

Abstract
Two-component flavoprotein monooxygenases are emerging biocatalysts that generally consist of a monooxygenase and a reductase component. Here we show that Rhodococcus opacus 1CP encodes a multifunctional enantioselective flavoprotein monooxygenase system composed of a single styrene monooxygenase (SMO) (StyA1) and another styrene monooxygenase fused to an NADH-flavin oxidoreductase (StyA2B). StyA1 and StyA2B convert styrene and chemical analogues to the corresponding epoxides at the expense of FADH2 provided from StyA2B. The StyA1/StyA2B system presents the highest monooxygenase activity in an equimolar ratio of StyA1 and StyA2B, indicating (transient) protein complex formation. StyA1 is also active when FADH2 is supplied by StyB from Pseudomonas sp. VLB120 or PheA2 from Rhodococcus opacus 1CP. However, in both cases the reductase produces an excess of FADH2, resulting in a high waste of NADH. The epoxidation rate of StyA1 heavily depends on the type of reductase. This supports that the FADH2-induced activation of StyA1 requires interprotein communication. We conclude that the StyA1/StyA2B system represents a novel type of multifunctional flavoprotein monooxygenase. Its unique mechanism of cofactor utilization provides new opportunities for biotechnological applications and is highly relevant from a structural and evolutionary point of view.
The environmentally harmful hydrocarbon styrene is readily biodegradable by various classes of microorganisms covering Gram-negative and Gram-positive bacteria as well as fungi (e.g., ascomycetes). Two major pathways for styrene mineralization have been described (reviewed in references 23, 26, and 33); of these, the most common one is initiated by a monooxygenase-catalyzed epoxidation of the vinyl side chain. Due to their biotechnological potential, the styrene monooxygenases (SMOs) involved in this reaction have received considerable attention. Most SMOs have been described for pseudomonads and were investigated for their biochemical properties (5, 13, 27, 31, 46) and their biotechnological applicability in cell-free (16, 17) or whole-cell systems (3, 12, 28, 29, 30, 32, 37). All SMOs investigated thus far convert styrene in a highly enantioselective manner to (S)-styrene oxide, which is a useful precursor for several chiral synthons and pharmaceuticals (2, 6, 14, 26, 34). Moreover, the relaxed substrate specificity of SMOs allows an enantioselective conversion of substituted styrene derivatives and structurally related compounds, like indene and dihydronaphthalene, as well as phenylalkylsulfides (Fig. 1) (17, 40, 45), thus increasing their biocatalytic potential.
FIG. 1.
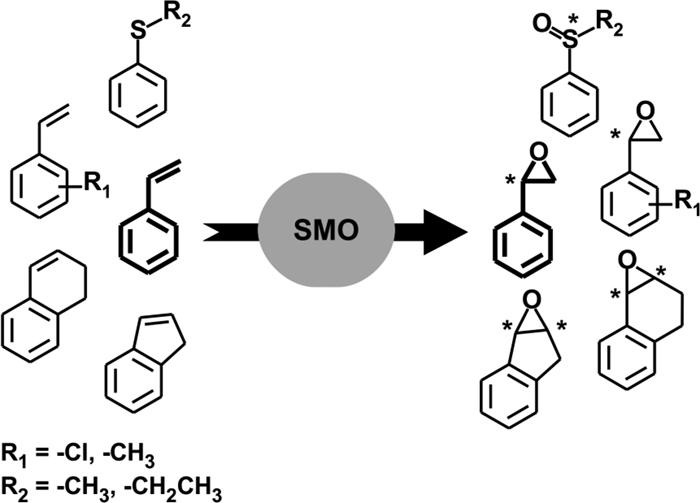
Enantioselective oxygenations catalyzed by styrene monooxygenases (SMOs). Each * indicates a chiral atom in the formed products.
Typical SMOs of pseudomonads consist of two enzymatically active protein components encoded by genes that are usually clustered adjacent to each other (styA and styB) (Fig. 2a) (26, 43, 46). The flavin reductase subunit (StyB) reduces flavin adenine dinucleotide (FAD) at the expense of NADH. The monooxygenase subunit (StyA) then utilizes the reduced flavin (FADH2) to activate molecular oxygen for styrene attack (Fig. 2b). The mechanism of FAD transfer between StyB and StyA is not yet fully understood. In vitro, StyB can be replaced by different flavin reductases from other organisms (e.g., oxidoreductases from Photobacterium fischeri or Geobacillus thermoglucosidasius) (27, 45), or FADH2 can be supplied electrochemically (16, 17). In both cases, StyA shows no significant loss in epoxidation capacity, indicating that no specific protein-protein interactions are needed for FADH2 transfer. Indications for a protein contact-based transfer of FADH2 were found by kinetic studies (19) and have also been proposed for other two-component flavoprotein monooxygenases (9, 10). Efficient interprotein transfer of the flavin cofactor will prevent FADH2 autooxidation and limit oxidative stress (21, 22). Preventing such stress is highly relevant for the biotechnological applicability of multicomponent oxygenases since it can severely improve long-term stability of whole-cell as well as cell-free transformation processes.
FIG. 2.

Schemes for genetic organization and proposed mechanism of StyA/StyB from Pseudomonas sp. VLB120 and StyA1/StyA2B from R. opacus 1CP. For simplicity, only monomers are shown. (a) For Pseudomonas sp. VLB120, gene cluster encoding the upper styrene degradation pathway: stySc and styR regulatory genes; styA and styB encoding a styrene monooxygenase, styC encoding a styrene oxide isomerase, and styD encoding phenylacetaldehyde dehydrogenase (31). For R. opacus 1CP, organization of investigated styrene-catabolic genes: styA1 for putative oxygenase component of a two-component SMO and styA2B for single-component SMO (40). (b) From left to right, cooperative mechanism of a typical two-component SMO of Pseudomonas (27), mechanism of the self-sufficient monooxygenase StyA2B of R. opacus 1CP (40), and hypothetical cooperation between StyA2B and StyA1. The FADH2 surplus of StyA2B is utilized by StyA1, yielding an increase of styrene-oxygenating activity. Dashed arrows indicate uncoupling-based FADH2 autooxidation leading to the formation of hydrogen peroxide.
