

Summary
Chloroquine resistance in Plasmodium falciparum is primarily conferred by mutations in pfcrt. Parasites resistant to chloroquine can display hypersensitivity to other antimalarials; however, the patterns of cross-resistance are complex, and the genetic basis has remained elusive. We show that stepwise selection for resistance to amantadine or halofantrine produced previously unknown pfcrt mutations (including S163R), which were associated with a loss of verapamil-reversible chloroquine resistance. This was accompanied by restoration of efficient chloroquine binding to hematin in these selected lines. This S163R mutation provides insight into a mechanism by which PfCRT could gate the transport of protonated chloroquine through the digestive vacuole membrane. Evidence for the presence of this mutation in a Southeast Asian isolate supports the argument for a broad role for PfCRT in determining levels of susceptibility to structurally diverse antimalarials.
Introduction
The malaria parasite Plasmodium falciparum is responsible for up to 2.7 million deaths annually, with children from sub-Saharan Africa bearing the brunt of this disease. Recent increases in mortality rates have been attributed to the spread of parasites resistant to chloroquine (CQ), still the most affordable and widely used antimalarial for many parts of Africa (Trape, 2001).
The importance of point mutations in pfcrt (P. falciparum chloroquine resistance transporter) in producing the chloroquine resistance (CQR) phenotype is beyond dispute (Sidhu et al., 2002). However, many questions remain regarding the CQR mechanism, the role of pfcrt mutations in crossresistance to other antimalarials, and the function of PfCRT. This protein contains ten putative transmembrane helices and is localized to the digestive vacuole (DV) membrane in the parasitized erythrocyte (Fidock et al., 2000). Such a location is consistent with our understanding of the mode of action of CQ, which is believed to interfere with heme detoxification by binding to hematin and preventing its sequestration into inert hemozoin (Fitch, 1970). Reduced CQ access to hematin has been observed in CQ-resistant parasites (Bray et al., 1998), but how PfCRT might account for this has not been established.
The treatment of mild to moderately severe falciparum malaria known or suspected to be CQ-resistant has included the use of the potent antimalarial halofantrine (HF). Widespread use of this phenanthrene methanol, however, has been restricted by high recrudescent rates, elevated costs, and cardiotoxicity (Gay et al., 1990; Bouchaud et al., 1994). Like CQ, HF is thought to bind to hematin and inhibit its incorporation into hemozoin (Egan et al., 1999). While little is known about the genetic basis of HF resistance, experiments selecting for resistance to the related antimalarial drug mefloquine (MFQ, a quinoline methanol) reported crossresistance to HF and increased sensitivity to CQ, and demonstrated amplification and overexpression of pfmdr1 (Wilson et al., 1989; Peel et al., 1993, 1994; Cowman et al., 1994).
Previously, we described the selection of a HF-resistant line, termed K1HF, from the CQ-resistant K1 isolate (Ritchie et al., 1996). K1HF exhibited crossresistance with MFQ, enhanced sensitivity to CQ, and loss of the verapamil (VP)-reversible component of CQR. A thorough analysis of K1HF, however, found no pfmdr1 amplification, overexpression, or sequence changes (Ritchie et al., 1996).
Moderate in vitro antimalarial activity has been reported with the antiviral agent amantadine (AM) (Evans and Havlik, 1993). Surprisingly, this drug, used for the treatment and prophylaxis of influenza A infection, is more effective against P. falciparum CQ-resistant strains even though it bears no structural similarity to any current antimalarial. AM binds to the M2 ion channel, a viral envelope protein that is activated by the acidic pH found in the endosomal compartment after endocytosis (Kelly et al., 2003). By blocking M2, AM prevents normal proton flow into the virion leading to the disruption of viral replication. Interestingly, AM resistance in the influenza virus can be readily selected and is associated with M2 point mutations that improve pH regulation in the presence of drug (Hay et al., 1985; Kelly et al., 2003).
We have tested the hypothesis that PfCRT might contribute to these intriguing inverse patterns of antimalarial susceptibility by using a combination of drug selection and allelic exchange approaches. These studies have led to the discovery of PfCRT mutations that implicate a defining role for this gene in antimalarial crossresistance. This leads us to propose a “charged drug leak” hypothesis to explain the CQR mechanism.
Results
CQ-Resistant PfCRT Haplotypes Confer Hypersensitivity to AM
To explore preliminary indications of an inverse CQ and AM response (Evans and Havlik, 1993, 1994), we assayed the AM response of the CQ-resistant lines Dd2 and 7G8 and the CQ-sensitive (CQS) lines 3D7 and GC03. Results showed much higher sensitivity to AM in Dd2 than in the CQS lines (Figure 1A). 7G8, which carries a mutant PfCRT haplotype that is distinct from Dd2 (Table 1), was intermediate. We also tested 106/1, a CQS Sudanese isolate that contains seven out of eight pfcrt mutations typically found in Asia but lacks the critical K76T polymorphism. Results were compared with J3D4, a CQ-resistant line carrying the PfCRT K76I mutation (on a Dd2-like pfcrt background) that had been selected and cloned after application of CQ pressure to 106/1 parasites (Fidock et al., 2000). Strikingly, the acquisition of CQR in J3D4 was accompanied by a dramatic increase in AM sensitivity, compared to the CQS (AM-resistant) parents (Figure 1A). These data suggested that PfCRT might constitute a key determinant of this differential pattern of CQ and AM susceptibility.
Figure 1. PfCRT Controls Parasite Susceptibility to Multiple Unrelated Drugs.
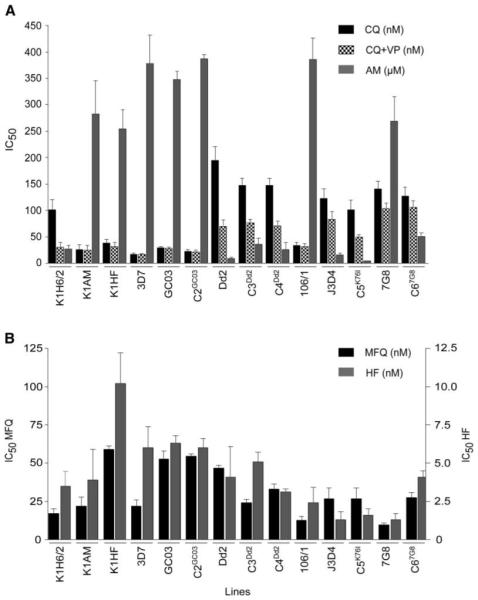
(A) CQ-resistant PfCRT haplotypes confer greater sensitivity to amantadine (AM). The data represent the mean + SEM of the IC50 values derived from at least three individual sensitivity assays for CQ ± VP and AM.
(B) CQ-resistant PfCRT haplotypes can also generate an increase in susceptibility to halofantrine (HF) and mefloquine (MFQ). Values represent means + SEM of the IC50 values derived from at least three individual assays.
Table 1.
