

Abstract
AML1-ETO is the chimeric protein product of the t(8;21) in acute myeloid leukemia. The ETO portion of the fusion protein includes the Nervy Homology Region 3 (NHR3) domain, which shares homology with A-Kinase Anchoring Proteins (AKAPs) and interacts with the regulatory subunit of type II cAMP-dependent Protein Kinase (PKA (RIIα)). We determined the solution structure of a complex between the AML1-ETO NHR3 domain and PKA (RIIα). On the basis of this structure, a key residue in AML1-ETO for PKA (RIIα) association was mutated. This mutation did not disrupt AML1-ETO’s ability to enhance the clonogenic capacity of primary mouse bone marrow cells or its ability to repress proliferation or granulocyte differentiation. Introduction of the mutation into AML1-ETO had minimal impact on in vivo leukemogenesis. Therefore, the NHR3 domain/PKA(RIIα) protein interaction does not appear to significantly contribute to AML1-ETO’s ability to induce leukemia.
Keywords: AML1-ETO, leukemia, NMR, AKAP, PKA(RIIα), NHR3
Introduction
AML1-ETO is a chimeric protein commonly observed in acute myeloid leukemia (AML) of the M2 subtype 1. This aberrant protein results from the t(8;21), which fuses the N-terminus of AML1 (encoded by RUNX1) with most of ETO (Eight twenty one, encoded by RUNXT1). AML1-ETO contains five highly conserved domains: the DNA and CBFβ binding region of AML1, known as the Runt domain, and four Nervy homology domains (NHR1-4) from ETO 2; 3; 4.
Multiple studies have examined the contribution of these five conserved domains to AML1-ETO’s activities following retroviral transduction into primary bone marrow (BM) cells. These studies have assessed a variety of AML1-ETO’s activities, including its ability to inhibit proliferation in ex vivo BM cultures, to impair granulocyte differentiation, to confer enhanced serial replating, and to cause leukemia in conjunction with secondary mutations. DNA- and CBFβ–binding by the Runt domain were demonstrated to be essential for all of these aforementioned activities 5; 6; 7; 8. Deletions of NHR1 (also known as the TATA box-binding protein association factor homology domain, or eTAFH), or mutations that impaired the interaction of NHR1 with E proteins, on the other hand, did not severely affect AML1-ETO’s activities 6; 8; 9. The NHR2 domain, also known as the hydrophobic heptad repeat (HHR), is an α-helical tetramer that acts as the ETO oligomerization domain, and is crucial for all of AML1-ETO’s activities 6; 8; 10; 11. NHR3 is an amphipathic α-helical rod that interacts with the regulatory domain of type II cyclic AMP-dependent protein kinase (PKA RIIα) 4. Deletion of NHR3 had no affect on AML1-ETO’s ability to confer serial replating activity 6. NHR4, or the myeloid-Nervy-DEAF-1 (MYND) domain is a member of the RING finger structural family and binds the SMRT and N-CoR co-repressors, as well as a putative RNA/DNA binding protein called SON 12; 13; 14; 15; 16. A C-terminal truncation of AML1-ETO that removed both the NHR3 and NHR4 domains actually conferred onto AML1-ETO the ability to cause leukemia in the absence of experimentally-induced secondary mutations, indicating that one or both domains actually interfere with some of the leukemogenic functions of AML1-ETO 17; 18. Deletion of NHR4 alone, or the introduction of mutations that disrupt its three dimensional structure also augmented AML1-ETO’s leukemogenic activity in mice 16. The contribution of NHR3 alone to AML1-ETO’s leukemogenic activity has not been assessed.
The NHR3 domain of AML1-ETO shares some sequence homology with A-kinase anchoring proteins (AKAP), which act as scaffolding proteins that associate with cyclic AMP-dependent protein kinase (PKA) 4; 19; 20; 21. AKAPs are believed to facilitate the compartmentalization of PKA for phosphorylation of specific targets in intracellular signal transduction pathways as well as incorporating cAMP signaling into different pathways and signaling events by dictating the formation of multi-protein complexes 19; 20; 22. Compartmentalization of PKA determines when and where a phosphorylation event takes place, emphasizing the importance of AKAPs in signal transduction. PKA is a holoenzyme comprised of two catalytic subunits (C) and a regulatory homodimer (RIIα) 19; 20; 21; 23. Phosphorylation of target substrates is catalyzed by the C subunits, while the localization of PKA is determined through the R subunit and its interaction with certain AKAPs 21; 24; 25; 26. Previous yeast-two-hybrid studies showed that AML1-ETO specifically interacts with PKA (RIIα) through the NHR3 domain, suggesting that the ETO domain acts as a novel AKAP 4.