An evolutionary strategy to overcome transport-based limitations is the generation of self-sufficient fusion proteins (24, 35, 38). The hemoflavoprotein P450 BM3 from Bacillus megaterium is a well-known prototype, and its high oxygenation efficiency has triggered activities of protein engineering in order to construct novel protein chimers by gene fusion (24, 25, 41). Recently, we reported on the identification of the first self-sufficient styrene monooxygenase from the nocardioform actinobacterium Rhodococcus opacus 1CP (40). This novel enzyme (StyA2B) harbors a monooxygenase and reductase unit in one polypeptide chain. The recombinant protein was biochemically characterized showing similar substrate specificity and enantioselectivity levels compared to those of two-component SMOs. However, the specific activity of the StyA2 oxygenase unit was found to be rather low. This observation contradicts the above-mentioned notion of a high-performance monooxygenase. A closer look at the genetic localization of styA2B indicated the presence of an open reading frame (styA1) encoding another putative styrene monooxygenase (Fig. 2a). Interestingly, a similar open reading frame is present directly upstream of other fused SMO genes in the genomes of Nocardia farcinica IFM10152 and Arthrobacter aurescens TC1 (40, 45). This structural conservation and feasibility of cotranscription may indicate a functional dependency (Fig. 2b). To address this issue, we studied the catalytic properties of the StyA1/StyA2B system. Our data indicate that this styrene-oxygenating system represents an unprecedented type of two-component flavoprotein monooxygenase.
MATERIALS AND METHODS
Chemicals and enzymes.
Oxygenation substrates, (S)- and (R)-styrene oxide, catalase, formate dehydrogenase, and cofactors were purchased from Sigma-Aldrich (Steinheim, Germany) and Carl Roth (Karlsruhe, Germany). Chlorostyrene oxides, 4-methylstyrene oxide, and dihydronaphthalene oxide were synthesized, purified, and structurally characterized as described previously (40).
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli BL21 strains were grown aerobically at 37°C under constant shaking in baffled shaking flasks with LB medium (100 μg ml−1 ampicillin or 20 μg ml−1 kanamycin, and 35 μg ml−1 chloramphenicol) (36).
TABLE 1.
Strains and plasmids
| Strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| E. coli BL21(DE3) (pLysS) | hsdSgal (λcIts857 ind1 Sam7 nin5lacUV5-T7 gene 1), pLysS (Cm) | Stratagene |
| E. coli BL21 Codon Plus RP | F−ompT hsdS(rB− mB−) dcm+ TetrgalendA Hte [argU proL Camr] | Stratagene |
| pSRoA2B_P01 | styA2B of R. opacus 1CP (1.722-kb NdeI/NotI-fragment) cloned in pET16bP | 40 |
| pSRoA1_P01 | styA1 of R. opacus 1CP (1.221-kb NdeI/NotI-fragment) cloned in pET16bP | 40 |
| pTEZ302 | pSPZ2 Not derivative harboring styB (Pseudomonas sp. VLB120) | 27 |
| pETP7Bα'1 | pET-derivative harboring pheA2 (R. opacus 1CP) | Unpublished |
Expression and purification of recombinant StyA1 and StyA2B (R. opacus 1CP).
Recombinant His10-StyA2B was expressed and purified as reported previously (40). The expression of His10-StyA1 was made in a bio-fermenter (7-l-Bioreactor, ADI 1030 Biocontroller; Applikon). For this purpose, 4 liters LB-medium (ampicillin and chloramphenicol) was inoculated with 50 ml of a preculture of E. coli BL21 (pSRoA1_P01), and cultivation was performed at 30°C (600 rpm, 0.3 standard liters of air per min) up to an optical density at 600 nm (OD600) of approximately 0.7. Temperature was then gradually decreased to 20°C, and induction was started by adding 0.05 to 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After 26 to 40 h of incubation (20°C), cells were harvested by centrifugation and disrupted in a precooled French press.
Purification of His10-StyA1 was achieved by Ni-chelate chromatography on a 1-ml HisTrap FF column, using an ÄKTA fast-performance liquid chromatographer (GE Healthcare). The chromatographic procedure was basically similar to that for His10-StyA2B. Fractions showing styrene-oxygenating activity were freed from imidazole and stored at −20°C as described previously (40). The achieved yields of active soluble His10-StyA1 were rather low, as described in Results. Attempts (change expression vector, host, or conditions) to improve the yield and turn expression from insoluble toward soluble protein failed (data not shown).
Expression, purification, and refolding of recombinant PheA2 (R. opacus 1CP).
The oxidoreductase PheA2 is the small component of a flavin-dependent two-component phenol monooxygenase from R. opacus 1CP (our unpublished data), and it reduces FAD to FADH2 at the expense of NADH.
The procedure for the expression of recombinant PheA2 was similar to the above-described procedure for StyA1. A volume of 60 ml overnight culture of E. coli BL21 Codon Plus RP (pETP7Bα'1) (our unpublished data) was used as the inoculum. Cultivation took place at 30°C to an OD600 of 0.6. Induction of the pET regulatory system was achieved by adding 0.2 mM IPTG, with further incubation for 20 h at 30°C.
PheA2 was expressed as inclusion bodies and purification/refolding according to the instructions of the QuickFoldTM protein refolding kit (Athena Environmental Sciences, Inc., Baltimore, MD). Inclusion body pellets resulting from crude extract preparation were subjected to four washing steps using 50 mM Tris HCl (pH 8.0), 4 M urea, 0.5 M NaCl, 1 mM EDTA, 0.1% (vol/vol) Triton X-100. Washed pellets were solubilized in 50 mM Tris HCl (pH 8.0), 8 M urea, 10 mM dithiothreitol (DTT) and incubated at 50°C until completely dissolved. Refolding of recombinant PheA2 was achieved by diluting ∼2 mg ml−1 solubilized enzyme dropwise 1:20 into 50 mM Tris HCl (pH 8.5), 240 mM NaCl, 10 mM KCl, 1 mM EDTA, 0.05% (wt/vol) PEG 4000, 1 mM glutathione reduced, 0.1 mM glutathione oxidized. The solution was carefully mixed by inversion and 20-min incubation at room temperature. Concentration of refolded protein was achieved by ammonium sulfate precipitation (80% final saturation). After 2.5 h of incubation on ice and subsequent centrifugation (20,000 × g, 20 min, 4°C), the protein precipitate was resuspended in 25 mM Tris HCl (pH 7.5), 1 M NaCl, 0.5 mM MgCl2, and 2 mM DTT and again centrifuged to remove remaining insoluble protein. Aliquots containing 50% (vol/vol) glycerol were stored at −20°C.
Expression, purification, and refolding of recombinant StyB (Pseudomonas sp. VLB120).
Expression of recombinant StyB was carried out according to Otto and coworkers (27) with the following modifications: (i) expression was carried out in shaking flasks with 100 ml to 300 ml LB medium and appropriate amounts of antibiotics, (ii) E. coli BL21 pLys was used as a host harboring pTEZ302, and (iii) induction of the alk regulatory system of the expression vector was accomplished by adding 0.05% (vol/vol) octane instead of dicyclopropylketone. Recombinant StyB was exclusively expressed as inclusion bodies.
