

Abstract
Naturally occurring cationic antimicrobial peptides (AMPs) and their mimics form a diverse class of antibacterial agents currently validated in preclinical and clinical settings for the treatment of infections caused by antimicrobial-resistant bacteria. Numerous studies with linear, cyclic, and diastereomeric AMPs have strongly supported the hypothesis that their physicochemical properties, rather than any specific amino acid sequence, are responsible for their microbiological activities. It is generally believed that the amphiphilic topology is essential for insertion into and disruption of the cytoplasmic membrane. In particular, the ability to rapidly kill bacteria and the relative difficulty with which bacteria develop resistance make AMPs and their mimics attractive targets for drug development. However, the therapeutic use of naturally occurring AMPs is hampered by the high manufacturing costs, poor pharmacokinetic properties, and low bacteriological efficacy in animal models. In order to overcome these problems, a variety of novel and structurally diverse cationic amphiphiles that mimic the amphiphilic topology of AMPs have recently appeared. Many of these compounds exhibit superior pharmacokinetic properties and reduced in vitro toxicity while retaining potent antibacterial activity against resistant and nonresistant bacteria. In summary, cationic amphiphiles promise to provide a new and rich source of diverse antibacterial lead structures in the years to come.
The rise in antibiotic resistance among pathogenic bacteria and the declining rate of novel drug discovery are common concerns in medicine (66), driving research into new antibacterial classes and novel drugs in order to maintain the existing ability to treat infectious diseases, especially those caused by multidrug-resistant (MDR) organisms (49, 51).
While the enzymatic inhibitors from which many of our strongest antibiotics are derived are highly effective in the microbial world, higher-order organisms do not appear to rely entirely on such selective inhibitors (27). These organisms instead produce a number of broad-range antimicrobial peptides (AMPs), which do not target any single molecule or process but instead associate with cellular membranes, resulting in depolarization, lysis, and cell death through a disruption of the membrane topology. A subset of these peptides is able to translocate into the cell and disrupt cellular processes, such as protein and DNA synthesis (33). AMPs play a key role in the human immune system, and mutations affecting their production and expression have been linked to diseases such as morbus Kostmann and Crohn's disease (56, 75).
Membrane targeting offers advantages over standard methods of drug design and antibiotic activity due to the wide variety of active structures and a reduced development of resistance mechanisms (78). Nevertheless, potential cytotoxicity to the host cells remains a major unsolved challenge (43). Mutants resistant to AMPs have been developed in the laboratory (54); however, such mutants may be hypersusceptible to conventional antibiotics as well as demonstrate reduced growth compared to wild-type strains (77). The lack of a specific cellular target is another significant advantage of AMPs, as activity toward Gram-positive and Gram-negative bacteria, fungi, and viruses has been reported (22, 26, 81, 82). The development of AMPs as pharmaceutical agents shows great promise, with a variety of natural and synthetic compounds currently in development (26). However, natural AMPs often suffer from a variety of pharmacokinetic shortcomings, including poor bioavailability, low metabolic stability, and formulation difficulties due to their size and the high number of amide bonds, which has driven research toward the creation of partially and wholly synthetic analogues. This review will examine recent research on AMPs and their mimics in an attempt to elucidate the underlying pharmacophore shared between them and highlight the current challenges in AMP-based drug design.
CURRENT RESEARCH IN NATURAL ANTIMICROBIAL PEPTIDES
The past 20 years have been a time of discovery for AMPs, with over 1,200 peptides in five structural classes cataloged in the antimicrobial peptide database (74). In the interest of brevity, only peptides that adopt an amphiphilic α-helical structure in their target membrane will be discussed in this review, as these most directly lead to an understanding of both AMPs and their mimics. These AMPs are between 10 and 50 residues long and contain a mixture of both cationic and hydrophobic amino acids, distributed to distinct regions or faces of the α-helix (17). While the pathways and thermodynamics of AMP binding are currently being investigated (9, 35, 45, 73), they will not be discussed in detail; rather, the focus is on the effects of sequence-specific modifications.
STABLE AMPHIPHILIC HELICES LEAD TO HEMOLYSIS
The secondary structure of many AMPs is highly dependent on their environment, modulating their activity. Folding into a stable α-helix separates the positive and hydrophobic amino acids, resulting in an overall amphiphilic structure. Association with negatively charged phospholipids may induce this folding, and studies using circular dichroism (CD) have demonstrated that many AMPs are structured in their target membranes but may be disordered in simple buffered solutions (35). Selectively disrupting the α-helix by replacing key amino acids with their d-enantiomers suggested that some prefolded AMPs are capable of inserting into neutral membranes, leading to hemolysis (17). This conclusion has been reaffirmed by work on V681, which forms stable α-helical conformations in both aqueous and lipid environments (13). Disrupting the hydrophobic region of V681 via the insertion of a polar lysine residue on the hydrophobic face was found to destabilize the α-helix in aqueous buffer, leading to a variant with over 30 times less hemolytic activity and unaltered antimicrobial activity (Table 1). The reverse transition has also been observed with the AMP RTA3, which contains a polar amino acid on the peptide's hydrophobic face (30). Substituting this residue to create RTA-R5L strengthened both the amphiphilic α-helix and the hemolytic effect. Disrupting the helix of V681 via d-amino acid substitutions created the less hemolytic peptides V13VD and V13KD, which retained the antimicrobial activity of V681 (14).
TABLE 1.
Antimicrobial peptide activity compared with alpha-helical stabilitya
| Peptide | Sequenceb | MIC (μM)c |
MHCd (μM) | Reference | |
|---|---|---|---|---|---|
| Gram-negative cells | Gram-positive cells | ||||
| Melittin | GIGAVLKVLTTGLPALISWIKRKRQQ-NH2 | 2 (EC) | 0.5 (SA) | 0.78 | 83 |
| Melittin peptoid F | GIGAVNfKVLTTGNfPALISWNfKRKRQQ-NH2 | 4 (EC) | 2 (SA) | >100 | 83 |
| RTA | RPAFRKAAFRVMRACV-NH2 | 4 (PA) | NP | >1,300 | 30 |
| RTA-F4W/ R5L | RPAWLKAAFRVMRACV-NH2 | 4 (PA) | NP | ∼7 | |
| V681 | Ac-KWKSFLKTFKSAVKTVLHTALKAISS-NH2 | 2.1 (EC) | NP | 5.2 | 13 |
| V681-V13K | Ac-KWKSFLKTFKSAKKTVLHTALKAISS-NH2 | 0.89 (EC) | NP | >88.6 | 13 |
| V681-V13VD | Ac-KWKSFLKTFKSAVDKTVLHTALKAISS-NH2 | 1.1 (EC) | NP | 22.2 | 14 |
| D-V681-V13K | Ac-KDWDSDFDLDKDTDFDKDSDADKDKDTDVDLDHDTDADLDKDADIDSDSD-NH2 | 1.1 (EC) | NP | >88.6 | 14 |
Bolding indicates peptides with high hemolytic activity, which also adopt stable alpha-helical structures in aqueous buffer.
Amino acids are denoted by their one-letter abbreviations. Nf, H5C6-CH2-NH-CH2-COOH. Ac, acetyl.
EC, Escherichia coli; PA, Pseudomonas aeruginosa; SA, Staphylococcus aureus; NP, not performed.