PfCRT Haplotype of Recombinant Clones, Wild-Type Lines, and Drug-Selected Lines
| Line | Parental Line |
Recombinant | Transformed with | CQ Phenotype |
PfCRT Haplotype | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 72 | 74 | 75 | 76 | 97 | 152 | 163 | 220 | 271 | 275 | 326 | 356 | 371 | |||||
| K1H6/2 | R | C | I | E | T | H | T | S | S | E | P | S | I | I | |||
| K1AM | K1H6/2 | S | C | I | E | T | H | T | R | S | E | P | S | V | I | ||
| K1HF | K1H6/2 | S | C | I | E | T | H | A | R | S | E | L | S | I | I | ||
| 3D7 | S | C | M | N | K | H | T | S | A | Q | P | N | I | R | |||
| GC03 | S | C | M | N | K | H | T | S | A | Q | P | N | I | R | |||
| C2GC03 | GC03 | Yes | pfcrt cDNA/GC03 | S | C | M | N | K | H | T | S | A | Q | P | N | I | R |
| Dd2 | R | C | I | E | T | H | T | S | S | E | P | S | T | I | |||
| C3D2 | GC03 | Yes | cDNA/all Dd2 mutations | R | C | I | E | T | H | T | S | S | E | P | S | T | I |
| C4Dd2 | GC03 | Yes | cDNA/all Dd2 mutations | R | C | I | E | T | H | T | S | S | E | P | S | T | I |
| 106/1 | S | C | I | E | K | H | T | S | S | E | P | S | I | I | |||
| J3D4 | Clone of 34-1/E | R | C | I | E | I | H | T | S | S | E | P | S | I | I | ||
| C5K761 | GC03 | Yes | cDNA/all Dd2 mutations | R | C | I | E | I | H | T | S | S | E | P | S | I | I |
| 7G8 | R | S | M | N | T | H | T | S | S | Q | P | D | L | R | |||
| C67G8 | GC03 | Yes | cDNA/all Dd2 mutations | R | S | M | N | T | H | T | S | S | Q | P | D | L | R |
| Pf164 | S | C | I | E | T | H | T | R | S | E | P | S | I | I | |||
K1AM and K1HF were CQS lines selected from K1H6/2 by applying drug pressure with AM and HF, respectively. These lines were found to encode novel pfcrt mutations (lighter gray shading). 3D7 is a CQS isolate that apparently originated from West Africa (Joy et al., 2003), and GC03 is a CQS progeny of the genetic cross between the CQ-resistant clone Dd2 (Indochina) and the CQS clone HB3 (Honduras) (Wellems et al., 1990). Dd2 and 7G8 (Brazil) represent common PfCRT haplotypes found in Asia/Africa and South America, respectively (Fidock et al., 2000). J3D4 is a clone of the CQ-resistant 34-1/E line, which was selected from 106/1 by applying increasing CQ concentrations, resulting in the generation of a full complement of pfcrt point mutations including a novel K76I mutation (Fidock et al., 2000). For allelic exchange the first round of transformation was performed with GC03 and used a plasmid containing the human dihydrofolate reductase selectable marker (Fidock and Wellems, 1997). The resulting clone, C1GC03 (Sidhu et al., 2002), shared the same CQ IC50 values and pfcrt coding sequence as GC03. Transformation of C1GC03 in the second round used plasmids containing the blasticidin S-deaminase (BSD) selectable marker (Mamoun et al., 1999) and resulted in the C2 to C6 clones that expressed wild-type or mutant forms of pfcrt. Dark-grey shading indicates previously-identified PfCRT mutations.
To test this hypothesis, we took advantage of recent work (Sidhu et al., 2002) in which the endogenous pfcrt allele of a CQS clone (GC03) was replaced by the pfcrt allele from various CQ-resistant lines (Table 1). Clones C3Dd2 and C4Dd2 contained the pfcrt allele from Dd2, clones C5K76I and C67G8 contained the pfcrt alleles from J3D4 and 7G8 respectively, and clone C2GC03 controlled for any basal effect of pfcrt allelic exchange without introducing point mutations. Each of the three CQR-associated pfcrt alleles was sufficient to increase the CQ IC50 from 29 nM for the parental GC03 line to 100–150 nM for the mutant pfcrt clones, a level of resistance close to that of nontransfected CQ-resistant lines (Figure 1A; Table 2). These mutant CQ-resistant clones also showed increased susceptibility to MFQ and HF (Figure 1B; Table 2).
Table 2.
In Vitro Drug Susceptibility of the K1H6/2 Parent Line and the K1AM and K1HF Mutant Lines
| P. falciparum line | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drug | K1H6/2 | K1AM | K1HF | 3D7 | GC03 | C2GC03 | Dd2 | C3Dd2 | C4Dd2 | 106/1 | J3D4 | C5K76I | 7G8 | C67G8 |
| AM | 27 ± 7 | 283 ± 63** | 254 ± 36** | 378 ± 55 | 348 ± 16 | 388 ± 7 | 9 ± 2 | 36 ± 11 | 26 ± 14 | 386 ± 41 | 16 ± 3 | 5 ± 1 | 269 ± 47 | 51 ± 7 |
| HF | 3.5 ± 1.3 | 3.9 ± 2.1 | 10.2 ± 2.5** | 6.2 ± 1.4 | 6.3 ± 0.5 | 6.1 ± 0.6 | 4.3 ± 2.0 | 5.1 ± 0.6 | 3.3 ± 0.3 | 2.4 ± 1.1 | 1.3 ± 0.5 | 1.6 ± 0.4 | 1.3 ± 0.4 | 4.1 ± 0.4 |
| CQ | 101 ± 19 | 26 ± 9** | 38 ± 7** | 17 ± 1 | 29 ± 2 | 23 ± 2 | 195 ± 25 | 147 ± 13 | 147 ± 13 | 33 ± 6 | 123 ± 17 | 101 ± 17 | 140 ± 15 | 127 ± 17 |
| CQ + VP | 31 ± 9 | 25 ± 9 | 32 ± 7 | 19 ± 2 | 28 ± 2 | 22 ± 3 | 70 ± 12 | 76 ± 8 | 71 ± 9 | 32 ± 5 | 83 ± 15 | 49 ± 5 | 103 ± 11 | 106 ± 12 |
| MFQ | 17 ± 3 | 22 ± 6 | 59 ± 2 | 22 ± 4 | 53 ± 5 | 54 ± 1 | 47 ± 2 | 24 ± 2 | 33 ± 3 | 13 ± 3 | 27 ± 7 | 31 ± 4 | 10 ± 1 | 27 ± 3 |
| QN | 177 ± 44 | 146 ± 44 | 418 ± 111 | 76 ± 5 | 228 ± 41 | 171 ± 28 | 492 ± 111 | 75 ± 11 | 93 ± 17 | 230 ± 56 | 22 ± 5 | 52 ± 5 | 136 ± 20 | 72 ± 14 |
| QN + VP | 38 ± 19 | 110 ± 54 | 82 ± 2 | 71 ± 5 | 220 ± 32 | 170 ± 27 | 166 ± 28 | 30 ± 4 | 36 ± 8 | 236 ± 68 | 60 ± 20 | 24 ± 3 | 104 ± 23 | 55 ± 7 |
CQ susceptibility was measured in the presence or absence of 5 μM VP. The data represent the mean ± SEM of the IC50 values derived from at least three individual sensitivity assays.
p < 0.01, test performed for K1AM and K1HF versus K1H6/2
Abbreviations: AM, amantadine; HF, halofantrine; CQ, chloroquine; MFQ, mefloquine; QN, quinine; VP, verapamil.
The C3Dd2,C4Dd2,C5K76I, and C67G8 clones displayed AM IC50 values of 36 ± 11, 26 ± 14, 5 ± 1, and 51 ± 7 μM, respectively (Figure 1A; Table 2). In comparison, both the initial CQS (wild-type) line GC03 and the negative control C2GC03 were relatively insensitive to AM, with IC50 values of 348 ± 16 and 388 ± 7 μM, respectively. These assays unequivocally demonstrated a major role for pfcrt in determining parasite sensitivity to CQ and AM. We note that the subtle differences in AM susceptibility between the mutant pfcrt clones mirrored differences between the nontransfected (parental) lines, providing further evidence that the degree of AM susceptibility observed in the CQ-resistant lines was largely dictated by the mutant PfCRT haplotypes (Figure 1A; Tables 1 and 2).
Stepwise Selection for AM Resistance Can Ablate CQR
We were interested to see if AM selection of CQ-resistant isolates might result in changes in CQ sensitivity. This study employed K1H6/2, a CQ-resistant P. falciparum clone of the Thailand isolate K1. The K1H6/2 is resistant to CQ (IC50 101 ± 19 nM), relatively hypersensitive to AM (IC50 27 ± 7 μM), and harbors the Dd2-type pfcrt allele typical of CQ-resistant Asian lines (Wootton et al., 2002; Table 1).