Purification, washing, and refolding procedures of StyB inclusion bodies were performed as described previously (27). Buffer exchange for storage (25 mM Tris HCl, pH 7.5, 1 M NaCl, 0.5 mM MgCl2, 0.5 mM DTT) and concentration of the renaturated protein was performed by means of a stirred ultrafiltration cell (type 8050, volume of 50 ml; Millipore Corp., Bedford, MA) equipped with a YM30 membrane (exclusion limit of 10,000 Da; Millipore). To remove insoluble protein, the concentrate was centrifuged and the supernatant separated and portioned. After adding glycerol to a final concentration of 5% (vol/vol), refolded active StyB was stored in aliquots at −20°C.
Protein quantification.
Protein concentrations were determined with the Bradford method (8) using the protein assay reagent (Bio-Rad). Bovine serum albumin (Sigma) served as a reference protein. Alternatively, His10-StyA2B concentrations were determined at 280 nm using a NanoDrop photometer (Peqlab).
Oxidoreductase activity determination.
Oxidoreductase activities of StyB from Pseudomonas sp. VLB120, PheA2 from R. opacus 1CP, and StyA2B from R. opacus 1CP were determined by measurement of NADH consumption at 340 nm. A typical assay (1-ml total volume) contained 20 μmol Tris HCl (pH 7.5), 0.05 to 0.15 μmol FAD, and a suitable amount of enzyme. After equilibration at 30°C for 10 min, the reaction was started by addition of 0.175 μmol NADH (40).
Oxygenase activity determination.
The oxygenase activity of recombinant StyA1 toward styrene and structurally similar substrates was measured by reversed-phase high-pressure liquid chromatography (RP-HPLC)-based quantification of the reaction product using the assay system described for StyA2B (40). Reaction rates (measured at least in triplicate) were determined from the initial slope of product formation, and relative velocities were calculated by referring the obtained specific activities to that for styrene (100%). From kinetic data, a standard error of <20% was observed. To regenerate trapped oxygen from hydrogen peroxide formed via uncoupling of the reductase-oxygenase system, catalase (650 U, from bovine liver) was added to all assays. Additionally, this prevents the SMOs from oxidative inactivation by hydrogen peroxide (40).
Formation of epoxides by means of whole-cell biotransformation.
Induced cells of E. coli BL21 (pSRoA2B_P01 or pSRoA1_P01) were harvested by centrifugation and were resuspended in 27 mM sodium-potassium phosphate (pH 7.3) to give a final OD600 of 20. Conversion of 1 mM styrene or 4-chlorostyrene to the corresponding oxides, solvent extraction of products, and concentration of combined organic phase was performed as described elsewhere (40). The final preparation of about 50 μl was analyzed by chiral HPLC.
Analysis of chiral products (HPLC).
Products from SMO oxygenations were analyzed by reversed-phase HPLC on a vertex column packed with Eurospher C18 (125-mm length by 4-mm internal diameter [i.d.], 5-μm particle size, 100-Å pore size; Knauer, Germany) as described previously (40). Additional experiments in order to separate (R)- and (S)-styrene oxide on a Nucleodex alpha-PM column (250-mm length by 4-mm i.d.; Macherey-Nagel, Germany) were performed in isocratic mode (40% [vol/vol] methanol; 0.1% [vol/vol] triethyl ammonium acetate [TEAA], pH 4.0; 0.7 ml min−1) (40).
The chromatographic behavior of products was outlined previously, except for 2-chlorostyrene oxide (net retention volume, 3.05 ml; 60% [vol/vol] acetonitrile) and 3-chlorostyrene oxide (2.76 ml; 60% [vol/vol] acetonitrile). Retention volumes as well as UV spectra (200 to 400 nm) of all products were compared to those of authentic standards. Absorbance at 210 nm was used for quantification and referred to multipoint calibrations of standards.
Detection of protein-bound flavins.
The content and identity of noncovalently bound flavin in StyA1 and StyA2B was determined by RP-HPLC (39). For that purpose, 35 μl of 3 mg ml−1 purified protein in 20 mM Tris HCl (pH 7.5) was treated with trichloroacetic acid (TCA) (final concentration, 75 mg ml−1), and after centrifugation (16,000 × g, 5 min, 4°C), the protein-freed supernatant was neutralized with 0.8 M K2HPO4. This sample containing the extracted flavin was subjected to a Eurospher C18 column (250-mm length by 4-mm i.d., 5-μm particle size, 100-Å pore size; Knauer, Germany). RP-HPLC was performed with 50 mM sodium acetate (pH 5.0), and flavins were eluted with a linear gradient of 30% to 60% methanol (flow, 0.7 ml min−1). A multipoint calibration for each flavin species was measured with authentic standards (net retention volume: 2.8 ml FAD, 4.3 ml flavin mononucleotide [FMN], 8.4 ml riboflavin) in a range from 0.25 μM to 25 μM. A similar control experiment was performed with bovine serum albumin (Roth).
Dissociation constants of apoenzyme-flavin complexes.
The apo forms of StyA1 and StyA2B were prepared by gel filtration (Bio-Gel P-6, 10 ml; Bio-Rad) using 50 mM Tris HCl (pH 7.5). Dissociation constants of protein-flavin complexes were determined from tryptophan fluorescence titration experiments (Cary Eclipse fluorescence spectrometer; Varian). Each apoenzyme (0.5 to 1.0 μM) was titrated at 15°C with 0 to 10 μM FAD following the tryptophan fluorescence signal at 340 nm from excitation at 290 nm (about 30 data points per titration step; standard error of <1%). The dissociation constants (Kd) for the apoenzyme-flavin complexes were obtained by fitting the fluorescence (F) over added flavin with a scaled Levenberg-Marquardt algorithm according to Bollen and coworkers (7).
Determination of protein size and subunit organization.
Discontinuous SDS-PAGE on 12% slab gels was performed to prove purity as well as subunit size of purified samples (36). Protein markers of the PagerRuler series (10 to 200 kDa; Fermentas) were used for size estimation. Proteins were checked for covalently bound flavins by fluorescence analysis after SDS-PAGE (39). The size of native His10-StyA1 was determined by size exclusion chromatography using a Superdex-200 HR16/60 column (GE Healthcare) running in 100 mM Tris HCl (pH 7.5), 150 mM NaCl. A low- and high-molecular-weight reference protein kit (Amersham Pharmacia) was used for size calibration.
Nucleotide sequence accession number.