MHC, minimal hemolytic concentration.
Other researchers have taken advantage of peptoid residues to disrupt helical AMPs. Peptoids share the same structure as the amino acids from which they are derived, with the exception that the side chain has been moved from the alpha carbon to the amide nitrogen. In one study, replacing three leucine and isoleucine residues in the hydrophobic region of melittin with peptoids such as Nf (Fig. 1) resulted in 2-fold-reduced antibacterial activity but concomitantly increased the minimal hemolytic concentration (MHC) from 0.78 μM to greater than 100 μM (Table 1) (83). It should be noted, however, that the peptoid residues also altered the hydrophobic moment.
FIG. 1.

Nonproteogenic amino acids used in AMP research. Peptoid Nf was used to reduce the hemolytic nature of melittin, while fluorinated leucine (Lf) can be used to increase the hydrophobic nature of some AMPs. The remaining nonproteogenic amino acids have all been used in short cationic AMP studies. BTF and 4-FPA increase the activity of small amphiphilic peptides by increasing peptide hydrophobicity, while TBW both increased activity and allowed Arg-TBW-Arg peptides to pierce biofilms. PPN was the most active hydrophobic substituent in a study on trypsin-resistant tripeptides, and ABM conferred trypsin resistance when attached to the C terminus.
PEPTIDE HYDROPHOBICITY AND CHARGE: TWIN WINDOWS OF ACTIVITY
Because AMPs interact nonspecifically with their target membranes through charge and hydrophobic interactions, varying a peptide's physical parameters allows optimization of the amino acid sequence. Positive charge is required for initial attraction to negatively charged bacterial membranes, whereas hydrophobic bulk guides insertion into, and disruption of, the membrane itself (17). Increasing hydrophobicity may increase antimicrobial activity, albeit often alongside an increase in hemolytic activity. For example, replacing two leucine residues with the more hydrophobic hexafluoro-leucine (Lf) residues (Fig. 1) in buforin II 10 enhanced antibacterial activity without significantly impacting the hemolytic activity (Table 2) (50). Similar experiments with an initially highly active antimicrobial peptide, magainin 2, created analogues with increased hemolytic activity and unaltered antibacterial activity, suggesting a window of effective hydrophobicity (46). Varying the hydrophobicity of V681 through amino acid substitutions revealed a similar window of activity (12). Peptides which are not significantly hydrophobic are both nonhemolytic and nonantimicrobial, and peptides with most of their hydrophilic amino acids replaced with more hydrophobic residues are highly hemolytic (Table 2). The significantly hydrophobic peptides self-associate, which may account for their reduced antimicrobial activity (13, 61).
TABLE 2.
The effect of charge and hydrophobicity on the activity of selected antimicrobial peptide series
| Peptide series | Sequencea | MIC (μM)b |
MHC (μM)c | Reference | |
|---|---|---|---|---|---|
| Gram-negative cells | Gram-positive cells | ||||
| Buforin II 10 | FPVGRVHRLLRK-H | >173 (EC) | >173 (BS) | >270 (HC50) | 46 |
| Buforin II 10-L9Lf/L10Lf | FPVGRVHRLfLfRK-H | 23 (EC) | 5.9 (BS) | >235 (HC50) | 46 |
| Magainin 2 | GIGKFLHAAKKFAKAFVAEIMNS-NH2 | 1.0 (EC) | 1.0 (BS) | 70.6 (HC50) | 46 |
| Magainin 2-L6Lf/I20Lf 2 | GIGKFLfHAAKKFAKAFVAELfMNS-NH2 | 0.92 (EC) | 0.92 (BS) | 7.4 (HC50) | 46 |
| Magainin 2-L6Lf/A9Lf/A13Lf/I20Lf | GIGKFLfHALfKKFLfKAFLfAELfMNS-NH2 | 13 (EC) | 3.3 (BS) | 3.6 (HC50) | 46 |
| Penetratin | RQIKIWFQNRRMKWKK-NH2 | 2 (EC) | 1 (SA) | >200 | 82 |
| Dual penetratin | (RQIKIWFQNRRMKWKK)2K-NH2 | 2 (EC) | 1 (SA) | 25 | 82 |
| V681-L6A/L21A | Ac-KWKSFAKTFKSAKKTVLHTAAKAISS-NH2 | 168 (PA) | NP | 336 | 12 |
| V681-A12L/A20L/A23L | Ac-KWSFLKTFKSLKKTVLHTLLKLISS-NH2 | 168 (PA) | NP | 1.3 | 12 |
| V681-V13K/T15K/T19K | Ac-KWKSFLKTFKSAKKKVLHKALKAISS-NH2 | 2.8 (EC) | 11 (BS) | <2.8 | 13 |
| D-V681-V13K | Ac-KDWDSDFDLDKDTDFDKDSDADKDKDTDVDLDHDTDADLDKDADIDSDSD-NH2 | 5.3 (PA) | NP | 88.6 | 14 |
Amino acids are denoted by their one-letter abbreviations. Lf, (CF3)2-CH-CH2-CH(NH2)-COOH. Ac, acetyl.
EC, Escherichia coli; PA, Pseudomonas aeruginosa; BS, Bacillus subtilis; SA, Staphylococcus aureus; NP, not performed.
MHC, minimal hemolytic concentration; HC50, concentration required for 50% hemolysis of blood cells.
Increasing the positive charge of an AMP by adding arginine, lysine, or histidine residues to the peptide sequence can also increase antibacterial activity. The increased charge raises electrostatic interactions between AMPs and the negatively charged bacterial membranes, without affecting interactions with the zwitterionic lipids found in mammalian membranes (44, 81). Systematically increasing the positive charge of V681-V13K revealed a threshold, past which hemolytic activity dramatically increases, with no significant change in activity against bacteria, as shown with V681-V13K/T15K/T19K in Table 2 (34). As many oligocationic molecules are cell penetrating, it may be that highly charged AMPs, drawn by the negative membrane potential inside the cell, are inserting into mammalian cells (67). In a second example of this effect, dimerizing a well-known cell-penetrating peptide, penetratin, created an AMP with similar antimicrobial activity but 8-fold-increased hemolytic activity (Table 2). The two peptides had nearly identical CD spectra, but only dual penetratin was found to lyse artificial liposomes (bacterial membrane mimics) composed of phosphatidyl ethanolamine and phosphatidyl glycerol, indicating that despite similar conformations, the longer dual penetratin had a different spectrum of activity (82). These results demonstrate that subtle modifications of AMPs can induce novel modes of antibacterial action, providing further evidence that AMPs can interact on multiple targets (56).
THE DEVELOPMENT OF SHORT CATIONIC AMPs
Unfortunately, the high costs associated with the synthesis of lengthy natural antimicrobial peptides, combined with their poor pharmacokinetic properties, limit their utility as pharmaceutical agents (43). Studies of the efficacy of peptides with 10 residues or fewer have been conducted, leading to the discovery of several highly effective short cationic antimicrobial peptides which rely on the activity of a few key amino acids.