While the structure of PKA (RIIα) as well as several PKA (RIIα)-AKAP complexes have been reported 27; 28, neither the structure of the AML1-ETO NHR3 domain nor of its complex with PKA (RIIα) have been determined. In order to better understand what role the PKA (RIIα)/AML1-ETO interaction might play in leukemogenesis, we determined the structure of this protein-protein complex. Based on the structure, a mutation that decreased binding of NHR3 to PKA (RIIα) by approximately 50 fold was identified. This mutation did not abolish AML1-ETO’s ability to enhance the clonogenic capacity of primary mouse BM cells, nor did it ameliorate its acute block on proliferation or granulocyte differentiation. This fifty-fold reduction in NHR3:PKA (RIIα) binding was also insufficient to impair AML1-ETO’s ability to cause leukemia in cooperation with an activated tyrosine kinase (TEL-PDGFβR).
Results
Solution structure of the NHR3 domain – PKA (RIIα) complex
Structural studies of the isolated NHR3 domain were complicated by the fact that much of the domain is unstructured. Circular dichroism (CD) spectroscopy showed that the NHR3 domain contains approximately twenty-five percent α-helical secondary structure with the remainder being random coil. This was confirmed with a 15N-1H HSQC experiment that revealed a poorly dispersed spectrum (Supplementary Data Figure 1A). However, when the NHR3 domain is titrated with PKA (RIIα), the signal dispersion improved, indicating that the NHR3 domain forms a defined structure only upon complex formation with PKA (RIIα) (Supplementary Data Figure 1A). Although there was improvement observed in the spectrum upon addition of PKA (RIIα), the quality of the NHR3 domain spectrum was not sufficient for assignment. Therefore we used a combination of limited proteolysis and mass spectroscopy to identify portions of the NHR3 domain that are not structured in the NHR3 domain - PKA (RIIα) complex. On the basis of this data, we produced a truncated construct of the NHR3 domain (A585-A615). While the spectrum of the NHR3(585-615) construct was still poorly dispersed, the spectrum of this construct in complex with PKA (RIIα) was very well dispersed and easily assignable (Supplementary Data Figure 1B).
Isothermal titration calorimetry (ITC) data showed that the binding of wildtype NHR3 domain to the PKA (RIIα) dimer occurs with high affinity (Kd = 67 nM) and a stoichiometry of 1:2 (data not shown). To verify that truncation of the domain to amino acids 585-615 did not impact binding, we performed ITC experiments on the truncated NHR3(585-615) protein fused to thioredoxin (Trx) for expression, solubility and stability purposes. We found that the Trx-NHR3(585-615) protein bound to PKA (RIIα) with slightly weaker affinity (Kd = 256 nM) than the entire NHR3 domain (Figure 1A). To determine whether the Trx fusion affected the binding interaction, we fused full-length NHR3 to Trx and measured binding using ITC. This construct bound 2-fold weaker (Kd = 128 nM) than the wildtype NHR3, indicating that the Trx fusion modestly reduces the binding affinity (Figure 1B). Overall, these results indicate that all the critical binding determinants are retained in the 585-615 construct we employed for structural studies.
Figure 1. The NHR3 domain of AML1-ETO specifically binds PKA (RIIα).
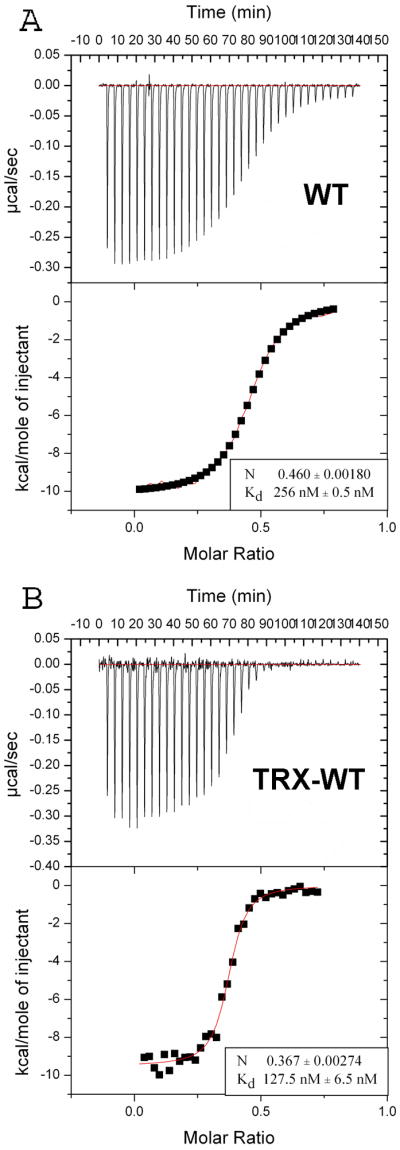
ITC measurements of the binding of Trx-NHR3 constructs to PKA (RIIα). In each panel, the top portion is the raw data and the bottom portion is a plot of the binding corrected for the dilution enthalpy (experimental data – squares; fit to a one-site binding model – line). The average N (stoichiometry) and Kd from two independent experiments (±SD) for each protein are shown in the box. (A) ITC of N-terminal Trx-NHR3 (585-615) construct. (B) ITC of the wild-type Trx-NHR3 (585-654) construct.