Parasite cultures were subjected to intermittent exposure to increasing AM concentrations (5.5–100 μM) over a 3 month period, after which time they exhibited normal growth rates in medium containing 100 μM AM. The AM IC50 increased significantly from 27 ± 7 μM in the K1H6/2 parent line to 283 ± 63 μM in the AM-resistant line, denoted K1AM (Table 2). Of particular interest was the 4-fold increase in CQ sensitivity of the AM-resistant line (CQ IC50 of 26 ± 9 nM and 101 ± 19 nM for K1AM and K1H6/2, respectively). This was accompanied by a complete loss of the VP-reversible component of CQR (Figure 1A; Table 2). The acquisition of AM resistance had no major effect on the parasite response to HF, MFQ, or QN compared to the CQ-resistant parent K1H6/2, although diminished VP-reversal was observed for QN (Figure 1B; Table 2).
The AM-resistant, CQS phenotype of K1AM was stable over at least 4 months of continuous culturing without drug (data not shown). To eliminate the possibility that we had selected a contaminating strain that displayed this particular drug-sensitivity profile, PCR fingerprinting was performed with the polymorphic microsatellite marker, PfRRM, previously shown to differentiate between laboratory lines (Su et al., 1998). This revealed an identical banding pattern for K1H6/2 and K1AM, contrasting with very different patterns for all other lines used in this study (Figure 2A). Further fingerprinting analysis with primers flanking variable regions of the genes MSA-1, MSA-2, and CSP (Wooden et al., 1992) yielded identical banding patterns for K1H6/2, K1AM, and K1, indicating that K1H6/2 and K1AM both originated from K1 (data not shown).
Figure 2. P. falciparum Microsatellite and Western Blot Analysis.
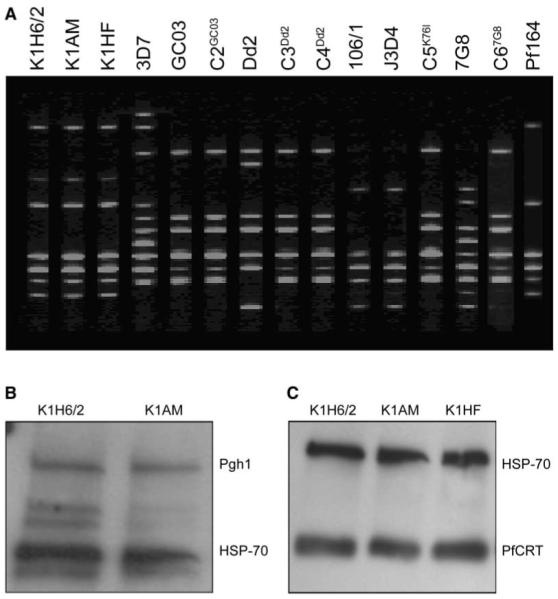
(A) PfRRM fingerprint analysis.
(B) Pgh1 expression in K1H6/2 and K1AM.
(C) PfCRT expression in K1H6/2, K1AM, and K1HF. Equal amounts of protein were separated on a 4%–15% gradient gel and incubated with antibodies raised against Pgh1 (160 kDa), PfCRT (44 kDa), or HSP-70 (70 kDa).
CQ Sensitivity in K1AM Is Not Attributed to pfmdr1
To explore the genetic basis of the restoration of CQ sensitivity in K1AM (selected prior to the discovery of pfcrt), we initially focused on pfmdr1. Until recently, pfmdr1 was considered by some investigators to be a primary candidate gene for CQR, in view of earlier reports indicating its potential involvement in P. falciparum multidrug resistance (Foote et al., 1990). The K1 pfmdr1 allele harbors the codon 86 asparagine to tyrosine substitution (N86Y), which has shown an association with CQR in some, but not all, parasite lines (Foote et al., 1990; Su et al., 1997; Peel, 2001).
Sequence analysis of the complete 4.3 kb pfmdr1 gene of K1AM and K1H6/2 revealed no change from K1. Also, there was no change in either pfmdr1 copy number, as determined by competitive PCR (data not shown) or expression of its product Pgh1 (Figure 2B). Therefore the increased sensitivity of K1AM to CQ was not related to pfmdr1 changes.
Loss of CQR Is Associated with Previously Unidentified pfcrt Mutations
Given the recent reports implicating pfcrt as the primary determinant of CQR (Fidock et al., 2000; Sidhu et al., 2002; Wootton et al., 2002), we searched for mutations in this gene. Note that the K1H6/2 parent contains the pfcrt mutations at codons 76 (K76T) and 220 (A220S) that are typically associated with CQR (Table 1). These mutations were detected in K1AM despite its complete loss of VP-reversible CQR (Table 2). To our knowledge, this is the first validated example of a culture-adapted line that harbors these critical pfcrt mutations and that is clearly and reproducibly CQS. Western blot analysis indicated equivalent PfCRT levels in the mutant and control lines (Figure 2C). Subsequent sequencing of the full-length pfcrt cDNA revealed two novel mutations in K1AM: a codon 163 serine to arginine substitution (S163R) and a codon 356 isoleucine to valine substitution (I356V). To our knowledge, pfcrt codon 163 mutations have never been previously reported. For codon 356, other polymorphisms (I356T and I356L) have previously been identified, implicating this as a residue with a role in drug-resistance phenotypes (Fidock et al., 2000; Wootton et al., 2002).
Previously we have described the K1HF line, which has been of particular interest because the selection for HF resistance resulted in a 3-fold increase in MFQ IC50 values and was accompanied by the loss of VP-reversible CQR (Table 2; Ritchie et al., 1996). As discussed earlier, this occurred independently of any change in pfmdr1 (Ritchie et al., 1996). Sequencing of the K1HF pfcrt cDNA revealed three polymorphisms: a codon 152 threonine to alanine substitution (T152A), S163R, and a codon 275 proline to leucine substitution (P275L). Thus the same mutation (S163R) was selected, accompanied by the same phenotypic change, in independent experiments that used two structurally unrelated drugs, both of which display inverse crossresistance with CQ (Oduola et al., 1988; Nateghpour et al., 1993; Evans and Havlik, 1993, 1994; Ritchie et al., 1996).
Loss of VP-Reversible CQR in K1AM Is Due to Increased CQ Access to Hematin
To determine the mechanistic basis of this altered CQ response, we assayed the CQ cellular accumulation ratio (CAR) and how this was affected by addition of the plasmepsin inhibitor Ro40-4388. This competitive protease inhibitor reversibly blocks hemoglobin digestion, prevents the release of hematin, and has been used to assess the extent of CQ-hematin binding in intact, infected erythrocytes (Moon et al., 1997; Bray et al., 1998, 1999).
The steady-state accumulation of [3H]-CQ was increased 3- to 4-fold in both K1AM and K1HF relative to the parental K1H6/2 line (Figure 3), consistent with the increase in CQ susceptibility (as measured by using [3H]-hypoxanthine assays; Figure 1A and Table 2). This was reversed in the presence of Ro40-4388, implying that the increased accumulation in the drug-selected lines was due to a restoration of CQ access to hematin (Figure 3). VP had no effect on the CAR for these mutant lines. In contrast, VP produced a 3-fold increase in the steady-state [3H]-CQ accumulation of K1H6/2 that was inhibited by Ro40-4388, suggesting that VP could restore access to hematin in this parental CQ-resistant line. Taken together, our data suggest that drug selection of both K1AM and K1HF resulted in a loss of a VP-sensitive mechanism that reduced the access of CQ to its hematin target.
Figure 3. K1AM and K1HF Display Increased Heme-Dependent Steady-State Uptake of CQ Compared to K1H6/2.
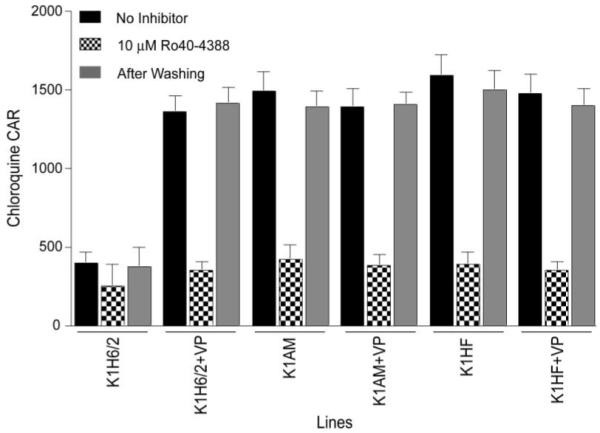
The reversible effect of the plasmepsin hemoglobinase inhibitor Ro40-4388 on steadystate uptake of [3H]-CQ is demonstrated in the presence or absence of 5 μM VP. The uptake data is presented as the cellular accumulation ratio (CAR). Ro40-4388 was removed by washing the infected cells with RPMI-1640 media, and this inhibitor’s effect on CQ accumulation was determined before and after washing. Data shown are mean + SEM from five individual experiments, each performed in triplicate.