In one study of the AMP Bac2A (RLARIVVIRVAR-NH2), every possible monosubstituted derivative was created, that is, every derivative that varied from Bac2A via the substitution of a single amino acid (31). Four amino acids were found to generally increase activity: cysteine, lysine, arginine, and tryptophan. Cysteine substitutions were proposed to result in Bac2A dimers via disulfide bridge formation, while the other residues increased either the hydrophobicity or the charge of Bac2A. Further substitutions determined that arginine and lysine substitutions were most effective when complemented with tryptophan substitutions, as tryptophan appeared to act as a hydrophobic moiety, balancing the cationic charge provided by either lysine or arginine. This result was reconfirmed by Fjell et al. in a more recent in silico analysis of over 100,000 nonameric peptides, which showed a strong preference for tryptophan residues in the top 50% of sequences, as organized by predicted activity, with the top three sequences all containing different combinations of the following residues: five tryptophans, one lysine, and three arginines (19). It is interesting that tryptophan was favored over the more hydrophobic amino acids leucine, isoleucine, and valine (36), possibly due to membrane perturbation resulting from the size of the indole ring.
Further reducing the length of AMPs demonstrated that simple arginine tryptophan (RW) repeats are capable of antimicrobial activity (37), despite their inability to form an α-helix (Table 3). The shortest active sequence is the amidated peptide RWR, and both antimicrobial activity and to a lesser extent hemolytic activity increase with the number of RW repeats (37). Esterification or amidation of the negatively charged C-terminal carboxylic acid moiety (68) and the attachment of discrete RW units to a molecular scaffold (Fig. 2) (38) are strategies which have been used to increase antimicrobial activity without impacting the MHC (Table 3).
TABLE 3.
Antibacterial and hemolytic activity of several arginine tryptophan (RW)-based short cationic antimicrobial peptides
| Peptide | Sequencea | MIC (μM)b |
MHC (μM)c | Reference | |
|---|---|---|---|---|---|
| Gram-negative cells | Gram-positive cells | ||||
| WRW-amide | WRW-NH2 | >436 (EC) | 218 (SA) | >500 μg/ml | 68 |
| WRW-ester | WRW-OBz | 140 (EC) | 9 (SA) | >500 μg/ml | 68 |
| (RW)3 | RWRWRW-NH2 | 16 (EC) | 8.0 (SA) | 210 (HD50) | 37 |
| (RW)4 | RWRWRWRW-NH2 | 9.6 (EC) | 5.1 (SA) | 100 (HD50) | 38 |
| (RW)5 | RWRWRWRWRW-NH2 | 6.2 (EC) | 3.6 (SA) | 76 (HD50) | 38 |
| RW4D | See Fig. 2 | 2.4 (EC) | 8.7 (SA) | 760 (HD50) | 38 |
Amino acids are denoted by their one-letter abbreviations. Bz, benzoyl.
EC, Escherichia coli; SA, Staphylococcus aureus.
MHC, minimal hemolytic concentration; HD50, concentration required for 50% hemolysis of blood cells. All values are in μM unless otherwise noted.
FIG. 2.
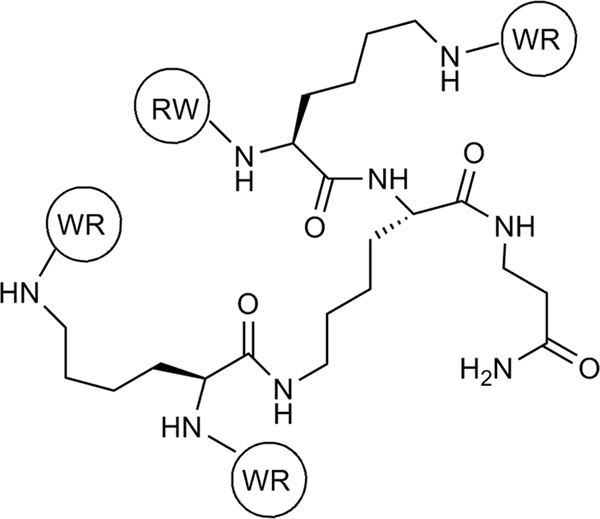
Structure of RW4D, a multivalent antimicrobial peptide, with arginine tryptophan repeats. Amino acids are attached at the N terminus and are denoted by their single-letter abbreviations.
INCREASING ACTIVITY
The short sequence of short cationic AMPs allows for facile incorporation of nonproteogenic amino acids, expanding the range of hydrophobicity beyond what can be obtained by leucine and tryptophan. One study using the fluorine-containing amino acid analogues BTF and 4-FPA (Fig. 1) found that raising the hydrophobicity of phenylalanine residues by replacing hydrogen atoms with fluorine atoms increased activity to be roughly equivalent to that for peptide sequences with tryptophan instead of 4-FPA. The most active peptides in this series contained two residues of BTF, the most heavily fluorinated and hydrophobic residue (23). All of the synthesized chains were reported to be nonhemolytic at 250 μg/ml, though no hemolysis data were provided.
Attempts to increase the steric bulk of the hydrophobic group have met with similar success. In one example, a short Arg-X-Arg peptide, with X equal to t-butylated tryptophan (TBW), is highly active and unlike many conventional antibiotics is able to penetrate biofilms (Fig. 1). As a result, this peptide is active against Staphylococcus epidermidis, a well-described biofilm producer associated with device-related infections (20, 29). Similar Arg-X-Arg molecules were created during research of protease susceptibility; the most active derivative contained a bulky para-phenyl naphthalene (PPN) substitution as the variable component (69).
REDUCING PROTEASE DEGRADATION
A significant drawback of AMPs is their protease susceptibility. This limits their oral bioavailability and also provides bacteria with a convenient route toward peptide inactivation by potentially overexpressing an endogenous peptidase (43, 55). Attempts to create protease-resistant AMPs have traditionally focused on the incorporation of d-amino acids, with some success. Complete substitution creates enantiomers which behave similarly to their l-amino acid counterparts, while diastereomeric sequences tend to be slightly less active against bacteria, though beneficial reductions in hemolytic activity have been observed (52, 65). Any loss of in vitro activity appears to be balanced by reduced susceptibility to in vivo degradation; one peptide sequence (LKLDLKKDLLDKDKLLKDLL-NH2) is known to be effective against infections caused by Gram-negative organisms in mice but only as a diastereomer (10).
An emerging method of conveying protease resistance is the incorporation of bulky side chains, which prevent the peptide from entering the peptidase active site (69). Combinatorial studies of a number of RWR analogues led to the discovery of molecules resistant to trypsin at a pH of 8.6 via the attachment of disubstituted amides, such as ABM (Fig. 1), to the C terminus (69). Susceptibility to another common protease, chymotrypsin, is currently being investigated.
LIPOPEPTIDES
Adding hydrophobic lipid tails to natural peptides increases bactericidal and fungicidal activity, likely by increasing association with the cell membrane. This effect has been observed with both naturally occurring AMPs (2) and short cationic AMPs (42) and may be used to impart antibacterial activity in otherwise inactive lysine/leucine sequences (42) (Table 4). The shortest cationic lipopeptides resemble antimicrobial surfactants (47) and are effective against fungi and Gram-positive and Gram-negative bacteria, with moderate hemolytic activity in the more active variants, such as C16-KKK (40) (Table 4). Studies have shown that membrane depolarization and calcein dye leakage correlate with activity, suggesting that either pore formation or cell lysis is the mode of action (2, 41, 42). This correlation can easily be observed with histidine-rich lipopeptides (Table 4), as these peptides depolarize cells primarily at a reduced pH, which corresponds to a large increase in antimicrobial activity (41).
