

Abstract
As a posttranslational protein modification, cysteine sulfenic acid (Cys-SOH) is well established as an oxidative stress-induced mediator of enzyme function and redox signaling. Data presented herein show that protein Cys-SOH forms spontaneously in air-exposed aqueous solutions of unfolded (disulfide-reduced) protein in the absence of added oxidizing reagents, mediating the oxidative disulfide bond formation process key to in vitro, non-enzymatic protein folding. Molecular oxygen (O2) and trace metals (e.g., copper (II)) are shown to be important reagents in the oxidative refolding process. Cys-SOH is also revealed to play a role in spontaneous disulfide-based dimerization of peptide molecules containing free cysteine residues. In total, the data presented expose a chemically ubiquitous role for Cys-SOH in solutions of free cysteine-containing protein exposed to air.
In the early 1960s Anfinsen and colleagues carried out seminal studies in protein disulfide bond formation and folding, showing that unfolded (disulfide reduced) proteins will spontaneously and completely refold, regenerating full protein activity, over a period of about a day in the absence of denaturants (1-6). For decades it has been known that free thiol-containing molecules will, over time, spontaneously form intra- and/or intermolecular disulfide bonds when stored exposed to air in aqueous solution. But thiols do not spontaneously oxidize one another to generate disulfide bonds in the absence of an oxidizing reagent (7). Moreover, an intermediate oxidized form of sulfur has not been clearly identified as a ubiquitous intermediate in the spontaneous formation of disulfide bonds in vitro. With the discovery that disulfide bond formation and protein folding in vivo is an enzyme catalyzed process that takes place in a matter of seconds, interest in identifying an oxidized sulfur intermediate relevant to non-enzymatic protein folding seems to have waned. Even so, such an intermediate remains important in peptide and protein research / production systems that, intentionally or not, involve non-enzyme mediated disulfide bond formation and protein folding. Knowledge of the identity of such an intermediate may allow for a better degree of control in manipulating in vitro protein systems.
Biologically, cysteine sulfenic acid (Cys-SOH) formation within folded protein molecules can serve as a means of regulating protein activity—helping to absorb oxidative insults (8, 9), informationally “register” such insults and/or alter protein activity (10-20), mediate redox signaling (16, 17, 21), and generally deflect (14-16, 22-26) what otherwise might have been injurious oxidative damage. In most studies on protein Cys-SOH, exogenous oxidants such as hydrogen peroxide are required to build up significant quantities of Cys-SOH within fully folded proteins.
In general, Cys-SOH is a transient intermediate that, until the past decade has been difficult to study. It has been understood that Cys-SOH likely renders the sulfur atom of cysteine electrophilic to the point of being susceptible to nucleophile attack (i.e., by a proximal thiol / thiolate anion) (10, 27). Yet, though they now appear to play a key role in the biochemical regulation of many different proteins, Cys-SOH modifications have, historically, been difficult to study due to their inherent instability outside of their native “cocoon-like” protein environment (9, 10, 28-31). Recent developments in analytical technologies and molecular probes—particularly those based on the specific alkylation (or irreversible “covalent trapping”) of Cys-SOH residues with 5,5-dimethyl-1,3-cyclohexanedione (dimedone) (10, 32-39) have produced a great deal of interest and a flurry of research activity centered on this unique protein modification.
The purpose of this study was to determine whether or not cysteine sulfenic acid serves as the oxidized sulfur intermediate that mediates air-induced disulfide bond formation and non-enzymatic protein folding. Using dimedone as a well established (10, 32-39) mass-shifting molecular probe of Cys-SOH (Scheme 1) and bovine ribonuclease A (RNAse A) as a model protein1, we report herein the observation that air and trace metal-generated Cys-SOH is a non-site-specific intermediate in the disulfide bond formation process that occurs as part of the in vitro, non-enzymatic protein folding process.
Scheme 1.
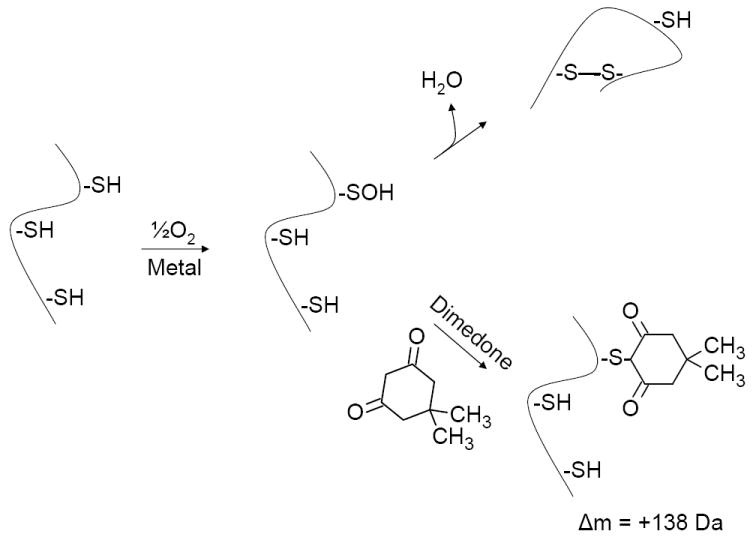
Proposed reaction pathway for in vitro generation of disulfide bonds and protein folding. Dimedone reacts covalently and irreversibly with Cys-SOH, making it an effective probe for the existence of Cys-SOH (10, 32-39). Detection of covalent dimedone-modified protein in the experiments described here indicates the production of protein Cys-SOH. By logical extension of previous studies on the reactivity of intra- and intermolecular sulfhydryl groups with Cys-SOH (7, 40), such detection suggests that Cys-SOH is an oxidized sulfur intermediate that mediates in vitro disulfide bond formation and protein folding. Early experiments by Anfinsen and colleagues (2-4, 6) showed that in vitro protein folding goes to completion and full restoration of protein activity.