We have previously shown that while the total CQ-hematin binding capacity is similar between lines, CQ-resistant lines exhibit a reduced apparent affinity of saturable CQ-hematin binding at equilibrium (Bray et al., 1999). Scatchard analysis clearly shows that K1AM exhibits an increased apparent affinity of CQ-hematin binding (Kd = 64.9 ± 5.4 nM) relative to K1H6/2 (Kd = 151.5 ± 18.3 nM) (Figure 4A). Similar data were obtained with K1HF (data not shown). Importantly, the binding capacity did not differ between the parental and drug-selected lines, consistent with the view that loss of VP-reversible CQR restored CQ access to hematin.
Figure 4. Novel Mutations in PfCRT Restore CQ Access to Hematin.

(A) Scatchard plot of equilibrium CQ binding in K1H6/2 and K1AM. Data points represent means of duplicate observations from three individual experiments. The derived Kd and capacity values respectively were 151.5 ± 18.3 nM and 40 ± 1.2 μM for K1H6/2 and 64.9 ± 5.4 nM and 40.7 ± 1.6 μM for K1AM.
(B) AM competes for CQ transport in CQ-resistant P. falciparum. Shown is the effect of increasing concentrations of AM on the steady-state uptake of CQ in K1H6/2 and K1AM. Data show the mean ± SEM of triplicate observations from five individual experiments.
The dichotomous relationship between CQ and AM prompted us to look for interactions between these two drugs. AM was observed to act similarly to VP in that it could increase CQ accumulation in K1H6/2 by 2- to 3-fold but was without significant effect on the level of CQ accumulation in K1AM (Figure 4B). One possibility is that AM might increase cellular accumulation of CQ in the parental line by competing for a VP-sensitive CQ transport process.
Detection of the S163R pfcrt Mutation in a Southeast Asian Field Isolate of P. falciparum
Given that the pfcrt S163R mutation arose independently in resistance-selection experiments with two unrelated drugs, we hypothesized that this mutation might also have arisen in field isolates. PCR screening for this mutation (which introduced a HinfI restriction enzyme site) in 44 geographically distinct P. falciparum field isolates identified S163R in a single isolate, Pf164, from Southeast Asia. This non-culture-adapted isolate had a CQIC50 value of 21 nM recorded with the WHO microtest, which classifies this as CQS. This mutation was confirmed by DNA sequence analysis of the genomic region flanking this codon. We then amplified and sequenced a 3.0 kb region of genomic DNA spanning all 13 pfcrt exons. This showed the S163R mutation to be the only codon that differed from the K1H6/2 sequence (Table 1). We note that sequencing of this region, as well as the corresponding regions from the K1AM and K1HF mutants, showed several codon differences (Table 1), indicating that the S163R mutation in Pf164 was not a product of contamination with either of those mutant lines. Molecular typing with PfRRM (Su et al., 1998) revealed similarity in the banding pattern between Pf164 and the K1 lineage; however, Pf164 clearly differed by the loss of a band shared between K1, K1AM, and K1HF (Figure 2A). Similar data indicating differences in microsatellite typing were found with the TA1 marker (Anderson et al., 1999; data not shown).
Discussion
This report provides evidence that point mutations in the transmembrane protein PfCRT can not only determine whether P. falciparum is resistant to CQ but can also significantly influence parasite susceptibility to structurally unrelated antimalarials. These results extend earlier observations on the role of PfCRT in contributing to parasite response to quinoline drugs (Cooper et al., 2002; Sidhu et al., 2002) and implicate this transporter as a key component underlying the complex cross-resistance patterns observed in this organism.
The once immeasurable benefit of CQ in treating P. falciparum malaria provided hope that its use, alongside DDT, would provide the foundation for the eradication of this disease (D’Alessandro and Buttiens, 2001). Characteristics unique to this drug included its rapid efficacy, minimal toxicity, ease of use, widespread availability, and, above all, affordability—a critical criterion for populations in malaria-endemic countries. The increasing ineffectiveness of CQ and its replacement drug sulfadoxine-pyrimethamine has dramatically compromised the treatment of uncomplicated malaria, shifting the clinical spectrum toward severe disease and increasing fatalities (Trape, 2001).
The genesis of CQR, driven by massive drug pressure and a noted preference for genetic outbreeding of this haploid parasite (Fidock et al., 1999), has been traced to a handful of mutational events that gave rise to region-specific PfCRT haplotypes (Chen et al., 2003; Lim et al., 2003; Mehlotra et al., 2001; Wootton et al., 2002). Common to all mutant haplotypes is the pfcrt mutation K76T, previously found only in lines displaying CQR. In vivo, this mutation has proven to be a valuable molecular marker for CQR (reviewed in Waller et al., 2004). For example, work from Mali found the presence of pfcrt alleles with the K76T mutation to be associated with a nearly 20-fold increase in CQ treatment failure (Djimde et al., 2001a). This increased risk was similar in regions with widely differing transmission rates, and K76T prevalence rates served to estimate rates of CQ treatment failure (an important consideration for regional treatment policy) (Djimde et al., 2001b).
Here, we report the isolation of mutant P. falciparum lines that retain the pfcrt K76T mutation yet have clearly lost CQR. This change was associated with the selection of point mutations in PfCRT domains not previously associated with modification in drug response (Figure 5A). Such mutations may explain the relatively rare occurrence of K76T in P. falciparum isolates that were thought to be CQS in vitro (Durand et al., 2001; Thomas et al., 2002). The possibility that additional pfcrt mutations exist outside of known polymorphisms should now be considered in molecular epidemiological investigations into P. falciparum drug resistance.
Figure 5. Predicted Structure of PfCRT and Proposed Model for CQR.

(A) Predicted protein structure of PfCRT, highlighting the ten transmembrane α helices and the intracellular and extracellular loops. Black filled circles indicate the positions of mutations frequently identified in the Eastern and Western hemispheres. The blue filled circle indicates the position of the newly identified mutation S163R present in both the amantadine-resistant and halofantrine-resistant lines K1AM and K1HF. The red and green closed circles indicate the positions of additional mutations identified exclusively in K1AM and K1HF, respectively. The S (serine) to R (arginine) change at codon 163 may be critical to the loss of chloroquine resistance (CQR) in P. falciparum. Adapted and reproduced from Carlton et al., (2001, Curr. Opin. Microbiol. 4, 415-420), with permission from Elsevier Science.
(B) Proposed mechanism of CQR. Allelic exchange studies have shown a definitive role for PfCRT in CQR. In the wild-type state (CQ-sensitive and encoding K76), the positively charged lysine residue may prevent the movement of diprotonated CQ through PfCRT (i). However, in the mutant state (CQ-resistant and encoding the K76T mutation) the loss of the positive charge at this position might allow the flux of CQ out of the digestive vacuole, thereby reducing the vacuolar CQ concentration and conferring resistance (ii). Verapamil (VP) may work by reintroducing a positive charge to a putative pore of the PfCRT protein and thus preventing the flux of CQ through PfCRT, resulting in an increased sensitivity to CQ (iii). This might explain why the VP effect is specific for CQ-resistant parasites. The selection for the S163R mutation (in lines that still encode K76T) may mimic the effects of both VP and the normal lysine residue at position 76, by introducing a positive charge to the putative PfCRT pore, thereby preventing the flux of CQ through this protein (iv).