TABLE 4.
Antifungal and antibacterial activity of selected (lipo)peptidesa
| Peptide | Sequenceb | MIC (μM) |
MHC (μM) | Reference | |
|---|---|---|---|---|---|
| Bacteria (E. coli) | Fungi (A. fumigatus) | ||||
| Magainin 2-F12W | GIGKFLHSAKKWGKAFVGEIMNS-NH2 | NP | >50 | NS | 2 |
| UA-magainin 2-F12W | CH3(CH2)9CO-GIGKFLHSAKKWGKAFVGEIMNS-NH2 | NP | 6.25 | NS | 2 |
| (LD)6K6 | LDKKLDLDLDKKLDLDKKLD-NH2 | >50 | 50 | NH at 25 | 42 |
| DDA-(LD)6K6 (pH 7.4) | CH3(CH2)10CO-LDKKLDLDLDKKLDLDKKLD-NH2 | 25c | 6.25 | NH at 25 | 42 |
| C16-KKK | CH3(CH2)14CO-KKK-NH2 | 1.56 | 3.1 | 100 (HC50) | 40 |
| DDA-(LD)6H6 (pH 7.4) | CH3(CH2)10CO-LDHHLDLDHHLDLDHHLD-NH2 | >100 | >100 | NH at 50 | 41 |
| DDA-(LD)6H6 (pH 5.5) | CH3(CH2)10CO-LDHHLDLDHHLDLDHHLD-NH2 | >100 | 6.2 | NP | 41 |
NP, not performed; MHC, minimal hemolytic concentration; NS, the compounds were described as either nonhemolytic or weakly hemolytic at bacterial MICs, but no data were provided; NH, not hemolytic; HC50, concentration required for 50% hemolysis of red blood cells.
Amino acids are denoted by their one-letter abbreviations.
Two strains tested.
As with the short cationic AMPs, the cationic, amphiphilic topology is more influential than the specific peptide sequence when considering lipopeptide antimicrobial activity. For instance, diastereomeric tetra-lipopeptides and tetra β-amino acid lipopeptides exhibit antibacterial activities similar to that of the standard l-amino acid-derived lipopeptides in vitro (64), with the diastereomeric lipopeptides effective against topical Aspergillus fumigatus infections in a mouse model (72).
OLIGO-ACYL-LYSINE CHAINS
Oligo-acyl-lysine chains (OAKs) represent a novel derivatization of the antimicrobial peptide backbone, linking lysine residues together with alkyl chains (Fig. 3). As an extensive review of these molecules was recently published (59), they will not be covered in detail. OAKs that display high selectivity for bacterial cells over red blood cells (RBCs) do not strongly absorb in some circular dichroism studies, suggesting that they do not adopt a rigid conformation in aqueous buffer or phosphatidylcholine/phosphatidylglycerol membranes (58). OAKs that do appear to adopt stable conformations in aqueous buffer, such as OAK B (Fig. 3), form visible aggregates and are hemolytic (concentration required for 50% hemolysis of blood cells [HD50], 4.1 μM) (57), with the ability to aggregate tied to the length of the acyl linkers. Chains that are 12 carbons long appear to fold upon themselves, creating a hydrophobic loop which absorbs circularly polarized light. When several loops come together, they then form a hydrophobic core, creating a stable, rigid superstructure. Shorter OAKs, such as OAK A, lack linkers long enough to form stable folds and do not display similar aggregation or hemolysis (<10% hemolysis at 100 μM) (58).
FIG. 3.

Two representative OAK molecules, based on the same scaffold. With a spacer equal to seven methylene units, OAK A is unable to form stable aggregates in aqueous buffer and is nonhemolytic, while the slightly longer acyl chains in OAK B allow aggregation and lead to hemolysis.
SYNTHETIC MIMICS OF AMPs
Work by the Gellman group has shown that the pharmacophore of AMPs can be mimicked by amphiphilic β-amide copolymers. Though no well-defined secondary structure was possible, these polymers selectively lysed Gram-positive and Gram-negative cells over mammalian cells, confirming the potential of wholly synthetic AMP mimics (48). Early synthetic mimics of AMPs began with an attempt to mimic the amphiphilic nature of the protein α-helix without the use of natural amino acids (63). Initial structures, exemplified by AMP mimetic A in Fig. 4, were equally active against both Gram-negative and Gram-positive bacteria and human erythrocytes (71). Reducing the flexibility of the molecule by establishing an extended hydrogen bonding system (shown in synthetic AMP mimetic B) had little effect on the hemolytic activity but significantly increased the activity against representative bacterial strains Escherichia coli and Bacillus subtilis (70). Dye leakage assays, a common method of assessing cell lysis (16), confirmed disruption of the target membranes, suggesting that a lytic or pore-forming effect resulting in enhanced permeability of the bacterial membrane is the mode of action.
FIG. 4.

Initial design of AMP mimetics and more advanced analogues. AMP mimetic A displayed roughly equivalent activities against Gram-positive and Gram-negative bacteria and red blood cells. AMP mimetic B contains an extended hydrogen-bonding network, as shown with dashed lines, and had significantly increased activity against representative Gram-positive and Gram-negative bacteria, with hemolytic activity similar to AMP mimetic A. Later research using mimetics C, D, and E probed the effect of charge on activity. The R groups of both the C and D series are the same, with the molecules varying only in their respective backbones. The AMP mimetic E has a unique guanidyl-derived R group but shares the same backbone as the D series. All compounds were prepared via polymeric techniques, with subunits linked by alkene groups and a molecular size equal to 3 kDa. Counterions are 2,2,2-trifluoroacetate.
CHARGE-BASED SELECTIVITY
As with the short cationic AMPs and lipopeptides, the net molecular charge is an important component to mimic AMP selectivity. Varying the number of amine functional groups in two AMP mimetic series, shown in Fig. 4, was found to modulate both hemolytic and antibacterial activity (Table 5) (1, 21). Hemolysis in the AMP mimetic C series decreased roughly 700-fold by an increase from 1 to 2 positive charges per residue, but no significant effect was observed with the relatively nonhemolytic D series. Instead, an increased charge led to a 16-fold decrease of the MIC against Staphylococcus aureus. Dye leakage assays with liposomes designed to mimic mammalian, Gram-negative, and Gram-positive cells found that leakage only partially correlated with activity, suggesting several modes of action (21). Further work indicated that a guanidine function was more effective in conferring selectivity to AMP mimetic E (21). The resulting AMP analogue did not appear to cause calcein dye leakage, but some AMPs and AMP mimetics associate with both negatively charged bacterial polysaccharides and with DNA and may therefore act internally (4, 11).
TABLE 5.
Activity of polymeric synthetic mimics of antimicrobial peptidesa
| SCAMP | MIC90 (μg/ml) |
HC50 (μg/ml) | Reference | |
|---|---|---|---|---|
| E. coli | S. aureus | |||
| C1 | 25 | 50 | 1 | 1 |
| C2 | 6 | 50 | 700 | 1 |
| C3 | 25 | 25 | 500 | 1 |
| D1 | >400 | >200 | >2,150 | 1 |
| D2 | 200 | 25 | 1,400 | 1 |
| D3 | 200 | 15 | 1,200 | 1 |
| E | 6 | 12 | 1,500 | 21 |
MIC90, MIC required to inhibit bacterial growth by 90%; HC50, concentration required for 50% hemolysis of red blood cells. All values are in μg/ml.