EXPERIMENTAL PROCEDURES (MATERIALS and METHODS)
Reagents
All chemicals including Ribonuclease A Type III-A from bovine pancreas were purchased from Sigma-Aldrich. Synthetic peptide “IGF 57-70” (H2N-ALLETYCATPAKSE-CO2H) was purchased from American Peptide. Amicon centrifugal concentration units were from Millipore. Endoproteinase Lys-C from Lysobacter enzymogenes was obtained from Roche. Spilfyter Hands-in-Bag atmospheric chambers were purchased from VWR.
Protein Reduction and Refolding
RNAse A was dissolved at 10 mg mL-1 in freshly prepared 8 M urea, pH 8.6 (adjusted with 5% (w/v) methylamine (2)), containing 50 mM dithiothreitol and incubated in a rotary shaker at 800 rpm and 37 °C for 1 hour. One hundred microliters of the reduced protein solution was adjusted to pH 3.5 with glacial acetic acid then loaded into an Amicon Ultra-4 5 kDa MWCO centrifugal concentration unit containing 4 mL of 0.1 M acetic acid. The sample was then centrifuged in a swing-bucket rotor for 20 minutes at 4,000 × g. The retentate was then re-diluted with 4 mL of 0.1 M acetic acid and the cycle of concentration and re-dilution was carried out a total of 5 times, resulting in a greater than one million fold dilution of urea and dithiothreitol.
The protein concentration of the final ~250-μL retentate was generally found to be ~120 μM by absorption spectrophotometry at 276 nm (ε = 9390 M-1cm-1 for reduced RNAse A (4)). To verify complete reduction, a 5-μL aliquot was removed and alkylated with 5 μL of 50 mM maleimide dissolved in 50 mM ammonium acetate, pH 5, and incubated at 50 °C for 15 minutes. The alkylated RNAse A was then analyzed intact by mass spectrometry to verify complete reduction by means of an anticipated 8 × 97 Da mass increase (Supporting Information Figure S1).
For each set of refolding conditions examined, two aliquots of purified, unfolded RNAse A were diluted to 0.075 mg mL-1 (5.5 μM), adjusted to pH 8 with a saturated solution of Tris base, and left to spontaneously refold at room temperature in the presence or absence of 50 mM dimedone. An additional negative control of native (folded) RNAse A was also incubated in the presence of 50 mM dimedone. When employed, additives such as metals (50 μM CuSO4 or Fe2(SO4)3) and metal chelators (1 mM EDTA and 0.1 mM DTPA) were added from 100-200x stock solutions (in 0.1 M acetic acid or tris acetate buffer) just prior to adjustment of solution pH and addition of protein. Final volumes were typically 1 mL. For incubations under controlled atmospheric conditions samples were placed into a Spilfyter Hands-in-Bag atmospheric chamber which was filled and continually pressurized with high purity nitrogen or oxygen. Samples were withdrawn with a gel loading pipette tip through a resealable pinhole in the top of the chamber.
Assessment of Refolding Progress
Refolding progress (in the absence of dimedone) was monitored by taking 5-μL aliquots of sample and mixing with 5 μL of 50 mM maleimide in 0.2 M acetic acid (giving a final pH ≤ 5). Samples were incubated at 50 °C for 15 minutes then diluted to 1 μM final concentration in starting LC solvent before immediately injecting 2 μL on trap. (Dimedone-containing samples were not alkylated, but simply diluted to 1 μM final concentration in starting LC solvent before 2 μL was injected on trap.)
Refolding progress was monitored by maleimide-alkylation of partially refolded protein and analysis of samples by ESI-MS. As demonstrated by the raw mass spectra (Supporting Information Figures S1 and S3), the procedure employed resulted in sulfhydryl-specific protein alkylation with essentially no alkylation of protein amino groups. To determine the precise degree to which proteins were refolded, deconvoluted ESI mass spectra were integrated. Peak areas were then summed and the fraction of protein in each folding state (i.e., 0, 1, 2, 3, or 4 disulfides—see Supporting Information Figure S3) determined by dividing the area of the appropriate representative peak by the total area. These fractions were then weighted by folding state according to the following equation:
Where Rp represents the percentage of protein refolded and FSSn represents the fraction of protein containing n disulfide bonds. Thus, for example, if two equal-area peaks were observed in a deconvoluted ESI mass spectrum representing protein molecules with 3 disulfides and protein molecules with 4 disulfides, the percentage of protein refolded would be reported as 87.5%. Notably, this summary method of reporting protein refolding progress as a single numerical value is useful for comparison with similar non-mass spectrometric techniques, but it mutes data on folding-state molecular statistics (which are available in the raw data) that might be considered informative toward certain kinetic models of folding (which were not under consideration here). In unmuted form—i.e., without applying the above equation—the raw data that are acquired using this technique contain more detailed information on protein disulfide status than is generally available with other non-mass spectrometric techniques.
Cysteine-containing Peptide Incubation with Dimedone
Three microliters of a 670 μM solution of a single-free-cysteine-containing peptide (H2N-ALLETYCATPAKSE-CO2H) (a.k.a. IGF 57-70) was added to 96 μL of freshly prepared 0.15 M ammonium bicarbonate, pH 7.1 and 1 μL of 1 M dimedone in ethanol, giving final peptide and dimedone concentrations of 20 μM and 10 mM, respectively. A dimedone-lacking control was prepared in parallel. The samples were then allowed to sit exposed to air for 20 hours in the dark at room temperature, at which point they were diluted 30-fold in MALDI matrix solution and analyzed by MALDI-MS and MALDI-MS/MS (TOF/TOF) as described below.
