doi: 10.1038/sj.bjp.0705789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Venderova K, Brown TM, Brotchie JM. Experimental Neurology. 2005;194:284. doi: 10.1016/j.expneurol.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 147.Sun Y, Alexander SPH, Garle MJ, Hewitt K, Murphy SP, Kendall DA, Bennett AJ. Br. J. Pharmacol. 2007;152:734. doi: 10.1038/sj.bjp.0707478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Laine K, Jarvinen K, Mechoulam R, Breuer A, Jarvinen T. Invest. Ophtalmol. Vis. Sci. 2002;43:3216. [PubMed] [Google Scholar]
- 149.Parkkari T, Salo OM, Huttunen KM, Savinainen JR, Laitinen JT, Poso A, Nevalainen T, Järvinen T. Bioorg. Med. Chem. 2006;14:2850. doi: 10.1016/j.bmc.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 150.Juntunen J, Vepsalainen J, Niemi R, Laine K, Jarvinen T. J. Med. Chem. 2003;46:5083. doi: 10.1021/jm030877j. [DOI] [PubMed] [Google Scholar]
- 151.Appendino G, Ligresti A, Minassi A, Daddario N, Bisogno T, Di Marzo V. Bioorg. Med. Chem. Lett. 2003;13:43. doi: 10.1016/s0960-894x(02)00839-9. [DOI] [PubMed] [Google Scholar]
- 152.Porter AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J, Nomikos GG, Carter P, Bymaster FP, Leese AB, Felder CC. J. Pharmacol. Exp. Ther. 2002;301:1020. doi: 10.1124/jpet.301.3.1020. [DOI] [PubMed] [Google Scholar]
- 153.Markey SP, Dudding T, Wang TC. J. Lipid Res. 2000;41:657. [PubMed] [Google Scholar]
- 154.Patricelli MP, Cravatt BF. Biochemistry. 1999;38:14125. doi: 10.1021/bi991876p. [DOI] [PubMed] [Google Scholar]
- 155.Kozlowska H, Baranowska M, Schlicker E, Kozlowski M, Laudanski J, Malinowska B. Br. J. Pharmacol. 2008;115:1034. doi: 10.1038/bjp.2008.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pertwee RG. Br. J. Pharmacol. 2007;152:984. doi: 10.1038/sj.bjp.0707464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bisogno T, Melck D, Bobrov MYU, Gretskaya NM, Berzuglov VV, De Petrocellis L, Di Marzo V. Biochem. J. 2000;351:817. [PMC free article] [PubMed] [Google Scholar]
- 158.Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezzo F, Tognotto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V. Proc. Natl. Acad. Sci. U.S.A. 2002;99:8400. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chu CJ, Huang SM, De Petrocellis L, Bisogno T, Ewig SA, Miller JD, Zipkin RE, Daddario T, Appendino G, Di Marzo V, Walker JM. J. Biol. Chem. 2003;278:13633. doi: 10.1074/jbc.M211231200. [DOI] [PubMed] [Google Scholar]
- 160.De Petrocellis L, Chu CJ, Moriello AS, Kellner JC, Walker JM, Di Marzo V. Br. J. Pharmacol. 2004;143:251. doi: 10.1038/sj.bjp.0705924. [DOI] [PMC free article] [PubMed] [Google Scholar]