Astute choices for malaria chemotherapy require a detailed understanding of the determinants and patterns of crossresistance. HF exhibits a degree of crossresistance with MFQ, associated with pfmdr1 amplification or polymorphisms (Cowman, 1991; Reed et al., 2000; Price et al., 2004). However, our HF-resistant line (K1HF) showed crossresistance to MFQ without detectable changes in pfmdr1 sequence or expression (Ritchie et al., 1996). Likewise, we found no pfmdr1 changes in our AM-resistant line (K1AM). The loss of CQR in both lines is consistent with earlier reports documenting an inverse crossresistance between these compounds and CQ (Basco et al., 1992; Evans and Havlik, 1993; Ritchie et al., 1996). Our finding of multiple pfcrt point mutations (T152A/S163R/P275L and S163R/I356V in K1HF and K1AM, respectively) strongly implicates this gene in contributing to these patterns of antimalarial drug susceptibility. The independent selection of the common S163R mutation also implicates this change as causing the loss of VP-reversible CQR in both these lines. Presumably, the other PfCRT mutations selected by AM or HF pressure (I356V and T152A/P275L, respectively) add some specificity to AM or HF resistance, respectively. Interestingly, a preliminary molecular screen of geographically diverse field isolates has identified one isolate, Pf164 from SE Asia, which harbors the pfcrt S163R mutation. This mutation possibly resulted from the widespread use of MFQ in SE Asia (Nosten et al., 1991; Price et al., 1999). Studies are in progress to further characterize its prevalence in malaria-endemic regions.
Formal proof of a causal relationship between these mutations and drug phenotypes requires long-term allelic exchange experiments, which have been initiated. We note from a statistical argument the extreme improbability that these mutations would have occurred by chance and not as a result of the selection procedure: in view of previous studies with both the antifolate drug-resistance determinant dhfr-ts and with pfcrt, which have estimated that single point mutations in P. falciparum occur at a frequency of <10−9 per codon (Paget-McNicol and Saul, 2001; Cooper et al., 2002). Just considering S163R, the probability of its random appearance in both lines would therefore be <10−18. Assuming instead that S163R resulted from the drug selection, one would predict that AM and HF resistance are mechanistically related to PfCRT function(s) and that the acquisition of resistance to both drugs is related to the loss of CQR. This argument is supported by several additional lines of evidence. (1) Studies with field isolates have always found an increased susceptibility to AM in lines with mutant PfCRT haplotypes (Table 2; Evans and Havlik, 1993). (2) Increased AM susceptibility was observed when mutant pfcrt was genetically engineered into CQS parasites (an experiment that used neither AM nor CQ during the allelic exchange approach). (3) The dramatic change in AM response observed in the CQ-selected J3D4 clone (generated from the CQS 106/1 clone), which had undergone a single PfCRT K76I mutation, was reproduced almost identically when this mutant allele was introduced via a CQ-independent transfection strategy into a CQS parasite (Table 2). For HF, similar arguments can apply in that earlier studies have noted that CQ-resistant parasites can display increased susceptibility to HF (Ritchie et al., 1996), and the introduction of mutant pfcrt into CQS parasites produced a consistent trend toward increased HF susceptibility (Figure 1B).
Previously, we demonstrated that CQ-resistant parasites exhibited reduced CQ access to hematin, characterized by a reduced apparent affinity (higher Kd) of CQ binding at equilibrium, and concluded that resistant parasites had evolved a mechanism to reduce the local CQ concentration in their DV (Bray et al., 1998). Our K1H6/2 parent line harbors the CQR-associated K76T and A220S mutations and exhibits a typically low apparent affinity of CQ binding (Kd 150 nM; Figure 4A). Yet the selection of these previously unidentified pfcrt mutations in the mutant lines (without loss of K76T and A220S) was associated with a significant increase in the apparent affinity of CQ binding. This change can be attributed to the loss of a VP-reversible mechanism that restricts CQ access to hematin (Table 2; Figure 4A).
In considering how this might occur, we note that CQ (with pKa values of 8.1 and 10.2) is thought to passively diffuse as an uncharged species into the acidic DV (whose pH is believed to be in the range of 5.0–5.5; Bennet et al., 2004; Waller et al., 2004). Inside the DV, protonation of CQ’s two nitrogen atoms would lead to trapping of positively charged, membrane-impermeable drug (Yayon et al., 1985). We hypothesize that the critical PfCRT K76T substitution in transmembrane domain I (Figure 5B) removes a positive charge from a putative transporter pore, allowing diprotonated CQ to escape from the DV along a massive electrochemical gradient. This would reduce the local DV concentration of CQ and the apparent affinity of CQ-hematin binding. Consistent with this model, CQR was produced in drug-pressured parasites that had undergone a lysine to isoleucine (K76I) or a lysine to asparagine (K76N) mutations in a variant pfcrt allele (Cooper et al., 2002). The relatively modest degree of CQR in parasites harboring PfCRT K76N argues for a predominantly physicochemical interaction of mutant PfCRT with CQ (Cooper et al., 2002). S163R substitutes a neutral for a positively charged residue in transmembrane domain 4, which we propose can restore a positive charge to the predicted pore involved in drug transport (Figure 5). This change may slow the outward leak of charged CQ, restoring hematin access and CQ sensitivity.
In the case of AM, this agent has a pKa of 9.0 and is almost fully ionized at pH 5.0 (Spector, 1988). The nonionized form of this drug is membrane permeable, and it is probable that AM, like CQ, can accumulate inside the DV via proton trapping. If so, the PfCRT K76T substitution might allow the escape of charged AM from the DV. Distribution of charged drug across this membrane should be strongly influenced by DV membrane potential, generated by an electrogenic V type ATPase located on both the plasma and DV membranes (Saliba et al., 2003; Allen and Kirk, 2004). The inside-positive potential of the DV could therefore accelerate the efflux of charged AM and CQ from the DV, while at the same time, the inside-negative potential of the cytoplasmic compartment would trap charged drug in the cytosol. Thus in parasites harboring K76T, we propose that the concentration of the charged forms of both drugs would be greatly reduced in the DV but moderately elevated in the cytoplasm.
Accordingly, the inverse pattern of sensitivity of AM and CQ suggests that the targets of AM and CQ reside in the cytoplasm and DV, respectively. In the case of the hematin binding drug HF, the inverse-sensitivity patterns are more difficult to explain, although it is possible that HF also exerts its effects on a target outside the DV. Alternatively, AM and HF may both target PfCRT itself: being lipophilic weak bases, both drugs might bind more readily to a hydrophobic region of mutant PfCRT harboring K76T. This might interfere with its function, providing a potential explanation for the hypersensitivity of mutant pfcrt parasites to these drugs. One could then rationalize the induction of resistance to AM and HF by a compensatory mutation (S163R) that restores a positive charge to the putative PfCRT substrate pore, making it more difficult for AM and HF to bind and block this transport.
Our model for CQR is based on the hypothesis that PfCRT can transport drug, an argument supported by a recent genome analysis implicating this protein as a member of the drug/metabolite transporter superfamily (Tran and Saier, 2004). Definitive evidence of this function has yet to be reported. Nonetheless, data presented in Figure 4B suggest that AM and CQ might act as competing transport substrates for a leak pathway whereby AM would increase the steady-state accumulation of CQ in the DV of the CQ-resistant K1H6/2 parental line, but not the CQS K1AM line. VP and other lipophilic, positively charged reversal agents might partition within the hydrophobic substrate pore, with their positive charge hindering the outward movement of protonated CQ (Figure 5B).
Alternatively, it has been suggested that PfCRT might influence drug distribution indirectly, by altering vacuolar ion gradients such as chloride, in turn affecting DV pH (Dzekunov et al., 2000; Zhang et al., 2002). This alternative hypothesis however, does not readily account for the inverse crossresistance patterns that are often seen between CQ and MFQ, QN, or HF. All these compounds are lipophilic weak bases that might be expected to exhibit crossresistance with CQ in the event of a change of resting vacuolar pH.