ESI-LC/MS
A trap-and-elute form of sample concentration / solvent exchange rather than traditional LC was employed. Samples were injected by a Spark Holland Endurance autosampler in microliter pick-up mode and loaded by an Eksigent nanoLC*1D at 10 μL min-1 (90/10 water/acetonitrile containing 0.1% (v/v) formic acid, Solvent A) onto a protein captrap (Michrom Bioresources, Auburn, CA) configured for unidirectional flow on a 6-port divert valve. After 2 minutes, the divert valve position was automatically toggled and flow over the cartridge changed to 1 μL min-1 Solvent A (running directly to the ESI inlet) which was immediately ramped over 5 minutes to 10/90 water/acetonitrile containing 0.1% (v/v) formic acid. By 7.2 minutes the run was completed and the flow back to 100% solvent A.
The bulk of the RNAse A eluted between 3.5 and 5 minutes into a Bruker MicrOTOF-Q (Q-TOF) mass spectrometer operating in positive ion, TOF-only mode, acquiring spectra in the m/z range of 50 to 3000. ESI settings for the Agilent G1385A capillary nebulizer ion source were as follows: End Plate Offset -500 V, Capillary -4500 V, Nebulizer nitrogen 2 Bar, Dry Gas nitrogen 3.0 L min-1 at 225 °C. Data were acquired in profile mode at a digitizer sampling rate of 2 GHz. Spectra rate control was by summation at 1 Hz.
One to two minutes of recorded spectra were averaged across the chromatographic peak apex of RNAse A elution. The ESI charge-state envelope was deconvoluted with Bruker DataAnalysis v3.4 software to a mass range of 1000 Da on either side of any identified peak.
Proteolytic Digestion of RNAse A
Reduced RNAse A that had been incubated in air for more than 20 hours in the absence or presence of dimedone was digested overnight at 37 °C and pH 7.5 with Lys-C at a protein to protease mass ratio of 3:1. One microliter of the resulting digestion mixture was added to 4 μL of MALDI matrix solution (see below) and allowed to dry on the MALDI target.
MALDI-MS
Peptides were mixed with MALDI matrix solution (33% (v/v) acetonitrile containing 0.4% (v/v) TFA and saturated with α-cyano-4-hydroxycinnamic acid) and spotted onto a gold-surfaced MALDI target and allowed to dry. Single stage and LIFT-TOF/TOF mass spectra were acquired on a Bruker Ultraflex MALDI-TOF/TOF instrument. Externally calibrated mass spectra were acquired in positive ion mode with the reflector engaged. For LIFT-TOF/TOF experiments, precursor ion selection width was set on an individual peptide basis to ensure that no undesired parent ions would contaminate the MS/MS spectra. Instrument voltages and other parameters were optimized for peptide resolution and sensitivity; at least 7,500 laser shots were acquired per sample to ensure excellent ion counting statistics.
RESULTS
Strategy
Scheme 1 depicts the hypothesized ambient oxygen and trace metal-induced formation of Cys-SOH in an unfolded protein molecule. Previous studies have demonstrated that in the absence of a trapping agent (which allows for detection of Cys-SOH) a facile reaction occurs between hydrogen peroxide-generated protein Cys-SOH and intramolecular thiols (40). (Intermolecular protein thiols are also reactive with peroxide-generated Cys-SOH (7, 41).) We therefore reasoned that if Cys-SOH were to be detected within a refolding protein, then it must be serving as an oxidized sulfur intermediate in the in vitro formation of intramolecular disulfide bonds.
Trapping of Cys-SOH During Protein Refolding
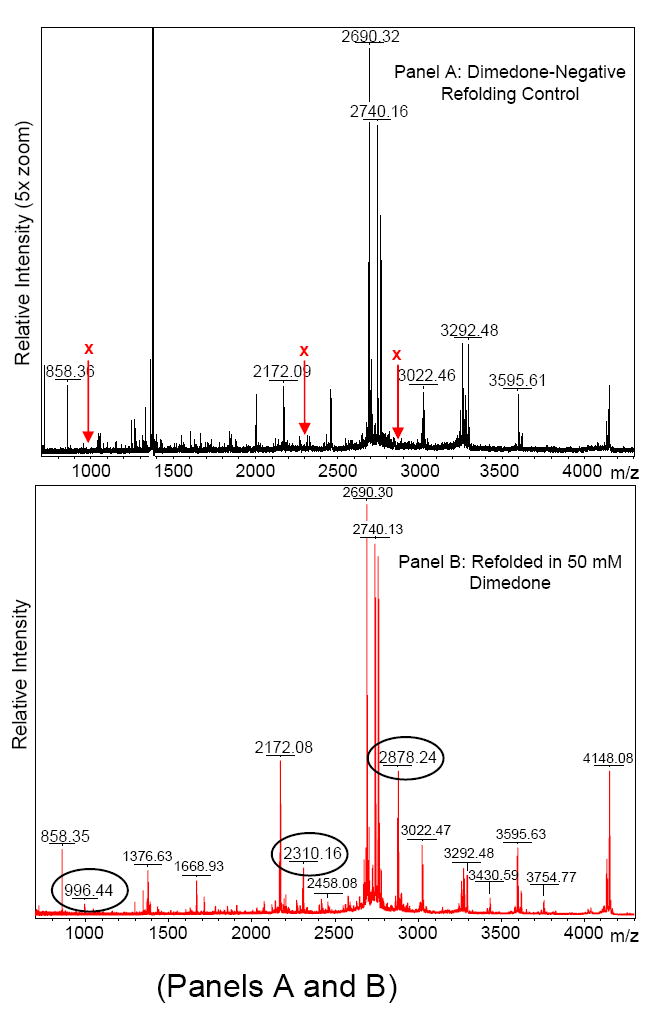
To determine the potential role of air-generated Cys-SOH in disulfide bond formation and in vitro, non-enzymatic protein folding, disulfide-reduced RNAse A was incubated at room temperature with exposure to air (analogous to Anfinsen and co-workers’ seminal experiments in protein folding (1-6)) in the presence of the Cys-SOH trapping reagent dimedone (32-39). Deconvoluted ESI-mass spectra of intact RNAse A incubated in the presence of dimedone were acquired (Figure 1). Mass shifts caused by covalent dimedone reaction products were present. Control experiments conducted in the absence of dimedone and, separately, in the presence of dimedone but with native (fully folded) RNAse A did not result in dimedone-modified RNAse A as demonstrated (Figure 1).