The 8,965-bp genomic fragment including the genes styA1 and styA2B from R. opacus 1CP has been deposited at GenBank under accession no. FJ403049 (40). The pheA2 sequence from strain 1CP has been deposited at GenBank under accession no. FN908432.
RESULTS
Expression and hydrodynamic properties of recombinant StyA1.
Recombinant His10-StyA1 was expressed in E. coli (pSRoA1_P01) mainly in the form of inclusion bodies. Active soluble protein (about 7.5 mg final yield) was obtained from a 4-liter bioreactor culture by keeping the temperature during growth phase at 30°C and by decreasing the temperature before induction to 20°C. SDS-PAGE of Ni-chelate-purified His10-StyA1 showed a single protein band with an expected molecular mass of ∼44 kDa (not shown; theoretical molecular weight [MW], 46,537). No covalently bound flavin was detected via fluorescence analysis after SDS-PAGE separation. Elution behavior of the native protein during size exclusion chromatography pointed to a hydrodynamic size of 80 ± 5 kDa, which corresponds to a homodimer. A similar native structure was reported for StyA from Pseudomonas sp. VLB120 (27) and for StyA2B from R. opacus 1CP (40).
Expression and purification of other monooxygenase components.
Recombinant His10-StyA2B showed a comparable FAD-reducing activity (3.3 U mg−1) and styrene oxygenase activity (0.02 U mg−1) after purification as previously described (40). Recombinant PheA2 from R. opacus 1CP, initially obtained as inclusion bodies, could be refolded to the active form with a specific FAD reductase activity of 20.7 U mg−1. The protein yielded a single band of about 17 kDa during SDS-PAGE and behaved as a homodimer in gel filtration. The recombinant StyB from Pseudomonas sp. VLB120 was successfully expressed, purified, and refolded as described elsewhere (27). With a specific FAD-reducing activity of 60 U mg−1, StyB appeared to be the most active reductase applied in this study.
Identification of the capability of StyA1 for styrene epoxidation.
Sequence analysis of StyA1 from R. opacus 1CP revealed the highest similarities to several oxygenases of two-component styrene monooxygenases from pseudomonads (40). A gene coding for a single flavin reductase component could not be identified close to styA1, an arrangement which is typical for these genes in pseudomonads (Fig. 2a) (26). Instead, an adjacent styB gene is part of styA2B (Fig. 2a), which encodes a self-sufficient SMO (40). Thus, the monooxygenase StyA1 might retrieve FADH2 from StyA2B, enabling its styrene epoxidation activity (Fig. 2b). Detection of StyA1 activity in the presence of StyA2B needs special consideration in view of the oxygenase activity of bifunctional StyA2B. Therefore, we independently determined the oxygenase activity of StyA1 in the presence of the StyB reductase from Pseudomonas sp. VLB120. This approach was chosen since the specificity between oxygenases and FAD reductases seems rather broad, especially, for SMOs (27, 45).
Equal molar amounts of both proteins (StyA1/StyB) were subjected to the standard oxygenase assay using styrene as the oxygenase substrate as well as FAD and NADH as the substrates for StyB. Styrene epoxidation activity of 0.35 U mg−1 was determined for His10-StyA1, clearly proving its function as a monooxygenase. As expected, a lack of StyB prevented any styrene conversion, whereas a molar StyB/StyA1 ratio of 1:2 did not bring about any loss of StyA1 activity (0.36 U mg−1). This observation reflects the fact that even under these conditions the StyB activity exceeds that of His10-StyA1 (see below) (Table 2), and a 2-fold molar excess of StyB resulted in a 45% reduction of the styrene epoxidation rate (0.19 U mg−1). This was most likely caused by the high StyB activity (271 mU) resulting in overproduction of FADH2, which may act as an oxygen trap (22). Another explanation would be that StyA1 is inactivated by the reactive oxygen species generated during the reoxidation of FADH2 (4, 40).
TABLE 2.
StyA1 oxygenase activity in dependence of different FAD reductases
| FAD reductase | Molar amt (pmol) | FAD-reducing activity (mU)a | StyA1 oxygenase (molar amt [pmol]) | Epoxidation activity (mU)a | Relative activity (%)b | Efficiency of oxygenation |
|
|---|---|---|---|---|---|---|---|
| Styrene oxide per FADH2c | Relative efficiency (%)b | ||||||
| StyA2B | 62.5 | 13.2 | 125 | 0.58 (0.66) | 46 | 0.044 | 90 |
| 125 | 26.4 | 1.28 (1.43) | 100 | 0.049 | 100 | ||
| 250 | 52.8 | 1.22 (1.52) | 96 | 0.023 | 47 | ||
| StyB | 62.5 | 67.2 | 125 | 2.06 | 161 | 0.031 | 63 |
| 125 | 135.5 | 2.03 | 159 | 0.015 | 31 | ||
| 250 | 271 | 1.11 | 87 | 0.004 | 8 | ||
| PheA2 | 250 | 87 | 250d | 0.18 | 7 | 0.002 | 4 |
All experiments were performed in triplicate, and for oxygenations, a standard error of <20% was observed. Values in parentheses refer to experimentally obtained data comprising additional epoxidation activity of bifunctional StyA2B (0.019 U mg−1).
Normalized values set to 100% for equimolar StyA1/StyA2B reaction (for 125 pmol StyA1).
Efficiency is defined as the ratio of oxygenase-generated epoxide per reductase-generated FADH2.
With PheA2 as reductase, only higher amounts of StyA1 yielded reliable epoxidation rates.
Activity of StyA1 with different FAD reductases.
StyA1 activity showed a remarkable dependence on the type of FAD reductase. Comparison of equimolar ratios of both enzymes showed the highest activity for the StyA1/StyB system (2.03 mU; this value corresponds to a specific activity of 0.35 U mg−1), followed by StyA1/StyA2B (1.28 mU) and StyA1/PheA2 (0.09 mU) for 125 pmol of each protein (Table 2). Reliable epoxidation rates from the last combination were yielded only from 250 pmol of each component. The combinations of 125 pmol StyA1 and 125 or 250 pmol PheA2 did not yield a measurable amount of epoxide.
In view of the different specific activities of the reductases, the term epoxidation efficiency was introduced. This efficiency indicates the amount of FADH2 cofactor necessary for the incorporation of one atom of oxygen in the process of styrene epoxidation. The very low activity of StyA1 in the presence of equimolar amounts of PheA2 points to a low degree of cooperation between these proteins. StyA2B- and StyB-containing systems with lower or similar FAD-reducing capacities show a considerably higher epoxidation efficiency with StyA1 (Table 2). The StyA1/StyA2B couple is clearly the most efficient epoxidation system, utilizing approximately 20 molecules of FADH2 for the epoxidation of one molecule styrene. Both StyA2B surplus as well as StyA2B deficit result in a decrease of oxygenation activity compared to the equimolar mixture, thus indicating a highly balanced system. The observation that an increase of StyA2B does not bring about a further increase of StyA1 activity suggests that FADH2 transfer is induced by a direct contact between the monooxygenase components rather than occurring as free diffusion. In this case, a higher StyA2B ratio should not lead to remarkable FADH2 overproduction which traps oxygen and limits StyA1 activity, because StyA2B has rather low FAD-reducing activity compared to StyB (Table 2).