The solution structure of the NHR3(585-615) – PKA (RIIα) complex was calculated using NOEs, chemical shifts, and residual dipolar couplings (RDCs) without any significant constraint violations (Table 1). An ensemble of fifteen lowest-energy structures reveals a well-defined NHR3(585-615) and PKA (RIIα) (Figure 2A). An r.m.s.d. value of 0.58 Å was obtained for backbone residues 5-42 of protomers A and B and backbone residues 589-613 of NHR3 (Table 1). The NHR3(585-615) subunit forms an amphipathic α-helix when in complex with the PKA(RIIα) dimer (Figure 2B–D). NHR3(585-615) begins with an extended N-terminal region of five residues including A585-V589. The structure then continues into a turn dictated by a proline at position 590, and then into a straight helical rod spanning residues E593-Q613, with a two residue unstructured region at the C-terminus. The RIIα domain of PKA in complex with NHR3(585-615) forms an X-type four-helix bundle (Figure 2B–D) as shown previously for PKA(RIIa) alone as well as bound to Ht31 or AKAP79 27; 28. Each protomer (protomer A: 2-44; protomer B: 2-44) begins with a short extended region of 4 residues (2-5, A and B) at the N-terminus followed by a turn generated by two sequential prolines. The structure then continues into a helix-turn-helix motif where the N-terminal helix is 14 residues in length (9-22, A and B), the turn, containing two sequential prolines, is 5 residues in length, and the C-terminal helix is 15 residues in length (28-42, A and B). The helices in each monomer are aligned nearly orthogonal to one another, with an interhelical angle of ~118°. The protomers of the PKA (RIIα) dimer are oriented anti-parallel to one another. More specifically, the N-terminal helix for protomer A is aligned anti-parallel to the N-terminal helix of protomer B with an interhelical angle of ~174°. The C-terminal helices of each protomer are also aligned anti-parallel, but with an interhelical angle of 135°. This orientation creates an extensive ordering of hydrophobic residues in the core of the four-helix bundle. In addition, a solvent exposed hydrophobic groove is created on the surface of the molecule between the N-terminal helices of both protomers (Figure 2C). Our structure of PKA (RIIα) in complex with NHR3(585-615) generally agrees with previously reported structures of PKA (RIIα) complexed with other AKAPs 27.
Table 1.
NMR and refinement statistics for NHR3(a.a. 585-615)/PKA(RIIα) complex protein structure‡.