Figure 1.

Charge deconvoluted “singly charged” ESI-mass spectra of A) Negative control in which reduced RNAse A was allowed to refold in the absence of dimedone at room temperature, pH 8, and exposure to air. “X” indicates the absence of a peak at the expected m/z value of dimedone-modified RNAse A; B) Negative control in which native (non-reduced / folded) RNAse A was exposed to refolding conditions in the presence of 50 mM dimedone; C) Reduced and purified RNAse A refolded in the presence of 50 mM dimedone. The calculated MH+ mass of fully reduced RNAse A is 13691.3 and that of fully folded RNAse A is 13683.2.
Verification of Covalent Dimedone-Protein Product Formation
To verify that dimedone was indeed covalently bound to RNAse A (due to its reaction with Cys-SOH), the samples described in Figure 1, panels A and C were digested with Lys-C and analyzed by MALDI-MS and MALDI-MS/MS (TOF/TOF). Mass spectra of the resulting peptide mixtures were acquired (Figure 2). With one exception, observed monoisotopic m/z values aligned to within 20 ppm of the calculated m/z values (Table 1). Average mass accuracy was 13.7 ppm (including the noted outlier). MS/MS (TOF/TOF) spectra for three of these peptides containing non-redundant cysteine residues were acquired; one of these spectra is shown as Panel C in Figure 2 (the others are available online as Supporting Information Figure S2). In agreement with the single-stage (peptide mapping) MALDI mass spectra, each of these MS/MS spectra confirm the covalent attachment of dimedone to the suspected cysteine residue.
Figure 2.
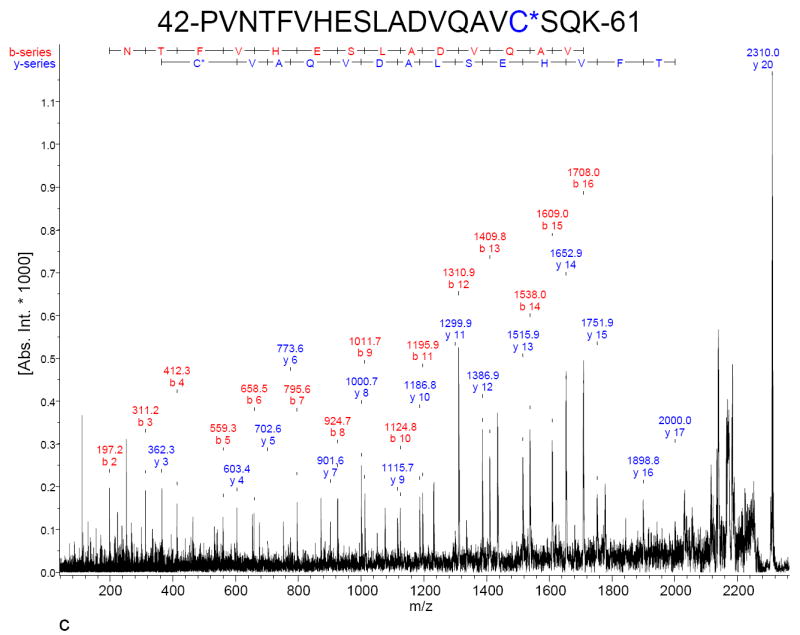
Reflector-mode MALDI mass spectra of Lys-C-digested RNAse A. A) RNAse A that was refolded in the absence of dimedone. “X” indicates the absence of peaks that are found when RNAse A is refolded in the presence of dimedone. B) RNAse A refolded in the presence of 50 mM dimedone. Circled m/z values indicate peaks representative of covalent dimedone-modified RNAse A peptides. These peptides are shifted up in mass by that of covalently attached dimedone (138.07 Da). (Non-covalently attached dimedone would shift the mass by 140.08 Da.) Table 1 provides a list of pertinent calculated and observed masses. Though not visible, isotopic clusters are baseline-resolved, hence m/z values indicated are monoisotopic. C) MALDI-MS/MS (TOF/TOF) spectrum of the peptide represented at m/z 2310 in Panel B. Assigned peaks are accurate to within 0.3 Da. The y-ion series indicates that the cysteine residue is shifted up in mass by 138 Da. MS/MS spectra for the dimedone-modified peptide ions represented by peaks at m/z 996 and 2878 in Panel B are available as Supporting Information.
Table 1.