Oxygenation specificity of StyA1.
The StyA1/StyA2B system was studied for its oxygenation specificity. For this purpose, both enzymes were mixed in equimolar amounts. Under these conditions, the epoxidation efficiency is maximal and StyA2B primarily acts as a FAD reductase (Table 2). Epoxidation was observed with styrene, 2-chlorostyrene, 3-chlorostyrene, 4-chlorostyrene, 4-methylstyrene, and dihydronaphthalene, while methylphenylsulfide is transformed into a sulfoxide (Table 3). Like StyA2B, StyA1 is able to convert a number of aromatic compounds. Comparison of relative StyA1 and StyA2B activities indicates a distinct selectivity which is especially obvious from the conversion of methylphenylsulfide, 3-chlorostyrene, and 2-chlorostyrene. The last compound is not a substrate for StyA1. As a consequence and depending on the type of substrate, StyA2B contributes different degrees of oxygenation activity to the StyA1/StyA2B system (Fig. 3). Compared to StyA2B alone, the StyA1/StyA2B system brings about a 10-fold increase of activity (with styrene as substrate), showing that the surplus of the FAD-reducing capacity of StyA2B can be efficiently used by StyA1.
TABLE 3.
Oxygenation specificity of StyA1 and other styrene monooxygenases
| Substrate/product | Specific oxygenase activity in U mg−1 (relative activity %) and, if determined, % enantiomeric excess (preferred enantiomer) for indicated oxygenase and reductaseb |
||||
|---|---|---|---|---|---|
| StyA1 of R. opacus 1CP and StyA2B of R. opacus 1CPa,c | StyA2B of R. opacus 1CP and StyA2B of R. opacus 1CPc | StyA1 + StyA2B of R. opacus 1CP and StyA2B of R. opacus 1CPc | StyA of Pseudomonas sp. VLB120 and electrochemical FAD reductiond | Unknown SMO, metagenome, and PheA2 of Geobacillus thermoglucosidasiuse | |
| Styrene/styrene oxide | 0.214 (100) >96 (S) | 0.019 (100) >94 (S) | 0.233 (100) >94 (S) | 0.204 (100) 98 (S) | 0.80 (100) >99 (S) |
| 2-Chlorostyrene/2-chlorostyrene oxide | <0.001 (<0.1) | 0.007 (37) | 0.007 (3) | ND | ND |
| 3-Chlorostyrene/3-chlorostyrene oxide | 0.162 (76) | 0.044 (232) | 0.206 (88) | ND | ND |
| 4-Chlorostyrene/4-chlorostyrene oxide | 0.078 (36) | 0.011 (58) >82 | 0.089 (38) | 0.312 (153) 98 | 0.23 (29) >99 (S) |
| 4-Methylstyrene/4-methylstyrene oxide | 0.102 (48) | 0.006 (32) | 0.108 (46) | 0.312 (153) | ND |
| Dihydronaphthalene/dihydronaphthalene oxide | 0.171 (80) | 0.011 (58) | 0.182 (78) | 0.376 (184) 99 | ND |
| Methylphenylsulfide/methylphenylsulfoxide | 0.325 (152) | 0.012 (63) | 0.337 (145) | 0.226 (111) 26 | 0.11 (14) 75 (R) |
FIG. 3.

Relative substrate specificities of the StyA1/StyA2B system and the extent of StyA2B oxygenation.
The stereospecificity of StyA1 (enantiomeric excess [e.e.], >96%) is comparable to that of StyA2B (e.e., >94%). Styrene is converted by both monooxygenases to the (S)-epoxide in a highly enantioselective manner, a property which is shared by all SMOs reported so far.
Flavin binding of monooxygenase components.
Styrene monooxygenases contain a Rossmann fold for binding FAD via its AMP moiety (27, 40, 42). This and the fact that FADH2 transfer is necessary for SMO activity raises the question of the protein-flavin interaction. TCA precipitation revealed that StyA1 and StyA2B both contain trace amounts of noncovalently bound FAD (1 to 4 moles percent [mol%]) (Fig. 4a). A similar result was obtained by spectrophotometrical analysis of StyA2B (40). This indicates that StyA1 and StyA2B have some affinity for oxidized FAD, since FAD was never added in any purification step.
FIG. 4.

Flavin binding to StyA1 and StyA2B. (a) Purified StyA1 and StyA2B were tested for noncovalently bound flavins by RP-HPLC as described in Materials and Methods. Bovine serum albumin (BSA) was used as a blank, and a multipoint calibration for each flavin standard allowed flavin determination up to a low μM range. (b) Normalized fluorescence emission (F/F0) from tryptophans of StyA1 (0.59 μM; +) or StyA2B (0.66 μM; •) measured at 340 nm (excitation 290 nm) during flavin titration with FAD to estimate dissociation constants of apoenzyme-flavin complexes.
To study the specificity of the protein-flavin interaction in more detail, the purified StyA1 and StyA2B apoproteins were titrated with FAD and analyzed for changes in tryptophan fluorescence. In the case of StyA1, the tryptophan fluorescence decreased considerably and clear binding of FAD was observed (Fig. 4b). From the titration curve and assuming 1:1 binding (0.59 μM StyA1), a Kd value for FAD [Kd(FAD)] of 0.23 ± 0.06 μM was estimated for the dissociation constant of the StyA1-FAD complex. In the case of StyA2B (0.66 μM; Fig. 4b), the tryptophan fluorescence decreased also upon FAD addition, but the curve shapes differed, and the final normalized tryptophan fluorescence was clearly lower than that from StyA1. This quenching behavior might result from the two flavin binding sites of StyA2B, one in the reductase and one in the oxygenase domain. Assuming one binding site, we estimated a Kd(FAD) of 5.05 ± 0.52 μM for the StyA2B-FAD complex. Assuming two independent binding sites, we estimated the following dissociation constants: Kd1(FAD) of 0.05 ± 0.17 μM and Kd2(FAD) of 7.19 ± 0.61 μM. These values have to be taken with care, since cooperativity cannot be excluded. Furthermore, the very small contribution of the first binding site (<5%) to the total decrease of the tryptophan fluorescence and the high standard error for Kd1(FAD) indicates another behavior. Thus, the dissociation constants given herein for StyA2B indicate only the μM range of both values.