| NMR Distance and Dihedral Constraints: | ||||||
|---|---|---|---|---|---|---|
| Distance constraints: | ||||||
| NOEs (total): 3094 | ||||||
| NHR3(a.a. 585-615) | PKA(RIIα) | |||||
| Monomer I | Monomer II | |||||
| Intra-residue | 379 | 454 | 454 | |||
| Inter-residue | ||||||
| Sequential (|i − j | = 1) | 173 | 258 | 258 | |||
| Medium-range (1 < |i − j | < 5) | 84 | 140 | 140 | |||
| Intramonomer | Intermonomer | Intramonomer | Intermonomer | |||
| Long-range (|i − j | > 4) | 6 | 20 | 88 | 52 | 79 | |
| Intermolecular | 294 (NHR3 → PKA(RIIα)) | 179 (PKA(RIIα) → NHR3) | ||||
| Ambiguous | N/A | 42 | ||||
| Total dihedral angle restraints: | ||||||
| Φ (total): 83 | ||||||
| Ψ (total): 83 | ||||||
| NHR3(a.a. 585-615) | PKA(RIIα) | |||||
| Φ | 21 | 62 (31 per monomer) | ||||
| Ψ | 21 | 62 (31 per monomer) | ||||
| Residual dipolar couplings: | ||||||
| NHR3(a.a. 585-615) | PKA(RIIα) | |||||
| Monomer I | Monomer II | |||||
| 1DHN (50 + M) | 18 | 22 | 17 | |||
| 1DNC′ (50 + M) | 21 | 21 | 21 | |||
| 1DC′Ca (50 + M) | 22 | 19 | 18 | |||
| 2DHNC′ (50 + M) | 19 | N/A | ||||
| 1DHN (50 ± MA) | 18 | N/A | ||||
| 1DNC′ (50 ± MA) | 21 | N/A | ||||
| 1DC′Ca (50 ± MA) | 18 | N/A | ||||
| 2DHNC′ (50 ± MA) | 19 | N/A | ||||
| Structure statistics: | ||
|---|---|---|
| Structures: | 15 | |
| Violations (mean and s.d.): | ||
| Distance constraints (Å) | 0.0110±0.0007 | |
| Dihedral angle constraints (°) | 0.2709±0.0333 | |
| Residual dipolar couplings (Hz): | ||
| HN (50 + M) | 1.7578±0.0707 | (complex) |
| NC′ (50 + M) | 0.7893±0.0364 | (complex) |
| C′Cα (50 + M) | 0.6587±0.0436 | (complex) |
| HNC′ (50 + M) | 2.4604±0.0865 | (NHR3 only) |
| HN (50 ± MA) | 1.3015±0.1680 | (NHR3 only) |
| NC′ (50 ± MA) | 0.3731±0.0411 | (NHR3 only) |
| C′Cα (50 ± MA) | 0.3997±0.0422 | (NHR3 only) |
| HNC′ (50 ± MA) | 1.3247±0.0700 | (NHR3 only) |
| Deviations from idealized geometry: | ||
| Bond lengths (Å) | 0.0027±0.0001 | |
| Bond angles (°) | 0.4223±0.0085 | |
| Impropers (°) | 0.2764±0.0152 | |
| Ramachandran plot statistics: | ||
| Rigid Regions* (%) | Complete Structure† (%) | |
| Residues in most favored regions | 93.2 | 85.8 |
| Residues in additionally allowed regions | 5.5 | 10.6 |
| Residues in generously allowed regions | 0.5 | 2.3 |
| Residues in disallowed regions | 0.8 | 1.3 |
| Average pairwise r.m.s. deviation: | ||
| Rigid Regions* (Å) | Complete Structure† (Å) | |
| Heavy | 1.45±0.09 | 2.09±0.18 |
| Backbone | 0.58±0.09 | 1.46±0.28 |
All statistics based on complex structure
Statistics applied in structure rigid regions (a.a. 11–48, 111–148, 212–236)
Statistics applied in total stucture (a.a. 8–50, 108–150, 208–238)
Figure 2. Solution structure of the NHR3 domain-PKA(RIIα) complex.

(A) Overlay of an ensemble of the 15 lowest energy NMR solution structures. The backbone residues A585-A615 of the NHR3 domain of AML1-ETO (red) and wild-type PKA (RIIα) protomer one (blue) and protomer two (green) are displayed after superimposing the structures using residues I11-R48 and I111-A148 of PKA (RIIα) and residues V589-Q613 of NHR3.
(B) Ribbon representation of the lowest energy structure. Blue and green represent promoter one and two, respectively, of PKA (RIIα), and red is the NHR3 domain
(C) Surface representation of the PKA (RIIα) dimer with a ribbon representation of NHR3 (white: non-polar residues, red: acidic residues, blue: basic residues). The white surface represents the solvent exposed hydrophobic residues of PKA (RIIα). These residues form a pocket where the hydrophobic residues from NHR3 (green) become buried and make contacts to PKA(RIIα).
(D) Surface representation of NHR3 with a ribbon representation of PKA (RIIα) (white: non-polar residues, red: acidic residues, blue: basic residues).