INCORPORATING THE PHARMACOPHORE OF AMPs INTO KNOWN DRUGS
In the last 2 years, a novel class of oligocationic amphiphiles, the aminoglycoside antibiotic-derived amphiphiles (AADAs), have emerged (Fig. 5) (3, 5-8, 79, 80). The most potent compound, AADA F, exhibits strong Gram-positive coccal activity against methicillin-resistant Staphylococcus epidermidis (MRSE) (MIC, 0.5 μg/ml) and methicillin-resistant Staphylococcus aureus (MRSA) (MIC, 1 μg/ml) and reduced activity against the Gram-negative E. coli (three strains; MIC, 16 to 32 μg/ml) (6). The AADAs A to J are believed to mimic the physicochemical and amphiphilic properties of AMPs by combining the cationic nature of aminoglycosides with hydrophobic side groups. The side groups are attached to auxiliary hydroxides via an amide bond, as in AADAs A to D, or either carbamate or ether linkages, as in AADAs E to H. Click chemistry has also been used to connect short hydrophobic peptide sequences to the aminoglycosides and test the potential for peptides to modulate activity (8). The AADAs may retain the RNA-binding properties of aminoglycoside antibiotics, and a dual warhead function has been hypothesized (5). Whether the conversion of highly cationic aminoglycosides into cationic amphiphiles results in synergistic effects between these two modes of action is unclear, but combination studies using AADA A with other antibiotics do show an enhancement over either agent alone (80).
FIG. 5.

Structures of aminoglycoside antibiotic-derived amphiphiles (AADAs) that exhibit potent antibacterial activity. Fmoc, 9-fluorenylmethoxy carbonyl.
COMBINING INFORMATION FROM MULTIPLE ANTIMICROBIAL CLASSES
As the interaction between bacterial membranes and AMPs and their mimics is based primarily on nonspecific interactions, such as hydrophobicity and electrostatics, it is not surprising that a large variety of sequences and structures have been found effective in the search for novel therapeutics. It is the common trends which unite these compounds that are most interesting, as they aid research toward ever more active and selective compounds and provide information toward the underlying role each molecule plays.
Notably, every molecule discussed has the ability to form an amphiphilic structure in the target membrane, either as a result of spatial separation of charges and hydrophobic region or by folding into a secondary structure. In several instances, such as with the natural AMPs RTA and V681 and the synthetic OAKs, the presence of a stable amphiphilic structure in aqueous buffer is linked to a large increase in hemolytic activity (13, 30, 61). This relationship is especially important when one considers the lipopeptides, as their long hydrophobic tails may promote separation from the aqueous solution into micelles and other aggregates (2).
At least two distinct modes of action are visible in the compounds discussed in this review. The first results in enhanced permeability, disruption, or perforation of the membrane and is exemplified by melittin, the lipopeptides, and others (40, 77, 83). This perturbation is observed via dye leakage assays and depolarization studies, which indicate the free transfer of small molecules through the lipid bilayer (16). In the second method of activity, characterized by the AMPs buforin and plectasin and the AMP mimetic E1 (21, 53, 62), the membrane does not appear to be perturbed, and antibacterial activity results from interactions with an unknown target(s), likely negatively charged molecules, such as nucleic acids and lipopolysaccharide (LPS) (4, 9, 24, 25, 32). These alternative AMP interactions may interfere with processes such as DNA replication, protein folding, and cell wall synthesis/cell division and septum formation (25). Modification of the AMP or AMP mimic may switch between these modes of action, as observed when lipid tails are added to the N terminus of magainin or in the modification of AMP mimetics D1 to E, and so it is likely that the balance between the polar and hydrophobic natures of AMPs determines which mode of action will result (2, 21). Indeed, it has been suggested that cationic antimicrobial peptides are “dirty drugs” in that they potentially have many targets due to their amphiphilic nature and cationic charge (55).
An initial concern involving AMPs was their poor bioavailability, which can result from nonspecific reactions with either serum or protein-containing media or from the digestive action of proteases. However, in vivo studies with short cationic AMPs and lipopeptides containing d-amino acids have found increased specificity for the bacterial membrane and resilience toward mammalian proteases (10, 72). The more synthetic molecules, such as the OAKs and AMP mimetics, were in part conceived to circumvent proteolysis and have also shown good in vivo activity (15, 60).
IN VIVO STUDIES WITH AMP MIMETICS
Only a limited number of in vivo and toxicology studies have been performed with cationic amphiphile-based AMP mimetics (15, 57, 58, 72). Using an E. coli mouse model, OAK compounds, such as those shown in Fig. 3, like conventional antibiotics, were able to prevent mortality. While antimicrobial peptides were degraded by plasma proteases, a 3-h preincubation in either mouse or human plasma had little effect on OAK activity. In vivo toxicology tests with these OAKs after a single intraperitoneal administration to neutropenic mice revealed no signs of adverse effects at dosages of 10 mg/kg of body weight (58). Similarly encouraging in vivo results have also been obtained with lipopeptides and AMP mimetics. A lipopeptide tetramer markedly increased the survival rate of Aspergillus fumigatus-infected mice, with tolerated doses of up to 10 mg/kg of body weight (72). Finally, arylamide AMP mimetics have been tested using a mouse thigh burden infection model, where CFU reductions comparable to that achieved with 30 mg/kg of vancomycin were observed (15).
CONCLUDING REMARKS
Research into the activity and design of AMPs and their mimics has produced several antimicrobial compounds with potent antibacterial activity and elucidated trends of increasing activity and specificity. Applications for this work are numerous, including antimicrobial surfaces (39, 76) and conjugates in targeted therapy (18, 28). It is expected that interest in AMPs will grow in the coming years, opening up new avenues in antimicrobial drug design.
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canadian Institutes of Health Research (CIHR), and the Manitoba Health Research Council (MHRC).
Footnotes
Published ahead of print on 9 August 2010.