DISCUSSION
The StyA1/StyA2B system reported herein represents a new type of FAD-dependent styrene monooxygenase (SMO). Recently, we described StyA2B from R. opacus 1CP as the first self-sufficient SMO (40). StyA2B comprises both styrene-oxygenating and FAD-reducing activity in one polypeptide and has a substrate pattern similar to those of conventional two-component SMOs (Table 3). However, the specific activity of StyA2B is rather low (0.02 U mg−1), and the FADH2 formed by the reductase domain, is inefficiently used by the fused oxygenase domain. These observations and the fact that an additional styA1 gene is located directly upstream from styA2B raised the hypothesis about a functional interaction between StyA1 and StyA2B (Fig. 2).
The His-tagged StyA1 protein was successfully expressed in E. coli and appeared to be a homodimer of about 80 kDa. Activity measurements with StyB from Pseudomonas sp. VLB120 showed that StyA1 is able to oxidize styrene under consumption of FADH2. When StyB was replaced by PheA2 from R. opacus 1CP, a drastic decrease in StyA1 activity occurred (Table 2). This is rather surprising because the FADH2 production rate of PheA2 (20.7 U mg−1) is similar to that of StyB (60 U mg−1). For the prototype StyA/StyB monooxygenase from Pseudomonas sp. VLB120, it was shown that the StyB reductase component can be replaced by another FAD reductase (27) or by an electrochemical reductant (Fig. 2b) (16, 17). From this it was argued that the FADH2 needed for styrene epoxidation is provided via free diffusion and that no specific interaction between StyA and StyB is needed. However, an equimolar amount of PheA2 achieved only 4 to 7% of StyA1 activity compared to that of assays with StyB or StyA2B. This suggests that free FADH2 is not feasible for StyA1 and that efficient epoxidation requires some type of interprotein communication. An alternative, less likely explanation is that the StyA1 activity is inhibited by PheA2.
With the StyA1/StyA2B system, the highest StyA1 activity was observed at an equimolar ratio of both components (Table 2). Bisection of the StyA2B amount and therewith reductase power yielded half of the StyA1 activity. Moreover, a surplus of the StyA2B amount and a similarly enhanced reductase power does not increase StyA1 activity. These observations support the suggestion that StyA1 is not active with free FADH2 and recognizes StyA2B as its natural partner. Similar transfer mechanisms based on transient interactions or complex formation have been proposed for other two-component flavin monooxygenases (1, 9, 10, 18). Based on the present findings, a putative StyA1/StyA2B reaction mechanism can be postulated (Fig. 2b). In this mechanism, both dimeric protein components form a transient complex in which FADH2 is channeled from StyA2B toward StyA1, resulting in epoxide formation. As side reactions, some epoxide is formed in the oxygenase-active site of StyA2B and H2O2 is produced due to auto-oxidation of FADH2. This reaction mechanism differs from that of conventional SMOs, especially in FADH2 transfer, since it is induced by protein cross talk. Nevertheless, it becomes obvious and must be kept in mind that the system of StyA1/StyA2B described herein is still less efficient in flavin transfer since 20 FADH2 molecules are necessary to form one molecule of epoxide (Table 2). Unfortunately, no data for the StyA1/StyA2B system in the natural host R. opacus 1CP are available.
Binding studies showed that StyA1 and StyA2B interact with oxidized FAD (Fig. 4). In the case of StyA2B, tryptophan fluorescence quenching pointed to two FAD binding sites with different cofactor affinities (strong, Kd1 = 0.05 ± 0.17 μM; and weak, Kd2 = 7.19 ± 0.61 μM). For StyA (Kd = 21.2 ± 2 μM) and StyB (Kd = 2.3 ± 0.3 μM) from Pseudomonas sp. VLB120, the reductase component had a higher affinity for FAD than the oxygenase component (27). Thus, it might be argued that the relatively strong interaction between StyA2B and FAD is caused by the reductase domain and not by the Rossmann fold of the oxygenase domain. This is further supported by the fact that the related PheA2 reductase from Geobacillus thermoglucosidasius binds FAD with nanomolar affinity (20, 44).
StyA1 from R. opacus 1CP binds oxidized FAD (Kd = 0.23 ± 0.06 μM) two orders of magnitude better than StyA from Pseudomonas sp. VLB120 (Kd = 21.2 ± 2 μM; 27). Nevertheless, the affinity of StyA1 to FAD is consistent with the fact that SMOs have a strong preference for binding FADH2 (19, 42). For StyA from Pseudomonas putida S12, it was reported that the reduced flavin cofactor binds about 8,000 times more tightly than the oxidized form (42). Based on this and structural considerations, a kinetic mechanism was proposed in which the reaction of oxygen with StyA-bound FADH2 precedes the binding of styrene. In the case of the StyA1/StyA2B system, we cannot exclude another sequence of events, but apparently, some transient interaction between the protein partners is needed to promote the proper binding of FADH2 to StyA1. One driving force for this might be the anionic nature of the isoalloxazine moiety of FADH2, which would bring cofactor and substrate together, as observed with flavoprotein aromatic hydroxylases (11). StyA1 shows a significant affinity for FAD and may bind FAD if no FADH2 is transferred (Fig. 4b). Binding of the oxidized flavin cofactor might be beneficial for the protein stability (7, 15), but it may also inhibit the monooxygenase activity by competing with FADH2 for the flavin binding site. The (transient) interaction between StyA1 and StyA2B might change the affinity of StyA1 for FAD(H2). However, as noted above, it is more likely that it supports the exchange and limits the uncoupling of epoxidation (Table 2).
In conclusion, StyA1/StyA2B from Rhodococcus opacus 1CP is a novel, unusual member of the family of two-component flavin-dependent monooxygenases (10, 43). The raised hypothesis of a functional interaction of both proteins was reinforced. StyA1 was identified as the major monooxygenase, and StyA2B turned out to function mainly as a FAD reductase with little oxygenating side activity. The StyA1/StyA2B system has an overall epoxidation activity of 0.24 U mg−1, which competes favorably with the activity of previously described SMOs (27, 45). Why has nature designed such a complex monooxygenase system? It cannot be excluded that the occurrence of the StyA2B fusion is a survival or intermediate from evolutionary events. No activity data of a comparable system are available yet. Only three sets of genes homologous to styA1-styA2B are known. All are adjacently located in genomes of Gram-positive actinobacteria (Arthrobacter, Nocardia, and Streptomyces) and share 50 to 70% amino acid sequence similarities (40). Indeed, the herein-described multifunctional monooxygenase StyA1/StyA2B needs further investigation and is an attractive target for optimization for biotechnological purposes.