In conclusion, we provide evidence that previously unknown PfCRT point mutations can be selected that totally abrogate VP-reversible CQR. We also show that PfCRT plays an important role in conferring hypersensitivity to AM and the antimalarial drug HF. These data support the view that sequence polymorphisms in PfCRT influence parasite response to an ever-increasing range of antimalarial compounds. The challenge will be to decipher the genetic and physiological effect of these individual mutations, as we strive to build a comprehensive understanding of P. falciparum susceptibility to heme binding antimalarials and search for ways to over-come resistance to this valuable family of drugs.
Experimental Procedures
P. falciparum Strains and Continuous Maintenance
K1 was kindly donated by Professor D. Walliker (University of Edinburgh) and was cloned by two rounds of limiting dilution (Rosario, 1981), resulting in K1H6/2. The J3D4 clone was similarly isolated from the 34-1/E line, a CQ-resistant mutant that had been selected after application of CQ pressure to 106/1 parasites and that had acquired the pfcrt K76I mutation (Fidock et al., 2000). All lines were cultured at 37°C in 2%–5% hematocrit in RPMI 1640 plus 23 mM NaHCO3, 25 mM HEPES, and 10% human AB serum (Trager and Jensen, 1976). Cultures were maintained under an atmosphere of 93% N2, 4% O2, and 3% CO2.
Selection for Amantadine or Halofantrine Resistance
This employed the intermittent drug exposure method described in (Nateghpour et al., 1993). Briefly, the parent CQ-resistant isolate K1H6/2 was exposed to AM or HF at the predetermined IC50 concentration until the parasitemia fell to less than 40% of control values (parasite lines cultured in the absence of drug). Medium-containing drug was then replaced with drug-free medium until the parasite multiplication rate returned to control values. This procedure was continued until parasite growth, equivalent to that seen in the control culture, was achieved in the drug-treated parasite line for 10 days or longer, at which time the drug concentration was increased to the newly established IC50 value. The resulting drug-resistant lines, obtained after at least three months of drug pressure, were designated K1AM and K1HF. The degree of genetic similarity of cultured lines and field isolates was assessed by using microsatellite methods (Su et al., 1998) and DNA fingerprinting (Wooden et al., 1992).
In Vitro Sensitivity and CQ Accumulation Assays
In vitro sensitivity assays were performed by using a [3H]-hypoxanthine method (Desjardins et al., 1979). For CQ accumulation assays, parasites were synchronized at the trophozoite stage and incubated in RPMI 1640 lacking serum and NaHCO3 and containing 3 nM [3H]-CQ (specific activity 50.4 Ci/mmol). Time course samples were processed as previously described (Bray et al., 1992). Steady-state CQ accumulation was measured over 1 hr in the presence or absence of VP (5 μM) or AM (0-400 μM). CQ accumulation was also determined in the presence of 10 μM Ro40-4388, an inhibitor of malarial plasmepsin aspartic proteases (Moon et al., 1997). The cellular accumulation ratio (CAR) was calculated as the amount of labeled drug in the infected cells divided by the amount of labeled drug in a similar volume of medium (Bray et al., 1996).
To determine CQ-hematin binding parameters, parasitized cells were suspended in growth medium containing 3 nM [3H]-CQ and 5 nM to 20 μM nonradioactive CQ. After 1 hr, the reaction was terminated and the cells processed for counting as described previously (Bray et al., 1996). The saturable uptake of CQ, represented with Scatchard plots, was calculated by subtracting nonsaturable CQ uptake from total CQ uptake. The apparent affinity of binding (Kd) was determined from the reciprocal of the slope and the amount of bound drug was given by the X intercept.
PCR Analysis and DNA Sequencing
The 4.3 kb pfmdr1 gene was PCR amplified and fully sequenced from both strands with internal primers. pfmdr1 gene copy number in K1H6/2 and K1AM was determined by a previously reported competitive PCR method (Ritchie et al., 1996). For pfcrt, the full-length open reading frame was PCR amplified from genomic DNA using the primers 5′-CTTTCCCAAGTTGTACTGCTTC-3′ and 5′-CCTTATAAAGTGTAATGCGATAGC-3′ with the following cycling conditions: 94°C for 2 min, 35 cycles of 94°C for 20 s, 59°C for 10 s, 54°C 20 s, and 62°C for 10 min, with a final extension of 62°C for 15 min. The resulting amplicon was cloned into the pCR 2.1-TOPO plasmid (Invitrogen) and sequenced with vector and gene-specific primers.
PfCRT and Pgh1 Protein Analysis
Rabbit PfCRT-specific antisera was raised against a peptide spanning amino acids 401–419 (KKMRNEENEDSEGELTNVDC) as described (Fidock et al., 2000). Rabbit antisera specific for Pgh1 (amino acids 2–19, GKEQKEKKDGNLSIKEEV) was kindly donated by Dr. Alan Cowman, Walter & Eliza Hall Institute, Melbourne, Australia. For Western blot analysis, P. falciparum trophozoites were prepared by saponin lysis, followed by resuspension of the parasite pellet in ice-cold PBS. A series of snap freezing and thawing of the parasite suspension was performed with liquid N2 prior to sample trituration through a 26G needle. For each sample, equal numbers of parasites (6 × 106 per well) were incubated with an equal volume of 2× loading buffer (60 mM Tris-Cl [pH 6.8], 2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.001% bromophenol blue) and heated at 65°C for 10 min prior to loading onto a 4%–15% gradient denaturing polyacrylamide gel (Biorad). Proteins were transferred to PVDF membranes (Immobilon-P 0.45 μm, Fisher Scientific, UK), and incubated with antisera specific for PfCRT or Pgh1 (diluted 1:1000 or 1:5000 respectively), followed by HRP-conjugated donkey anti-rabbit IgG (1:10,000 dilution, Amersham Pharmacia). Bands were detected with an Enhanced Chemiluminescence kit (ECL, Amersham Pharmacia). Sample loading was standardized by stripping the PVDF membranes and probing with anti-HSP-70 antibodies (1:100 dilution; Ritchie et al., 1996). Band intensities were analyzed with Syngene GeneTools software (Synoptics, UK).
Acknowledgments
D.J.J. gratefully acknowledges the financial support of a Medical Research Council (UK) Studentship and thanks Dr. Anthony Underwood (Public Health Laboratory Service, London, UK) for initial help with DNA sequence analysis. S.A.W. and P.G.B. were supported by the Wellcome Trust UK and BBSRC (BBS/B/05508). Funding for D.A.F. was provided by the National Institutes of Health (R01 AI50234), the Speaker’s Fund for Biomedical Research, and a Scholar in Global Infectious Disease Award from the Ellison Medical Foundation (Bethesda, MD).