Calculated and observed monoisotopic masses of cysteine-containing peptides and their corresponding covalent dimedone-modfied peptides that were observed upon MALDI-TOF analysis of Lys-C digested RNAse A that was refolded in the presence of 50 mM dimedone. IGF 57-70 is the synthetic peptide (H2N-ALLETYCATPAKSE-CO2H) incubated for 20 hours in the presence of dimedone (see Figure 4). Observed m/z values aligned to within 20 ppm of the calculated m/z values (with the single exception indicated).
| Calculated m/z | Observed m/z | |
|---|---|---|
| RNase A Y92 - K98 | ||
| Unmodified | 858.38 | 858.35a |
| Dimedone-Modified | 996.45 | 996.44 |
| RNase A P42 - K61 | ||
| Unmodified | 2172.08 | 2172.08 |
| Dimedone-Modified | 2310.14 | 2310.16 |
| RNase A F8 - K31 | ||
| Unmodified | 2740.12 | 2740.13 |
| Dimedone-Modified | 2878.19 | 2878.24 |
| IGF 57-70 Peptide | ||
| Unmodified | 1496.76 | 1496.78 |
| Dimedone-Modified | 1634.83 | 1634.86 |
| Disulfide-linked Dimer | 2990.49 | 2990.54 |
Mass accuracy outlier at 35 ppm.
Roles of Oxygen and Metals
Given its elemental composition, it is clear that formation of Cys-SOH requires oxygen. We hypothesized that diatomic oxygen from air contributes to Cys-SOH formation through the mediation of trace metals in solution. To elucidate the roles of ambient oxygen (O2) and trace metals on the rate of protein refolding, additional experiments were conducted under an oxygen atmosphere, under a nitrogen atmosphere, or in the presence of added copper (II), iron (III) or metal chelators (Figure 3). Samples of RNAse A were allowed to refold until either the refolding process was >95% complete or four days had passed—whichever came first. In the cases of refolding under a nitrogen atmosphere or refolding in the presence of metal chelators (in air), four days passed before 80% refolding was reached. It took just over two days for RNAse A to refold under air without added metals. The addition of 50 μM iron (III) had only a marginal effect on this rate. Refolding under a pure oxygen atmosphere was >95% complete within 24 hours and refolding in the presence of 50 μM copper (II) was >95% complete in less than 2 hours. For each sample described in Figure 3, parallel samples were refolded in the presence of dimedone. In each case covalent dimedone-RNAse A reaction products were discovered as described above. (A time course showing the degree of covalent incorporation of dimedone into refolding RNAse A over time is available as Supporting Information Figure S4.) Notably, none of the experimental alterations to the refolding environment resulted in formation of cysteine sulfinic acid (-SO2H).
Figure 3.
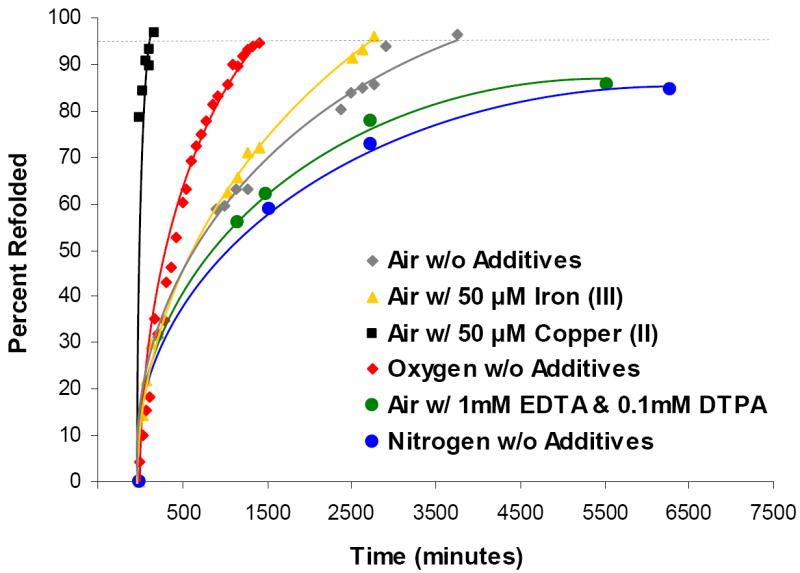
RNAase A refolding progress over time under different atmospheric and solution conditions. Grey diamonds represent RNAse A refolding under air without special additives.
Cys-SOH Mediates Spontaneous Formation of Peptide Dimers
The non-site-specific nature of Cys-SOH formation during RNAse A refolding suggested a generalized chemical phenomenon. To verify the hypothesis that Cys-SOH forms spontaneously on free thiols that are dissolved in neutral-to-slightly-alkaline aqueous solution and exposed to air, a 20 μM solution of the single-free-cysteine-containing peptide “IGF 57-70” (H2N-ALLETYCATPAKSE-CO2H) was placed under such conditions in the presence of dimedone for approximately 20 hours. MALDI-TOF mass spectra were acquired (Figure 4). Besides showing the expected formation of ample quantities of disulfide-linked homodimer, Panels B and C confirm that dimedone was found covalently attached to the cysteine residue of the monomeric peptide—indicating that Cys-SOH had formed under the mild conditions of neutral-to-slightly-alkaline pH and exposure to air.
Figure 4.