Acknowledgments
This work was supported by a predoctoral fellowship from the Deutsche Bundesstiftung Umwelt.
We thank A. Schmid (University of Dortmund) and coworkers for providing us with plasmids for expression of StyA/StyB and helpful discussions during the project. We are also grateful to A. H. Westphal (Laboratory of Biochemistry, Wageningen University) for guidance in fluorescence spectroscopy and M. Taubert (Helmholtz Centre for Environmental Research, Leipzig) for MS protein analysis.
Footnotes
Published ahead of print on 30 July 2010.
REFERENCES
- 1.Abdurachim, K., and H. R. Ellis. 2006. Detection of protein-protein interactions in the alkanesulfonate monooxygenase system from Escherichia coli. J. Bacteriol. 188:8153-8159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badone, D., and U. Guzzi. 1994. Synthesis of the potent and selective atypical beta-adrenergic agonist SR 59062 A. Bioorg. Med. Chem. Lett. 16:1921-1924. [Google Scholar]
- 3.Bae, J. W., S. Shin, S. M. Raj, S. E. Lee, S.-G. Lee, Y.-J. Jeong, and S. Park. 2008. Construction and characterization of a recombinant whole-cell biocatalyst of Escherichia coli expressing styrene monooxygenase under the control of arabinose promoter. Biotechnol. Bioprocess Eng. 13:69-76. [Google Scholar]
- 4.Ballou, D., G. Palmer, and V. Massey. 1969. Direct demonstration of superoxide anion production during the oxidation of reduced flavin and of its catalytic decomposition by erythrocuprein. Biochem. Biophys. Res. Commun. 36:898-904. [DOI] [PubMed] [Google Scholar]
- 5.Beltrametti, F., A. M. Marconi, G. Bestetti, E. Galli, M. Ruzzi, and E. Zennaro. 1997. Sequencing and functional analysis of styrene catabolism genes from Pseudomonas fluorescens ST. Appl. Environ. Microbiol. 63:2232-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Besse, P., and H. Veschambre. 1994. Chemical and biological synthesis of chiral epoxides. Tetrahedron 50:8885-8927. [Google Scholar]
- 7.Bollen, Y. J. M., S. M. Nabuurs, W. J. H. van Berkel, and C. P. M. van Mierlo. 2005. Last in, first out: the role of cofactor binding in flavodoxin folding. J. Biol. Chem. 280:7836-7844. [DOI] [PubMed] [Google Scholar]
- 8.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 9.Chakraborty, S., M. Ortiz-Maldonado, B. Entsch, and D. P. Ballou. 2010. Studies on the mechanism of p-hydroxyphenylacetate 3-hydroxylase from Pseudomonas aeruginosa: a system composed of a small flavin reductase and a large flavin-dependent oxygenase. Biochemistry 49:372-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellis, H. R. 2010. The FMN-dependent two-component monooxygenase systems. Arch. Biochem. Biophys. 497:1-12. [DOI] [PubMed] [Google Scholar]
- 11.Entsch, B., and W. J. H. van Berkel. 1995. Structure and mechanism of para-hydroxybenzoate hydroxylase. FASEB J. 9:476-483. [DOI] [PubMed] [Google Scholar]
- 12.Gursky, L., J. Nikodinovic-Runic, K. Feenstra, and K. O'Connor. 2010. In vitro evolution of styrene monooxygenase from Pseudomonas putida CA-3 for improved epoxide synthesis. Appl. Microbiol. Biotechnol. 85:995-1004. [DOI] [PubMed] [Google Scholar]
- 13.Hartmans, S., M. J. Van der Werf, and J. A. M. De Bont. 1990. Bacterial degradation of styrene involving a novel flavin adenine dinucleotide-dependent styrene monooxygenase. Appl. Environ. Microbiol. 56:1347-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hattori, K., M. Nagano, T. Kato, I. Nakanishi, K. Imai, T. Kinoshita, and K. Sakane. 1995. Asymmetric synthesis of FR165914: a novel beta-3-adrenergic agonist with a benzocycloheptene structure. Bioorg. Med. Chem. Lett. 5:2821-2824. [Google Scholar]
- 15.Hefti, M. H., J. Vervoort, and W. J. H. van Berkel. 2003. Deflavination and reconstitution of flavoproteins. Eur. J. Biochem. 270:4227-4242. [DOI] [PubMed] [Google Scholar]
- 16.Hollmann, F., K. Hofstetter, T. Habicher, B. Hauer, and A. Schmid. 2005. Direct electrochemical regeneration of monooxygenase subunits for biocatalytic asymmetric epoxidation. J. Am. Chem. Soc. 127:6540-6541. [DOI] [PubMed] [Google Scholar]
- 17.Hollmann, F., P.-C. Lin, B. Witholt, and A. Schmid. 2003. Stereospecific biocatalytic epoxidation: the first example of direct regeneration of a FAD-dependent monooxygenase for catalysis. J. Am. Chem. Soc. 125:8209-8217. [DOI] [PubMed] [Google Scholar]
- 18.Jeffers, C. E., J. C. Nichols, and S.-C. Tu. 2003. Complex formation between Vibrio harveyi luciferase and monomeric NADPH:FMN oxidoreductase. Biochemistry 42:529-534. [DOI] [PubMed] [Google Scholar]
- 19.Kantz, A., F. Chin, N. Nallamothu, T. Nguyen, and G. T. Gassner. 2005. Mechanism of flavin transfer and oxygen activation by the two-component flavoenzyme styrene monooxygenase. Arch. Biochem. Biophys. 442:102-116. [DOI] [PubMed] [Google Scholar]
- 20.Kirchner, U., A. H. Westphal, R. Müller, and W. J. H. van Berkel. 2003. Phenol hydroxylase from Bacillus thermoglucosidasius A7, a two-protein component monooxygenase with a dual role for FAD. J. Biol. Chem. 278:47545-47553. [DOI] [PubMed] [Google Scholar]
- 21.Lee, K. 1999. Benzene-induced uncoupling of naphthalene dioxygenase activity and enzyme inactivation by production of hydrogen peroxide. J. Bacteriol. 181:2719-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massey, V. 1994. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 269:22459-22462. [PubMed] [Google Scholar]
- 23.Mooney, A., P. G. Ward, and K. E. O'Connor. 2006. Microbial degradation of styrene: biochemistry, molecular genetics, and perspectives for biotechnological applications. Appl. Microbiol. Biotechnol. 72:1-10. [DOI] [PubMed] [Google Scholar]
- 24.Munro, A. W., H. M. Girvan, and K. J. McLean. 2007. Cytochrome P450-redox partner fusion enzymes. Biochim. Biophys. Acta 1770:345-359. [DOI] [PubMed] [Google Scholar]