Footnotes
References
- Allen RJ, Kirk K. The membrane potential of the intraerythrocytic malaria parasite Plasmodium falciparum. J. Biol. Chem. 2004;279:11264–11272. doi: 10.1074/jbc.M311110200. [DOI] [PubMed] [Google Scholar]
- Anderson TJ, Su X.-z., Bockarie M, Lagog M, Day KP. Twelve microsatellite markers for characterization of Plasmodium falciparum from finger-prick blood samples. Parasitology. 1999;119:113–125. doi: 10.1017/s0031182099004552. [DOI] [PubMed] [Google Scholar]
- Basco LK, Gillotin C, Gimenez F, Farinotti R, Le Bras J. Antimalarial activity in vitro of the N-desbutyl derivative of halofantrine. Trans. R. Soc. Trop. Med. Hyg. 1992;86:12–13. doi: 10.1016/0035-9203(92)90416-a. [DOI] [PubMed] [Google Scholar]
- Bennett TN, Kosar AD, Ursos LM, Dzekunov S, Sidhu ABS, Fidock DA, Roepe PD. Drug resistance-associated PfCRT mutations confer decreased Plasmodium falciparum digestive vacuolar pH. Mol. Biochem. Parasitol. 2004;133:99–114. doi: 10.1016/j.molbiopara.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Bouchaud O, Basco LK, Gillotin C, Gimenez F, Ramiliarisoa O, Genissel B, Bouvet E, Farinotti R, Le Bras J, Coulaud JP. Clinical efficacy and pharmacokinetics of micronized halofantrine for the treatment of acute uncomplicated falciparum malaria in nonimmune patients. Am. J. Trop. Med. Hyg. 1994;51:204–213. doi: 10.4269/ajtmh.1994.51.204. [DOI] [PubMed] [Google Scholar]
- Bray PG, Howells RE, Ritchie GY, Ward SA. Rapid chloroquine efflux phenotype in both chloroquine-sensitive and chloroquine-resistant Plasmodium falciparum. A correlation of chloroquine sensitivity with energy-dependent drug accumulation. Biochem. Pharmacol. 1992;44:1317–1324. doi: 10.1016/0006-2952(92)90532-n. [DOI] [PubMed] [Google Scholar]
- Bray PG, Hawley SR, Ward SA. 4-Aminoquinoline resistance of Plasmodium falciparum: insights from the study of amodiaquine uptake. Mol. Pharmacol. 1996;50:1551–1558. [PubMed] [Google Scholar]
- Bray PG, Mungthin M, Ridley RG, Ward SA. Access to hematin: the basis of chloroquine resistance. Mol. Pharmacol. 1998;54:170–179. doi: 10.1124/mol.54.1.170. [DOI] [PubMed] [Google Scholar]
- Bray PG, Janneh O, Raynes KJ, Mungthin M, Ginsburg H, Ward SA. Cellular uptake of chloroquine is dependent on binding to ferriprotoporphyrin IX and is independent of NHE activity in Plasmodium falciparum. J. Cell Biol. 1999;145:363–376. doi: 10.1083/jcb.145.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton J, Fidock DA, Djimde A, Plowe CV, Wellems TE. Conservation of a novel vacuolar transporter in Plasmodium species and its role in chloroquine resistance in falciparum but not vivax malaria. Curr. Opin. Microbiol. 2001;4:415–420. doi: 10.1016/s1369-5274(00)00228-9. [DOI] [PubMed] [Google Scholar]
- Chen N, Kyle DE, Pasay C, Fowler EV, Baker J, Peters JM, Cheng Q. pfcrt allelic types with two novel amino acid mutations in chloroquine-resistant Plasmodium falciparum isolates from the Philippines. Antimicrob. Agents Chemother. 2003;47:3500–3505. doi: 10.1128/AAC.47.11.3500-3505.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RA, Ferdig MT, Su X.-z., Ursos LM, Mu J, Nomura T, Fujioka H, Fidock DA, Roepe PD, Wellems TE. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol. Pharmacol. 2002;61:35–42. doi: 10.1124/mol.61.1.35. [DOI] [PubMed] [Google Scholar]
- Cowman AF. The P-glycoprotein homologs of Plasmodium falciparum—are they involved in chloroquine resistance. Parasitol. Today. 1991;7:70–76. doi: 10.1016/0169-4758(91)90197-v. [DOI] [PubMed] [Google Scholar]
- Cowman AF, Galatis D, Thompson JK. Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. Proc. Natl. Acad. Sci. USA. 1994;91:1143–1147. doi: 10.1073/pnas.91.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro U, Buttiens H. History and importance of antimalarial drug resistance. Trop. Med. Int. Health. 2001;6:845–848. doi: 10.1046/j.1365-3156.2001.00819.x. [DOI] [PubMed] [Google Scholar]
- Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979;16:710–718. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djimde A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourte Y, Dicko A, Su X.-z., Nomura T, Fidock DA, et al. A molecular marker for chloroquine-resistant falciparum malaria. N. Engl. J. Med. 2001a;344:257–263. doi: 10.1056/NEJM200101253440403. [DOI] [PubMed] [Google Scholar]
- Djimde A, Doumbo OK, Steketee RW, Plowe CV. Application of a molecular marker for surveillance of chloroquine-resistant falciparum malaria. Lancet. 2001b;358:890–891. doi: 10.1016/S0140-6736(01)06040-8. [DOI] [PubMed] [Google Scholar]
- Durand R, Jafari S, Vauzelle J, Delabre JF, Jesic Z, Le Bras J. Analysis of pfcrt point mutations and chloroquine susceptibility in isolates of Plasmodium falciparum. Mol. Biochem. Parasitol. 2001;114:95–102. doi: 10.1016/s0166-6851(01)00247-x. [DOI] [PubMed] [Google Scholar]
- Dzekunov SM, Ursos LM, Roepe PD. Digestive vacuolar pH of intact intraerythrocytic P. falciparum either sensitive or resistant to chloroquine. Mol. Biochem. Parasitol. 2000;110:107–124. doi: 10.1016/s0166-6851(00)00261-9. [DOI] [PubMed] [Google Scholar]
- Egan TJ, Hempelmann E, Mavuso WW. Characterisation of synthetic beta-haematin and effects of the antimalarial drugs quinidine, halofantrine, desbutylhalofantrine and mefloquine on its formation. J. Inorg. Biochem. 1999;73:101–107. doi: 10.1016/S0162-0134(98)10095-8. [DOI] [PubMed] [Google Scholar]
- Evans SG, Havlik I. Plasmodium falciparum: effects of amantadine, an antiviral, on chloroquine-resistant and -sensitive parasites in vitro and its influence on chloroquine activity. Biochem. Pharmacol. 1993;45:1168–1170. doi: 10.1016/0006-2952(93)90264-w. [DOI] [PubMed] [Google Scholar]
- Evans SG, Havlik I. In vitro drug interaction between amantadine and classical antimalarial drugs in Plasmodium falciparum infections. Trans. R. Soc. Trop. Med. Hyg. 1994;88:683–686. doi: 10.1016/0035-9203(94)90229-1. [DOI] [PubMed] [Google Scholar]
- Fidock DA, Wellems TE. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc. Natl. Acad. Sci. USA. 1997;94:10931–10936. doi: 10.1073/pnas.94.20.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock DA, Su X.-z., Deitsch KW, Wellems TE. Genetic approaches to the determinants of drug response, pathogenesis and infectivity in Plasmodium falciparum malaria. In: Wahlgren M, Perlmann P, editors. Malaria: Molecular and Clinical Analysis. Harwood Academic Publishers; Amsterdam: 1999. pp. 217–248. [Google Scholar]
- Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch CD. Plasmodium falciparum in owl monkeys: drug resistance and chloroquine binding capacity. Science. 1970;169:289–290. doi: 10.1126/science.169.3942.289. [DOI] [PubMed] [Google Scholar]
- Foote SJ, Kyle DE, Martin RK, Oduola AM, Forsyth K, Kemp DJ, Cowman AF. Several alleles of the multidrug-resistance gene are closely linked to chloroquine resistance in Plasmodium falciparum. Nature. 1990;345:255–258. doi: 10.1038/345255a0. [DOI] [PubMed] [Google Scholar]
- Gay F, Bustos DG, Diquet B, Rojas Rivero L, Litaudon M, Pichet C, Danis M, Gentilini M. Cross-resistance between mefloquine and halofantrine. Lancet. 1990;336:1262. doi: 10.1016/0140-6736(90)92884-k. [DOI] [PubMed] [Google Scholar]
- Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, Krettli AU, Ho M, Wang A, White NJ, Suh E, et al. Early origin and recent expansion of Plasmodium falciparum. Science. 2003;300:318–321. doi: 10.1126/science.1081449. [DOI] [PubMed] [Google Scholar]
- Kelly ML, Cook JA, Brown-Augsburger P, Heinz BA, Smith MC, Pinto LH. Demonstrating the intrinsic ion channel activity of virally encoded proteins. FEBS Lett. 2003;552:61–67. doi: 10.1016/s0014-5793(03)00851-2. [DOI] [PubMed] [Google Scholar]