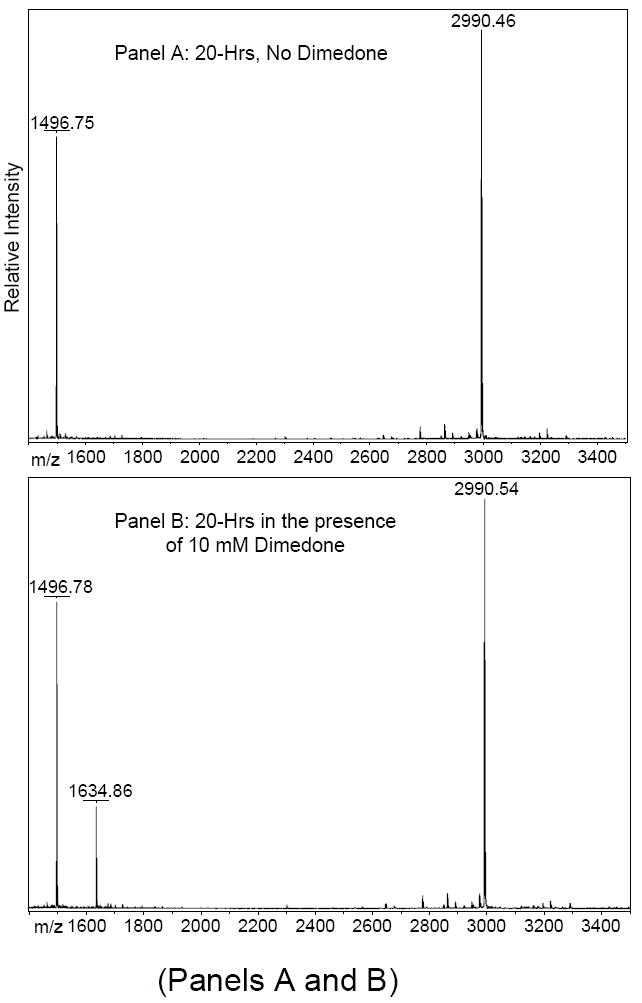
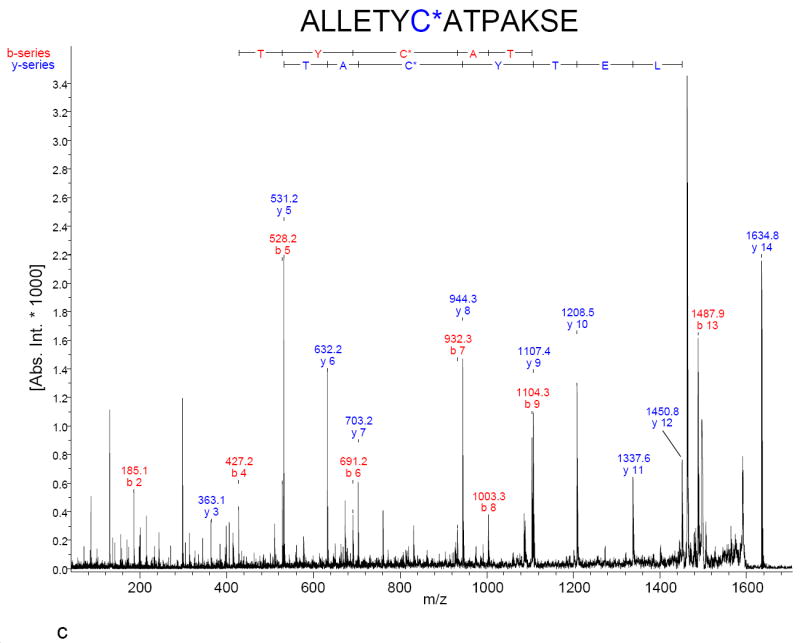
MALDI mass spectra of synthetic peptide “IGF 57-70” (H2N-ALLETYCATPAKSE-CO2H) incubated for 20 hours at room temperature, pH 7.1 in the A) absence of dimedone and B) presence of 10 mM dimedone. Isotopic clusters are baseline resolved and indicated m/z values are monoisotopic. The final rows in Table 1 provide a list of pertinent calculated and observed masses. C) MALDI-MS/MS (TOF/TOF) mass spectrum of the dimedone-modified peptide at m/z 1634. The b- and y-ion series indicate that the cysteine residue is shifted up in mass by 138 Da.
DISCUSSION
The data shown here demonstrate air and trace metal induced formation of Cys-SOH as part of the reduced RNAse A protein molecule. When allowed to proceed for a long enough period of time, the refolding process completes and returns full activity to the RNAse A molecule (2-4, 6). Notably, arsenite is considered a reducing agent specific for Cys-SOH that does not affect protein disulfides (13, 42). As such, if Cys-SOH is involved in the formation of protein disulfide bonds, arsenite should inhibit in vitro protein folding. Such inhibition was recently well documented by Ramadan et al (43). Thus, considering 1) the detection of covalent dimedone-protein reaction products on cysteine residues within the unfolded protein in the absence of added oxidants, 2) that arsenite inhibits oxidative protein folding in vitro (43), 3) that no detectable Cys-SOH forms within a folded RNAse A molecule (Figure 1b), and 4) the known reactivity of free sulfhydryl groups with Cys-SOH (7, 8, 10, 21, 40, 41, 44-46), it is logical to conclude that ambient oxygen-induced Cys-SOH serves as an intermediate in the spontaneous disulfide bond formation process that takes place as part of in vitro, non-enzymatic protein folding. In 2007, Johansson and Lundberg (45) presented evidence for the spontaneous formation of protein-glutathione mixed disulfides. Based on inhibition of the process with dimedone and (separately) arsenite, they suggested that the mixed disulfides were formed via a Cys-SOH intermediate. Here we extend their studies by physically trapping and directly analyzing a spontaneously formed Cys-SOH intermediate which mediates intramolecular disulfide bond formation and in vitro protein folding. Additionally, we show that such natural Cys-SOH species mediate disulfide bond formation between free cysteine-containing peptides, suggesting a ubiquitous role for Cys-SOH in the spontaneous, frequently undesirable formation of disulfide-linked intermolecular dimers.
The data presented in Figure 3 demonstrate that molecular oxygen and trace metals are important reagents in the spontaneous in vitro generation of Cys-SOH. Compared to a “natural” folding rate of about two days in air (grey diamonds), refolding under nitrogen or in the presence of metal chelators (EDTA and DTPA) in air takes several days—but refolding under an oxygen atmosphere takes about one day and refolding in the presence of 50 μM copper (II) takes just a couple of hours. Notably, Anfinsen observed complete refolding of RNAse A in air in about 20-24 hours (3, 5). The differences between Anfinsen’s results and those reported here may be due to the fact that we were able to utilize highly pure deionized water and virgin polypropylene test tubes which are likely to minimize trace metal contamination compared to reagents and reaction vessels available several decades ago.