- 25.Munro, A. W., D. G. Leys, K. J. McLean, K. R. Marshall, T. W. B. Ost, S. Daff, C. S. Miles, S. K. Chapman, D. A. Lysek, C. C. Moser, C. C. Page, and P. L. Dutton. 2002. P450 BM3: the very model of a modern flavocytochrome. Trends Biochem. Sci. 27:250-257. [DOI] [PubMed] [Google Scholar]
- 26.O'Leary, N. D., K. E. O'Connor, and A. D. W. Dobson. 2002. Biochemistry, genetics and physiology of microbial styrene degradation. FEMS Microbiol. Rev. 26:403-417. [DOI] [PubMed] [Google Scholar]
- 27.Otto, K., K. Hofstetter, M. Roethlisberger, B. Witholt, and A. Schmid. 2004. Biochemical characterization of StyAB from Pseudomonas sp. strain VLB120 as a two-component flavin-diffusible monooxygenase. J. Bacteriol. 186:5292-5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panke, S., V. De Lorenzo, A. Kaiser, B. Witholt, and M. G. Wubbolts. 1999. Engineering of a stable whole-cell biocatalyst capable of (S)-styrene oxide formation for continuous two-liquid-phase applications. Appl. Environ. Microbiol. 65:5619-5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Panke, S., M. Held, M. G. Wubbolts, B. Witholt, and A. Schmid. 2002. Pilot-scale production of (S)-styrene oxide from styrene by recombinant Escherichia coli synthesizing styrene monooxygenase. Biotechnol. Bioeng. 80:33-41. [DOI] [PubMed] [Google Scholar]
- 30.Panke, S., A. Meyer, C. M. Huber, B. Witholt, and M. G. Wubbolts. 1999. An alkane-responsive expression system for the production of fine chemicals. Appl. Environ. Microbiol. 65:2324-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panke, S., B. Witholt, A. Schmid, and M. G. Wubbolts. 1998. Towards a biocatalyst for (S)-styrene oxide production: characterization of the styrene degradation pathway of Pseudomonas sp. strain VLB120. Appl. Environ. Microbiol. 64:2032-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park, M. S., J. W. Bae, J. H. Han, E. Y. Lee, S.-G. Lee, and S. Park. 2006. Characterization of styrene catabolic genes of Pseudomonas putida SN1 and construction of a recombinant Escherichia coli containing styrene monooxygenase gene for the production of (S)-styrene oxide. J. Microbiol. Biotechnol. 16:1032-1040. [Google Scholar]
- 33.Prenafeta-Boldú, F. X., R. Summerbell, and G. S. de Hoog. 2006. Fungi growing on aromatic hydrocarbons: biotechnology's unexpected encounter with biohazard? FEMS Microbiol. Rev. 30:109-130. [DOI] [PubMed] [Google Scholar]
- 34.Rao, A. V. R., M. K. Gurjar, and V. Kaiwar. 1992. Enantioselective catalytic reductions of ketones with new four membered oxazaborolidines: application to (S)-tetramisole. Tetrahedron Asymmetry 3:859-862. [Google Scholar]
- 35.Roberts, G. A., G. Grogan, A. Greter, S. L. Flitsch, and N. J. Turner. 2002. Identification of a new class of cytochrome P450 from a Rhodococcus sp. J. Bacteriol. 184:3898-3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambrook, J., E. Fritsch, and T. Maniatis. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 37.Schmid, A., K. Hofstetter, H.-J. Feiten, F. Hollmann, and B. Witholt. 2001. Integrated biocatalytic synthesis on gram scale: the highly enantioselective preparation of chiral oxiranes with styrene monooxygenase. Adv. Synth. Catal. 343:732-737. [Google Scholar]
- 38.Srivastava, D., J. P. Schuermann, T. A. White, N. Krishnan, N. Sanyal, G. L. Hura, A. Tan, M. T. Henzl, D. F. Becker, and J. J. Tanner. 2010. Crystal structure of the bifunctional proline utilization A flavoenzyme from Bradyrhizobium japonicum. Proc. Natl. Acad. Sci. U. S. A. 107:2878-2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao, M., M. S. Casutt, G. Fritz, and J. Steuber. 2008. Oxidant-induced formation of a neutral flavosemiquinone in the Na+-translocating NADH:quinone oxidoreductase (Na+-NQR) from Vibrio cholerae. Biochim. Biophys. Acta 1777:696-702. [DOI] [PubMed] [Google Scholar]
- 40.Tischler, D., D. Eulberg, S. Lakner, S. R. Kaschabek, W. J. H. van Berkel, and M. Schlömann. 2009. Identification of a novel self-sufficient styrene monooxygenase from Rhodococcus opacus 1CP. J. Bacteriol. 191:4996-5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torres Pazmiño, D. E., R. Snajdrova, B.-J. Baas, M. Ghobrial, M. D. Mihovilovic, and M. W. Fraaije. 2008. Self-sufficient Baeyer-Villiger monooxygenases: effective coenzyme regeneration for biooxygenation by fusion engineering. Angew. Chem. Int. Ed. Engl. 47:2275-2278. [DOI] [PubMed] [Google Scholar]
- 42.Ukaegbu, U. E., A. Kantz, M. Beaton, G. T. Gassner, and A. C. Rosenzweig. 2010. Structure and ligand binding properties of the epoxidase component of styrene monooxygenase. Biochemistry 49:1678-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Berkel, W. J. H., N. M. Kamerbeek, and M. W. Fraaije. 2006. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 124:670-689. [DOI] [PubMed] [Google Scholar]
- 44.van den Heuvel, R. H. H., A. H. Westphal, A. J. R. Heck, M. A. Walsh, S. Rovida, W. J. H. van Berkel, and A. Mattevi. 2004. Structural studies on flavin reductase PheA2 reveal binding of NAD in an unusual folded conformation and support novel mechanism of action. J. Biol. Chem. 279:12860-12867. [DOI] [PubMed] [Google Scholar]
- 45.van Hellemond, E. W., D. B. Janssen, and M. W. Fraaije. 2007. Discovery of a novel styrene monooxygenase originating from the metagenome. Appl. Environ. Microbiol. 73:5832-5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Velasco, A., S. Alonso, J. L. Garcia, J. Perera, and E. Diaz. 1998. Genetic and functional analysis of the styrene catabolic cluster of Pseudomonas sp. strain Y2. J. Bacteriol. 180:1063-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]