REFERENCES
- 1.Al-Badri, Z. M., A. Som, S. Lyon, C. F. Nelson, K. Nusslein, and G. N. Tew. 2008. Investigating the effect of increasing charge density on the hemolytic activity of synthetic antimicrobial polymers. Biomacromolecules 9:2805-2810. doi: 10.1021/bm800569x. [DOI] [PubMed] [Google Scholar]
- 2.Avrahami, D., and Y. Shai. 2002. Conjugation of a magainin analogue with lipophilic acids controls hydrophobicity, solution assembly, and cell selectivity. Biochemistry (NY) 41:2254-2263. doi: 10.1021/bi011549t. [DOI] [PubMed] [Google Scholar]
- 3.Baussanne, I., A. Bussiere, S. Halder, C. Ganem-Elbaz, M. Ouberai, M. Riou, J. M. Paris, E. Ennifar, M. P. Mingeot-Leclercq, and J. L. Decout. 2010. Synthesis and antimicrobial evaluation of amphiphilic neamine derivatives. J. Med. Chem. 53:119-127. doi: 10.1021/jm900615h. [DOI] [PubMed] [Google Scholar]
- 4.Beckloff, N., D. Laube, T. Castro, D. Furgang, S. Park, D. Perlin, D. Clements, H. Tang, R. W. Scott, G. N. Tew, and G. Diamond. 2007. Activity of an antimicrobial peptide mimetic against planktonic and biofilm cultures of oral pathogens. Antimicrob. Agents Chemother. 51:4125-4132. doi: 10.1128/AAC.00208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bera, S., G. G. Zhanel, and F. Schweizer. 2008. Design, synthesis, and antibacterial activities of neomycin-lipid conjugates: polycationic lipids with potent gram-positive activity. J. Med. Chem. 51:6160-6164. doi: 10.1021/jm800345u. [DOI] [PubMed] [Google Scholar]
- 6.Bera, S., G. G. Zhanel, and F. Schweizer. 2010. Antibacterial activities of aminoglycoside antibiotics-derived cationic amphiphiles. Polyol-modified neomycin B-, kanamycin A-, amikacin-, and neamine-based amphiphiles with potent broad spectrum antibacterial activity. J. Med. Chem. doi: 10.1021/jm1000437. [DOI] [PubMed]
- 7.Bera, S., G. G. Zhanel, and F. Schweizer. 2010. Antibacterial activity of guanidinylated neomycin B- and kanamycin A-derived amphiphilic lipid conjugates. J. Antimicrob. Chemother. doi: 10.1093/jac/dkq083. [DOI] [PubMed]
- 8.Bera, S., G. G. Zhanel, and F. Schweizer. 2010. Evaluation of amphiphilic aminoglycoside-peptide triazole conjugates as antibacterial agents. Bioorg. Med. Chem. Lett. 20:3031-3035. [DOI] [PubMed] [Google Scholar]
- 9.Bhunia, A., A. Ramamoorthy, and S. Bhattacharjya. 2009. Helical hairpin structure of a potent antimicrobial peptide MSI-594 in lipopolysaccharide micelles by NMR spectroscopy. Chem. Eur. J. 15:2036-2040. doi: 10.1002/chem.200802635. [DOI] [PubMed] [Google Scholar]
- 10.Braunstein, A., N. Papo, and Y. Shai. 2004. In vitro activity and potency of an intravenously injected antimicrobial peptide and its DL amino acid analog in mice infected with bacteria. Antimicrob. Agents Chemother. 48:3127-3129. doi: 10.1128/AAC.48.8.3127-3129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brogden, K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238-250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 12.Chen, Y., M. T. Guarnieri, A. I. Vasil, M. L. Vasil, C. T. Mant, and R. S. Hodges. 2007. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 51:1398-1406. doi: 10.1128/AAC.00925-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, Y., C. T. Mant, S. W. Farmer, R. E. Hancock, M. L. Vasil, and R. S. Hodges. 2005. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 280:12316-12329. doi: 10.1074/jbc.M413406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen, Y., A. I. Vasil, L. Rehaume, C. T. Mant, J. L. Burns, M. L. Vasil, R. E. Hancock, and R. S. Hodges. 2006. Comparison of biophysical and biologic properties of alpha-helical enantiomeric antimicrobial peptides. Chem. Biol. Drug Des. 67:162-173. doi: 10.1111/j.1747-0285.2006.00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi, S., A. Isaacs, D. Clements, D. Liu, H. Kim, R. W. Scott, J. D. Winkler, and W. F. DeGrado. 2009. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. U. S. A. 106:6968-6973. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chongsiriwatana, N. P., and A. E. Barron. 2010. Comparing bacterial membrane interactions of antimicrobial peptides and their mimics. Methods Mol. Biol. 618:171-182. doi: 10.1007/978-1-60761-594-1_12. [DOI] [PubMed] [Google Scholar]
- 17.Dathe, M., and T. Wieprecht. 1999. Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1462:71-87. doi: 10.1016/S0005-2736(99)00201-1. [DOI] [PubMed] [Google Scholar]
- 18.Ellerby, H. M., D. E. Bredesen, S. Fujimura, and V. John. 2008. Hunter-killer peptide (HKP) for targeted therapy. J. Med. Chem. 51:5887-5892. doi: 10.1021/jm800495u. [DOI] [PubMed] [Google Scholar]
- 19.Fjell, C. D., H. Jenssen, K. Hilpert, W. A. Cheung, N. Pante, R. E. Hancock, and A. Cherkasov. 2009. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 52:2006-2015. doi: 10.1021/jm8015365. [DOI] [PubMed] [Google Scholar]
- 20.Flemming, K., C. Klingenberg, J. P. Cavanagh, M. Sletteng, W. Stensen, J. S. Svendsen, and T. Flaegstad. 2009. High in vitro antimicrobial activity of synthetic antimicrobial peptidomimetics against staphylococcal biofilms. J. Antimicrob. Chemother. 63:136-145. doi: 10.1093/jac/dkn464. [DOI] [PubMed] [Google Scholar]
- 21.Gabriel, G. J., A. E. Madkour, J. M. Dabkowski, C. F. Nelson, K. Nusslein, and G. N. Tew. 2008. Synthetic mimic of antimicrobial peptide with nonmembrane-disrupting antibacterial properties. Biomacromolecules 9:2980-2983. doi: 10.1021/bm800855t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge, Y., D. L. MacDonald, K. J. Holroyd, C. Thornsberry, H. Wexler, and M. Zasloff. 1999. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob. Agents Chemother. 43:782-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giménez, D., C. Andreu, M. D. Olmo, T. Varea, D. Diaz, and G. Asensio. 2006. The introduction of fluorine atoms or trifluoromethyl groups in short cationic peptides enhances their antimicrobial activity. Bioorg. Med. Chem. 14:6971-6978. doi: 10.1016/j.bmc.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 24.Giuliani, A., G. Pirri, and A. C. Rinaldi. 2010. Antimicrobial peptides: the LPS connection. Methods Mol. Biol. 618:137-154. doi: 10.1007/978-1-60761-594-1_10. [DOI] [PubMed] [Google Scholar]
- 25.Hale, J. D., and R. E. Hancock. 2007. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti Infect. Ther. 5:951-959. doi: 10.1586/14787210.5.6.951. [DOI] [PubMed] [Google Scholar]