The apparent plateau phases reached by samples folding under nitrogen or in the presence of metal chelators (Figure 3) may indicate that the extremely low quantities of available oxygen and trace metals, respectively, were effectively depleted during the refolding process—and that because of this the refolding process may never fully complete under these conditions. The mechanisms and rate laws governing formation of Cys-SOH are the subject of future investigation.
Notably, the means by which the folding process was monitored in these studies using maleimide alkylation and monitoring by mass spectrometry provides a unique viewpoint into the protein folding process by allowing for direct determination of the relative number of protein molecules in each state of disulfide bond formation at any given point in time. Though it is not obvious by the way the data are plotted in Figure 3, as explained in the experimental section, each data point was gleaned via a mass spectrum in which information on the relative abundance of protein molecules with 0, 1, 2, 3, or 4 disulfide bonds was unambiguously provided (see Supporting Information Figure S3 for as series of illustrative mass spectra). By examining these relative populations of protein folding states over time, it is clear that formation of the last disulfide bond is the slowest step in the refolding of RNAse A. For example, the sample containing copper (II) was nearly 80% folded within one minute, but took almost 2 hours to reach >95%. These observations support the RNAse A disulfide folding mechanism asserted by Creighton over 30 years ago (47, 48).
Conclusions
The data presented here demonstrate that oxygen and trace metal-generated Cys-SOH is a ubiquitous modification of cysteine residues in solution and an intermediate that mediates in vitro, non-enzymatic disulfide bond formation and protein folding.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Matthew Schaab for helpful discussions.
ABBREVIATIONS
- Cys-SOH
Cysteine sulfenic acid
- RNAse A
bovine ribonuclease A
Footnotes
This research was supported by the National Institutes of Health Grant R21RR024440
Bovine RNAse A (UniProtKB/Swiss-Prot accession number P61823) is a 124-amino acid single-chain protein containing 8 cysteine residues, all of which are involved in intramolecular disulfide bonds. It was employed as a model protein in the seminal protein folding studies conducted by Anfinsen and colleagues in the early 1960s (1-6). To the greatest degree possible, the folding conditions employed by Anfinsen have been replicated here.
This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.White FH, Jr, Anfinsen CB. Some relationships of structure to function in ribonuclease. Ann N Y Acad Sci. 1959;81:515–523. doi: 10.1111/j.1749-6632.1959.tb49333.x. [DOI] [PubMed] [Google Scholar]
- 2.Anfinsen CB, Haber E. Studies on the reduction and re-formation of protein disulfide bonds. J Biol Chem. 1961;236:1361–1363. [PubMed] [Google Scholar]
- 3.Anfinsen CB, Haber E, Sela M, White FH., Jr The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc Natl Acad Sci U S A. 1961;47:1309–1314. doi: 10.1073/pnas.47.9.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White FH., Jr Regeneration of native secondary and tertiary structures by air oxidation of reduced ribonuclease. J Biol Chem. 1961;236:1353–1360. [PubMed] [Google Scholar]
- 5.Haber E, Anfinsen CB. Side-chain interactions governing the pairing of half-cystine residues in ribonuclease. J Biol Chem. 1962;237:1839–1844. [PubMed] [Google Scholar]
- 6.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 7.Turell L, Botti H, Carballal S, Ferrer-Sueta G, Souza JM, Duran R, Freeman BA, Radi R, Alvarez B. Reactivity of sulfenic acid in human serum albumin. Biochemistry. 2008;47:358–367. doi: 10.1021/bi701520y. [DOI] [PubMed] [Google Scholar]
- 8.Carballal S, Radi R, Kirk MC, Barnes S, Freeman BA, Alvarez B. Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry. 2003;42:9906–9914. doi: 10.1021/bi027434m. [DOI] [PubMed] [Google Scholar]
- 9.Carballal S, Alvarez B, Turell L, Botti H, Freeman BA, Radi R. Sulfenic acid in human serum albumin. Amino Acids. 2006 doi: 10.1007/s00726-006-0430-y. [DOI] [PubMed] [Google Scholar]
- 10.Allison WS. Formation and reactions of sulfenic acids in proteins. Accts of Chem Res. 1976;9:293–299. [Google Scholar]
- 11.Allison WS, Connors MJ. The activation and inactivation of the acyl phosphatase activity of glyceraldehyde-3-phosphate dehydrogenase. Arch Biochem Biophys. 1970;136:383–391. doi: 10.1016/0003-9861(70)90209-2. [DOI] [PubMed] [Google Scholar]
- 12.You KS, Benitez LV, McConachie WA, Allison WS. The conversion of glyceraldehyde-3-phosphate dehydrogenase to an acylphosphatase by trinitroglycerin and inactivation of this activity by azide and ascorbate. Biochim Biophys Acta. 1975;384:317–330. doi: 10.1016/0005-2744(75)90033-9. [DOI] [PubMed] [Google Scholar]
- 13.Lin WS, Armstrong DA, Gaucher GM. Formation and repair of papain sulfenic acid. Canadian journal of biochemistry. 1975;53:298–307. doi: 10.1139/o75-042. [DOI] [PubMed] [Google Scholar]
- 14.Claiborne A, Miller H, Parsonage D, Ross RP. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. Faseb J. 1993;7:1483–1490. doi: 10.1096/fasebj.7.15.8262333. [DOI] [PubMed] [Google Scholar]
- 15.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, 3rd, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 16.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 17.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, Wouters EF, Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Georgiou G. How to flip the (redox) switch. Cell. 2002;111:607–610. doi: 10.1016/s0092-8674(02)01165-0. [DOI] [PubMed] [Google Scholar]