- Lim P, Chy S, Ariey F, Incardona S, Chim P, Sem R, Denis MB, Hewitt S, Hoyer S, Socheat D, et al. pfcrt polymorphism and chloroquine resistance in Plasmodium falciparum strains isolated in Cambodia. Antimicrob. Agents Chemother. 2003;47:87–94. doi: 10.1128/AAC.47.1.87-94.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamoun CB, Gluzman IY, Goyard S, Beverley SM, Goldberg DE. A set of independent selectable markers for transfection of the human malaria parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. USA. 1999;96:8716–8720. doi: 10.1073/pnas.96.15.8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlotra RK, Fujioka H, Roepe PD, Janneh O, Ursos LM, Jacobs-Lorena V, McNamara DT, Bockarie MJ, Kazura JW, Kyle DE, et al. Evolution of a unique Plasmodium falciparum chloroquine-resistance phenotype in association with pfcrt polymorphism in Papua New Guinea and South America. Proc. Natl. Acad. Sci. USA. 2001;98:12689–12694. doi: 10.1073/pnas.221440898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RP, Tyas L, Certa U, Rupp K, Bur D, Jacquet C, Matile H, Loetscher H, Grueninger-Leitch F, Kay J, et al. Expression and characterisation of plasmepsin I from Plasmodium falciparum. Eur. J. Biochem. 1997;244:552–560. doi: 10.1111/j.1432-1033.1997.00552.x. [DOI] [PubMed] [Google Scholar]
- Nateghpour M, Ward SA, Howells RE. Development of halofantrine resistance and determination of cross-resistance patterns in Plasmodium falciparum. Antimicrob. Agents Chemother. 1993;37:2337–2343. doi: 10.1128/aac.37.11.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosten F, ter Kuile F, Chongsuphajaisiddhi T, Luxemburger C, Webster HK, Edstein M, Phaipun L, Thew KL, White NJ. Mefloquine-resistant falciparum malaria on the Thai-Burmese border. Lancet. 1991;337:1140–1143. doi: 10.1016/0140-6736(91)92798-7. [DOI] [PubMed] [Google Scholar]
- Oduola AM, Milhous WK, Weatherly NF, Bowdre JH, Desjardins RE. Plasmodium falciparum: induction of resistance to mefloquine in cloned strains by continuous drug exposure in vitro. Exp. Parasitol. 1988;67:354–360. doi: 10.1016/0014-4894(88)90082-3. [DOI] [PubMed] [Google Scholar]
- Paget-McNicol S, Saul A. Mutation rates in the dihydrofolate reductase gene of Plasmodium falciparum. Parasitology. 2001;122:497–505. doi: 10.1017/s0031182001007739. [DOI] [PubMed] [Google Scholar]
- Peel SA. The ABC transporter genes of Plasmodium falciparum and drug resistance. Drug Resist. Updat. 2001;4:66–74. doi: 10.1054/drup.2001.0183. [DOI] [PubMed] [Google Scholar]
- Peel SA, Merritt SC, Handy J, Baric RS. Derivation of highly mefloquine-resistant lines from Plasmodium falciparum in vitro. Am. J. Trop. Med. Hyg. 1993;48:385–397. doi: 10.4269/ajtmh.1993.48.385. [DOI] [PubMed] [Google Scholar]
- Peel SA, Bright P, Yount B, Handy J, Baric RS. A strong association between mefloquine and halofantrine resistance and amplification, overexpression, and mutation in the P-glycoprotein gene homolog (pfmdr) of Plasmodium falciparum in vitro. Am. J. Trop. Med. Hyg. 1994;51:648–658. doi: 10.4269/ajtmh.1994.51.648. [DOI] [PubMed] [Google Scholar]
- Price RN, Cassar C, Brockman A, Duraisingh M, van Vugt M, White NJ, Nosten F, Krishna S. The pfmdr1 gene is associated with a multidrug-resistant phenotype in Plasmodium falciparum from the western border of Thailand. Antimicrob. Agents Chemother. 1999;43:2943–2949. doi: 10.1128/aac.43.12.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RN, Uhlemann A-C, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004;364:438–447. doi: 10.1016/S0140-6736(04)16767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000;403:906–909. doi: 10.1038/35002615. [DOI] [PubMed] [Google Scholar]
- Ritchie GY, Mungthin M, Green JE, Bray PG, Hawley SR, Ward SA. In vitro selection of halofantrine resistance in Plasmodium falciparum is not associated with increased expression of Pgh1. Mol. Biochem. Parasitol. 1996;83:35–46. doi: 10.1016/s0166-6851(96)02746-6. [DOI] [PubMed] [Google Scholar]
- Rosario V. Cloning of naturally occurring mixed infections of malaria parasites. Science. 1981;212:1037–1038. doi: 10.1126/science.7015505. [DOI] [PubMed] [Google Scholar]
- Saliba KJ, Allen RJ, Zissis S, Bray PG, Ward SA, Kirk K. Acidification of the malaria parasite’s digestive vacuole by a H+-ATPase and a H+-pyrophosphatase. J. Biol. Chem. 2003;278:5605–5612. doi: 10.1074/jbc.M208648200. [DOI] [PubMed] [Google Scholar]
- Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–213. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector R. Transport of amantadine and rimantadine through the blood-brain barrier. J. Pharmacol. Exp. Ther. 1988;244:516–519. [PubMed] [Google Scholar]
- Su X.-z., Kirkman LA, Fujioka H, Wellems TE. Complex polymorphisms in an approximately 330 kDa protein are linked to chloroquine-resistant P. falciparum in Southeast Asia and Africa. Cell. 1997;91:593–603. doi: 10.1016/s0092-8674(00)80447-x. [DOI] [PubMed] [Google Scholar]
- Su X.-z., Carucci DJ, Wellems TE. Plasmodium falciparum: parasite typing by using a multicopy microsatellite marker, PfRRM. Exp. Parasitol. 1998;89:262–265. doi: 10.1006/expr.1998.4299. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Ndir O, Dieng T, Mboup S, Wypij D, Maguire JH, Wirth DF. In vitro chloroquine susceptibility and PCR analysis of pfcrt and pfmdr1 polymorphisms in Plasmodium falciparum isolates from Senegal. Am. J. Trop. Med. Hyg. 2002;66:474–480. doi: 10.4269/ajtmh.2002.66.474. [DOI] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Tran CV, Saier MH., Jr. The principal chloroquine resistance protein of Plasmodium falciparum is a member of the drug/metabolite transporter superfamily. Microbiol. 2004;150:1–3. doi: 10.1099/mic.0.26818-0. [DOI] [PubMed] [Google Scholar]
- Trape JF. The public health impact of chloroquine resistance in Africa. Am. J. Trop. Med. Hyg. 2001;64:12–17. doi: 10.4269/ajtmh.2001.64.12. [DOI] [PubMed] [Google Scholar]
- Waller KL, Lee S, Fidock DA. Molecular and cellular biology of chloroquine resistance in Plasmodium falciparum. In: Waters AP, Janse CJ, editors. Malaria Parasites: Genomes and Molecular Biology. Caister Academic Press; Wymondham, UK: 2004. pp. 501–540. [Google Scholar]
- Wellems TE, Panton LJ, Gluzman IY, do Rosario VE, Gwadz RW, Walker-Jonah A, Krogstad DJ. Chloroquine resistance not linked to mdr-like genes in a Plasmodium falciparum cross. Nature. 1990;345:253–255. doi: 10.1038/345253a0. [DOI] [PubMed] [Google Scholar]
- Wilson CM, Serrano AE, Wasley A, Shanker AH, Wirth DF. Amplification of a gene related to mammalian mdr genes in drug-resistant Plasmodium falciparum. Science. 1989;244:1184–1186. doi: 10.1126/science.2658061. [DOI] [PubMed] [Google Scholar]
- Wooden J, Gould EE, Paull AT, Sibley CH. Plasmodium falciparum: a simple polymerase chain reaction method for differentiating strains. Exp. Parasitol. 1992;75:207–212. doi: 10.1016/0014-4894(92)90180-i. [DOI] [PubMed] [Google Scholar]
- Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Magill AJ, Su X.-z. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418:320–323. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- Yayon A, Cabantchik ZI, Ginsburg H. Susceptibility of human malaria parasites to chloroquine is pH dependent. Proc. Natl. Acad. Sci. USA. 1985;82:2784–2788. doi: 10.1073/pnas.82.9.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Howard EM, Roepe PD. Analysis of the antimalarial drug resistance protein Pfcrt expressed in yeast. J. Biol. Chem. 2002;277:49767–49775. doi: 10.1074/jbc.M204005200. [DOI] [PubMed] [Google Scholar]