- 26.Hancock, R. E., and H. G. Sahl. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24:1551-1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 27.Hancock, R. E., and M. G. Scott. 2000. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. U. S. A. 97:8856-8861. doi: 10.1073/pnas.97.16.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansel, W., C. Leuschner, and F. Enright. 2007. Conjugates of lytic peptides and LHRH or betaCG target and cause necrosis of prostate cancers and metastases. Mol. Cell. Endocrinol. 269:26-33. doi: 10.1016/j.mce.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 29.Haug, B. E., W. Stensen, M. Kalaaji, O. Rekdal, and J. S. Svendsen. 2008. Synthetic antimicrobial peptidomimetics with therapeutic potential. J. Med. Chem. 51:4306-4314. doi: 10.1021/jm701600a. [DOI] [PubMed] [Google Scholar]
- 30.Hawrani, A., R. A. Howe, T. R. Walsh, and C. E. Dempsey. 2008. Origin of low mammalian cell toxicity in a class of highly active antimicrobial amphipathic helical peptides. J. Biol. Chem. 283:18636-18645. doi: 10.1074/jbc.M709154200. [DOI] [PubMed] [Google Scholar]
- 31.Hilpert, K., R. Volkmer-Engert, T. Walter, and R. E. Hancock. 2005. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol. 23:1008-1012. doi: 10.1038/nbt1113. [DOI] [PubMed] [Google Scholar]
- 32.Jadhav, V., S. Maiti, A. Dasgupta, P. K. Das, R. S. Dias, M. G. Miguel, and B. Lindman. 2008. Effect of the head-group geometry of amino acid-based cationic surfactants on interaction with plasmid DNA. Biomacromolecules 9:1852-1859. doi: 10.1021/bm8000765. [DOI] [PubMed] [Google Scholar]
- 33.Jenssen, H., P. Hamill, and R. E. Hancock. 2006. Peptide antimicrobial agents. Clin. Microbiol. Rev. 19:491-511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang, Z., A. I. Vasil, J. D. Hale, R. E. Hancock, M. L. Vasil, and R. S. Hodges. 2008. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 90:369-383. doi: 10.1002/bip.20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klocek, G., T. Schulthess, Y. Shai, and J. Seelig. 2009. Thermodynamics of melittin binding to lipid bilayers. Aggregation/pore formation. Biochemistry 48:2586-2596. doi: 10.1021/bi802127h. [DOI] [PubMed] [Google Scholar]
- 36.Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105-132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 37.Liu, Z., A. Brady, A. Young, B. Rasimick, K. Chen, C. Zhou, and N. R. Kallenbach. 2007. Length effects in antimicrobial peptides of the (RW)n series. Antimicrob. Agents Chemother. 51:597-603. doi: 10.1128/AAC.00828-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu, Z., A. W. Young, P. Hu, A. J. Rice, C. Zhou, Y. Zhang, and N. R. Kallenbach. 2007. Tuning the membrane selectivity of antimicrobial peptides by using multivalent design. Chembiochem 8:2063-2065. doi: 10.1002/cbic.200700502. [DOI] [PubMed] [Google Scholar]
- 39.Madkour, A. E., J. M. Dabkowski, K. Nusslein, and G. N. Tew. 2009. Fast disinfecting antimicrobial surfaces. Langmuir 25:1060-1067. doi: 10.1021/la802953v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makovitzki, A., J. Baram, and Y. Shai. 2008. Antimicrobial lipopolypeptides composed of palmitoyl di- and tricationic peptides: in vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 47:10630-10636. doi: 10.1021/bi8011675. [DOI] [PubMed] [Google Scholar]
- 41.Makovitzki, A., and Y. Shai. 2005. pH-dependent antifungal lipopeptides and their plausible mode of action. Biochemistry 44:9775-9784. doi: 10.1021/bi0502386. [DOI] [PubMed] [Google Scholar]
- 42.Malina, A., and Y. Shai. 2005. Conjugation of fatty acids with different lengths modulates the antibacterial and antifungal activity of a cationic biologically inactive peptide. Biochem. J. 390:695-702. doi: 10.1042/BJ20050520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marr, A. K., W. J. Gooderham, and R. E. Hancock. 2006. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr. Opin. Pharmacol. 6:468-472. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Matsuzaki, K. 2008. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 1788:1687-1692. doi: 10.1016/j.bbamem.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 45.Melo, M. N., R. Ferre, and M. A. Castanho. 2009. Antimicrobial peptides: linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 7:245-250. doi: 10.1038/nrmicro2095. [DOI] [PubMed] [Google Scholar]
- 46.Meng, H., and K. Kumar. 2007. Antimicrobial activity and protease stability of peptides containing fluorinated amino acids. J. Am. Chem. Soc. 129:15615-15622. doi: 10.1021/ja075373f. [DOI] [PubMed] [Google Scholar]
- 47.Mitra, R. N., A. Shome, P. Paul, and P. K. Das. 2009. Antimicrobial activity, biocompatibility and hydrogelation ability of dipeptide-based amphiphiles. Org. Biomol. Chem. 7:94-102. doi: 10.1039/b815368j. [DOI] [PubMed] [Google Scholar]
- 48.Mowery, B. P., S. E. Lee, D. A. Kissounko, R. F. Epand, R. M. Epand, B. Weisblum, S. S. Stahl, and S. H. Gellman. 2007. Mimicry of antimicrobial host-defense peptides by random copolymers. J. Am. Chem. Soc. 129:15474-15476. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- 49.Mulvey, M. R., and A. E. Simor. 2009. Antimicrobial resistance in hospitals: how concerned should we be? CMAJ 180:408-415. doi: 10.1503/cmaj.080239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naarmann, N., B. Bilgicer, H. Meng, K. Kumar, and C. Steinem. 2006. Fluorinated interfaces drive self-association of transmembrane alpha helices in lipid bilayers. Angew. Chem. Int. Ed. Engl. 45:2588-2591. doi: 10.1002/anie.200503567. [DOI] [PubMed] [Google Scholar]
- 51.Otter, J. A., and G. L. French. 2006. Nosocomial transmission of community-associated methicillin-resistant Staphylococcus aureus: an emerging threat. Lancet Infect. Dis. 6:753-755. doi: 10.1016/S1473-3099(06)70636-3. [DOI] [PubMed] [Google Scholar]
- 52.Papo, N., and Y. Shai. 2004. Effect of drastic sequence alteration and D-amino acid incorporation on the membrane binding behavior of lytic peptides. Biochemistry 43:6393-6403. doi: 10.1021/bi049944h. [DOI] [PubMed] [Google Scholar]
- 53.Park, C. B., H. S. Kim, and S. C. Kim. 1998. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 244:253-257. doi: 10.1006/bbrc.1998.8159. [DOI] [PubMed] [Google Scholar]
- 54.Perron, G. G., M. Zasloff, and G. Bell. 2006. Experimental evolution of resistance to an antimicrobial peptide. Proc. Biol. Sci. 273:251-256. doi: 10.1098/rspb.2005.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peschel, A., and H. G. Sahl. 2006. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4:529-536. doi: 10.1038/nrmicro1441. [DOI] [PubMed] [Google Scholar]
- 56.Putsep, K., G. Carlsson, H. G. Boman, and M. Andersson. 2002. Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet 360:1144-1149. doi: 10.1016/S0140-6736(02)11201-3. [DOI] [PubMed] [Google Scholar]
- 57.Radzishevsky, I. S., T. Kovachi, Y. Porat, L. Ziserman, F. Zaknoon, D. Danino, and A. Mor. 2008. Structure-activity relationships of antibacterial acyl-lysine oligomers. Chem. Biol. 15:354-362. doi: 10.1016/j.chembiol.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 58.Radzishevsky, I. S., S. Rotem, D. Bourdetsky, S. Navon-Venezia, Y. Carmeli, and A. Mor. 2007. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat. Biotechnol. 25:657-659. doi: 10.1038/nbt1309. [DOI] [PubMed] [Google Scholar]