- 19.Fuangthong M, Helmann JD. The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc Natl Acad Sci U S A. 2002;99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helmann JD. OxyR: a molecular code for redox sensing? Sci STKE. 2002;2002:PE46. doi: 10.1126/stke.2002.157.pe46. [DOI] [PubMed] [Google Scholar]
- 21.Poole LB, Nelson KJ. Discovering mechanisms of signaling-mediated cysteine oxidation. Current opinion in chemical biology. 2008;12:18–24. doi: 10.1016/j.cbpa.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeh JI, Claiborne A, Hol WG. Structure of the native cysteine-sulfenic acid redox center of enterococcal NADH peroxidase refined at 2.8 A resolution. Biochemistry. 1996;35:9951–9957. doi: 10.1021/bi961037s. [DOI] [PubMed] [Google Scholar]
- 23.Choi HJ, Kang SW, Yang CH, Rhee SG, Ryu SE. Crystal structure of a novel human peroxidase enzyme at 2.0 A resolution. Nat Struct Biol. 1998;5:400–406. doi: 10.1038/nsb0598-400. [DOI] [PubMed] [Google Scholar]
- 24.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 25.Seth D, Rudolph J. Redox regulation of MAP kinase phosphatase 3. Biochemistry. 2006;45:8476–8487. doi: 10.1021/bi060157p. [DOI] [PubMed] [Google Scholar]
- 26.Becker K, Savvides SN, Keese M, Schirmer RH, Karplus PA. Enzyme inactivation through sulfhydryl oxidation by physiologic NO-carriers. Nat Struct Biol. 1998;5:267–271. doi: 10.1038/nsb0498-267. [DOI] [PubMed] [Google Scholar]
- 27.Toennies G. Oxidation of cysteine in non-aqueous media: The “sulfenic acid” as the primary oxidation product. J Biol Chem. 1937;122:27–47. [Google Scholar]
- 28.Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D. Structural, redox, and mechanistic parameters for cysteine-sulfenic acid function in catalysis and regulation. Adv Protein Chem. 2001;58:215–276. doi: 10.1016/s0065-3233(01)58006-7. [DOI] [PubMed] [Google Scholar]
- 29.Poole LB, Ellis HR. Identification of cysteine sulfenic acid in AhpC of alkyl hydroperoxide reductase. Methods Enzymol. 2002;348:122–136. doi: 10.1016/s0076-6879(02)48632-6. [DOI] [PubMed] [Google Scholar]
- 30.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 31.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 32.Benitez LV, Allison WS. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J Biol Chem. 1974;249:6234–6243. [PubMed] [Google Scholar]
- 33.Reddie KG, Carroll KS. Expanding the functional diversity of proteins through cysteine oxidation. Current opinion in chemical biology. 2008;12:746–754. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 34.Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poole LB, Zeng BB, Knaggs SA, Yakubu M, King SB. Synthesis of chemical probes to map sulfenic acid modifications on proteins. Bioconjug Chem. 2005;16:1624–1628. doi: 10.1021/bc050257s. [DOI] [PubMed] [Google Scholar]
- 36.Takanishi CL, Ma LH, Wood MJ. A genetically encoded probe for cysteine sulfenic acid protein modification in vivo. Biochemistry. 2007;46:14725–14732. doi: 10.1021/bi701625s. [DOI] [PubMed] [Google Scholar]
- 37.Charles RL, Schroder E, May G, Free P, Gaffney PR, Wait R, Begum S, Heads RJ, Eaton P. Protein sulfenation as a redox sensor: proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics. 2007;6:1473–1484. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci U S A. 2009;106:16163–16168. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS chemical biology. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 40.Conway ME, Poole LB, Hutson SM. Roles for cysteine residues in the regulatory CXXC motif of human mitochondrial branched chain aminotransferase enzyme. Biochemistry. 2004;43:7356–7364. doi: 10.1021/bi0498050. [DOI] [PubMed] [Google Scholar]
- 41.Regazzoni L, Panusa A, Yeum KJ, Carini M, Aldini G. Hemoglobin glutathionylation can occur through cysteine sulfenic acid intermediate: electrospray ionization LTQ-Orbitrap hybrid mass spectrometry studies. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3456–3461. doi: 10.1016/j.jchromb.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 42.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci U S A. 2004;101:17982–17987. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramadan D, Rancy PC, Nagarkar RP, Schneider JP, Thorpe C. Arsenic(III) species inhibit oxidative protein folding in vitro. Biochemistry. 2009;48:424–432. doi: 10.1021/bi801988x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carballal S, Alvarez B, Turell L, Botti H, Freeman BA, Radi R. Sulfenic acid in human serum albumin. Amino Acids. 2007;32:543–551. doi: 10.1007/s00726-006-0430-y. [DOI] [PubMed] [Google Scholar]
- 45.Johansson M, Lundberg M. Glutathionylation of beta-actin via a cysteinyl sulfenic acid intermediary. BMC biochemistry. 2007;8:26. doi: 10.1186/1471-2091-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagy P, Lemma K, Ashby MT. Reactive sulfur species: kinetics and mechanisms of the reaction of cysteine thiosulfinate ester with cysteine to give cysteine sulfenic acid. J Org Chem. 2007;72:8838–8846. doi: 10.1021/jo701813f. [DOI] [PubMed] [Google Scholar]
- 47.Creighton TE. Kinetics of refolding of reduced ribonuclease. J Mol Biol. 1977;113:329–341. doi: 10.1016/0022-2836(77)90145-0. [DOI] [PubMed] [Google Scholar]
- 48.Creighton TE. Intermediates in the refolding of reduced ribonuclease A. J Mol Biol. 1979;129:411–431. doi: 10.1016/0022-2836(79)90504-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.