- 59.Rotem, S., and A. Mor. 2009. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta 1788:1582-1592. doi: 10.1016/j.bbamem.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 60.Rotem, S., I. S. Radzishevsky, D. Bourdetsky, S. Navon-Venezia, Y. Carmeli, and A. Mor. 2008. Analogous oligo-acyl-lysines with distinct antibacterial mechanisms. FASEB J. 22:2652-2661. doi: 10.1096/fj.07-105015. [DOI] [PubMed] [Google Scholar]
- 61.Sarig, H., S. Rotem, L. Ziserman, D. Danino, and A. Mor. 2008. Impact of self-assembly properties on antibacterial activity of short acyl-lysine oligomers. Antimicrob. Agents Chemother. 52:4308-4314. doi: 10.1128/AAC.00656-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schneider, T., T. Kruse, R. Wimmer, I. Wiedemann, V. Sass, U. Pag, A. Jansen, A. K. Nielsen, P. H. Mygind, D. S. Raventos, S. Neve, B. Ravn, A. M. Bonvin, L. De Maria, A. S. Andersen, L. K. Gammelgaard, H. G. Sahl, and H. H. Kristensen. 2010. Plectasin, a fungal defensin, targets the bacterial cell wall precursor lipid II. Science 328:1168-1172. doi: 10.1126/science.1185723. [DOI] [PubMed] [Google Scholar]
- 63.Scott, R. W., W. F. DeGrado, and G. N. Tew. 2008. De novo designed synthetic mimics of antimicrobial peptides. Curr. Opin. Biotechnol. 19:620-627. doi: 10.1016/j.copbio.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serrano, G. N., G. G. Zhanel, and F. Schweizer. 2009. Antibacterial activity of ultrashort cationic lipo-beta-peptides. Antimicrob. Agents Chemother. 53:2215-2217. doi: 10.1128/AAC.01100-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shai, Y., and Z. Oren. 2001. From “carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Peptides 22:1629-1641. doi: 10.1016/S0196-9781(01)00498-3. [DOI] [PubMed] [Google Scholar]
- 66.Song, J. 2008. What's new on the antimicrobial horizon? Int. J. Antimicrob. Agents 32:S207-S213. doi: 10.1016/S0924-8579(09)70004-4. [DOI] [PubMed] [Google Scholar]
- 67.Stewart, K. M., K. L. Horton, and S. O. Kelley. 2008. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 6:2242-2255. doi: 10.1039/b719950c. [DOI] [PubMed] [Google Scholar]
- 68.Strøm, M. B., B. E. Haug, M. L. Skar, W. Stensen, T. Stiberg, and J. S. Svendsen. 2003. The pharmacophore of short cationic antibacterial peptides. J. Med. Chem. 46:1567. doi: 10.1021/jm0340039. [DOI] [PubMed] [Google Scholar]
- 69.Svenson, J., W. Stensen, B. O. Brandsdal, B. E. Haug, J. Monrad, and J. S. Svendsen. 2008. Antimicrobial peptides with stability toward tryptic degradation. Biochemistry 47:3777-3788. doi: 10.1021/bi7019904. [DOI] [PubMed] [Google Scholar]
- 70.Tang, H., R. J. Doerksen, T. V. Jones, M. L. Klein, and G. N. Tew. 2006. Biomimetic facially amphiphilic antibacterial oligomers with conformationally stiff backbones. Chem. Biol. 13:427-435. doi: 10.1016/j.chembiol.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 71.Tew, G. N., D. Liu, B. Chen, R. J. Doerksen, J. Kaplan, P. J. Carroll, M. L. Klein, and W. F. DeGrado. 2002. De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. U. S. A. 99:5110-5114. doi: 10.1073/pnas.082046199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vallon-Eberhard, A., A. Makovitzki, A. Beauvais, J. P. Latge, S. Jung, and Y. Shai. 2008. Efficient clearance of Aspergillus fumigatus in murine lungs by an ultrashort antimicrobial lipopeptide, palmitoyl-lys-ala-DAla-lys. Antimicrob. Agents Chemother. 52:3118-3126. doi: 10.1128/AAC.00526-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van den Bogaart, G., J. V. Guzman, J. T. Mika, and B. Poolman. 2008. On the mechanism of pore formation by melittin. J. Biol. Chem. 283:33854-33857. doi: 10.1074/jbc.M805171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang, G., X. Li, and Z. Wang. 2009. APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 37:D933-D937. doi: 10.1093/nar/gkn823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wehkamp, J., and E. F. Stange. 2006. A new look at Crohn's disease: breakdown of the mucosal antibacterial defense. Ann. N. Y. Acad. Sci. 1072:321-331. doi: 10.1196/annals.1326.030. [DOI] [PubMed] [Google Scholar]
- 76.Willcox, M. D., E. B. Hume, Y. Aliwarga, N. Kumar, and N. Cole. 2008. A novel cationic-peptide coating for the prevention of microbial colonization on contact lenses. J. Appl. Microbiol. 105:1817-1825. doi: 10.1111/j.1365-2672.2008.03942.x. [DOI] [PubMed] [Google Scholar]
- 77.Yang, L., V. D. Gordon, D. R. Trinkle, N. W. Schmidt, M. A. Davis, C. DeVries, A. Som, J. E. Cronan, Jr., G. N. Tew, and G. C. Wong. 2008. Mechanism of a prototypical synthetic membrane-active antimicrobial: efficient hole-punching via interaction with negative intrinsic curvature lipids. Proc. Natl. Acad. Sci. U. S. A. 105:20595-20600. doi: 10.1073/pnas.0806456105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zasloff, M. 2002. Antimicrobial peptides in health and disease. N. Engl. J. Med. 347:1199-1200. doi: 10.1056/NEJMe020106. [DOI] [PubMed] [Google Scholar]
- 79.Zhang, J., F. I. Chiang, L. Wu, P. G. Czyryca, D. Li, and C. W. Chang. 2008. Surprising alteration of antibacterial activity of 5"-modified neomycin against resistant bacteria. J. Med. Chem. 51:7563-7573. doi: 10.1021/jm800997s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang, J., K. Keller, J. Y. Takemoto, M. Bensaci, A. Litke, P. G. Czyryca, and C. W. Chang. 2009. Synthesis and combinational antibacterial study of 5"-modified neomycin. J. Antibiot. (Tokyo) 62:539-544. doi: 10.1038/ja.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu, W. L., H. Lan, I. S. Park, J. I. Kim, H. Z. Jin, K. S. Hahm, and S. Y. Shin. 2006. Design and mechanism of action of a novel bacteria-selective antimicrobial peptide from the cell-penetrating peptide Pep-1. Biochem. Biophys. Res. Commun. 349:769-774. doi: 10.1016/j.bbrc.2006.08.094. [DOI] [PubMed] [Google Scholar]
- 82.Zhu, W. L., and S. Y. Shin. 2009. Antimicrobial and cytolytic activities and plausible mode of bactericidal action of the cell penetrating peptide penetratin and its lys-linked two-stranded peptide. Chem. Biol. Drug Des. 73:209-215. doi: 10.1111/j.1747-0285.2008.00769.x. [DOI] [PubMed] [Google Scholar]
- 83.Zhu, W. L., Y. M. Song, Y. Park, K. H. Park, S. T. Yang, J. I. Kim, I. S. Park, K. S. Hahm, and S. Y. Shin. 2007. Substitution of the leucine zipper sequence in melittin with peptoid residues affects self-association, cell selectivity, and mode of action. Biochim. Biophys. Acta 1768:1506-1517. doi: 10.1016/j.bbamem.2007.03.010. [DOI] [PubMed] [Google Scholar]