Basis for PKA (RIIα) binding to the NHR3 domain
The interaction of the NHR3 domain with PKA (RIIα) is driven principally by hydrophobic interactions with no apparent salt bridge formation (Figure 2C–D). In addition, there are several polar contacts formed at the binding interface. The hydrophobic groove located on the surface of the N-terminal helices of PKA (RIIα) is formed by L9 of protomer A as well as residues T10, L13, Q14, T17, V18, L21, and R22 of each protomer (Figure 3A). There are additional contacts made through the N-terminal extended regions of PKA (RIIα). Specifically, I3 and I5 from both protomers make contacts to NHR3. I3 of protomer A makes hydrophobic contacts to I593 and K596 of NHR3, while I5 makes contacts to K596 and A600 and is highly ordered. I3 of protomer B makes contacts to M609 and L612 and I5 makes contacts with M609 and K605, but does not have the same kind of ordering as I5 in protomer A. Residues forming intermolecular interactions from the amphipathic helix of NHR3(585-615) to both N-terminal helices of PKA (RIIα) include I593, W594, A597, A600, V601, V604, A609, and L612 (Figure 2C–D). In particular, residues V601 and V604 are deeply imbedded in the hydrophobic pocket of PKA (RIIα) (Figure 3A). The methyl groups of V601 are aligned nearly parallel to the N-terminal helix of PKA protomer B, allowing them to make strong hydrophobic contacts with the methyl groups of T10 and T17, the methyl groups of L13, as well as with the aliphatic protons from the side chains in Q14, all of which reside on the N-terminal helix of protomer B. Meanwhile, V604 is aligned nearly perpendicular to the N-terminal helix of protomer A and makes similar extensive contacts to the same residues in protomer A of the dimer, suggesting that the two valines are equally important to the interaction (Figure 3A). V589 from the N-terminal extended region also makes contacts with the N-terminus of one of the two PKA subunits. In addition, the charged and hydrophilic residues K596, K605, and Q613 are also in proximity to PKA (RIIα). While Q613 only make contacts through the α and β positions, hydrogen bonds form between the side-chains of K596 and I3 (not shown) of protomer A and between the side-chains of K605 and T10 of protomer B. A hydrogen bond is also formed between the side-chain of residue R22 of protomer B and the backbone oxygens of residues G587 and Y588 (not shown), which are located in the extended region of NHR3, explaining the significance of this region in helping to stabilize the interaction. Hydrogen bonds are also observed between the side-chains of E598 and Q14 of protomer B, the side-chains of Q607 and Q14 of protomer A, as well as between the backbone oxygen of A597 and the side-chain of T17 of protomer B (Figure 3B). The remaining residues form the hydrophilic side of the amphipathic helix and make no contacts with the PKA (RIIα) dimer. A total of ~1059 Å2 of surface area on PKA (RIIα) as well as NHR3(585-615) is buried upon binding of the NHR3 domain. This is comparable to the interaction between Ht31/AKAP79 and PKA(RIIα) where 800 Å2 of surface area on PKA (RIIα) is buried, as well as the interaction between D-AKAP2 and PKA(RIα) where ~710 Å2 of the hydrophobic groove located on PKA(RIα) is buried 27; 29. While a smaller area of the PKA(RIα) dimer is buried by the D-AKAP2, which has a similarly sized helix as NHR3, the decrease in binding surface may be explained by the fact that NHR3 makes contacts to PKA(RIIα) not only through the helix, but also through residues located in the N-terminal extended region. The D-AKAP2 interaction with PKA(RIα) occurs at a comparable binding affinity (Kd=48 nM) to that of the interaction between NHR3 and PKA(RIIα), which is consistent with the similarity in binding surface30. However, D-AKAP2 binds PKA(RIIα) with a Kd of 2 nM, ~30-fold higher than NHR3, suggesting that specific residues involved in the AKAP sequences are important to PKA isoform specificity30. The flexible unstructured N-terminal regions in RIIα allow for higher affinity binding as well as the ability to bind multiple AKAP sequences31. In contrast, the rigid N-terminal helices found on RIα allow high affinity binding of only certain AKAP sequences and at lower affinity compared to RIIα31.
Figure 3. Residues important for NHR3/PKA (RIIα) binding.

(A) Surface representation of the PKA (RIIα) dimer (blue: protomer one, green: protomer two) with a line representation of NHR3 (red). Residues that form hydrophobic pockets in PKA (RIIα) are shown in dark blue and dark green line representations. Residues V601 and V604 of NHR3 are shown in orange and yellow, respectively.
(B) Line representation of PKA (RIIα) dimer (blue: protomer one, green: protomer two) with a line representation of NHR3 (red). Hydrogen bonds formed between residues of PKA (RIIα) and NHR3 are shown as yellow dotted lines.
(C) Surface representation of the PKA (RIIα) dimer (blue: protomer one, green: protomer two) with a ribbon representation of NHR3 (red). Residues in NHR3 selected for alanine mutations are highlighted.
(D) Alignment of AKAP homologues including the NHR3 domain of ETO and other ETO homologs. Residues important to the PKA (RIIα) interaction shown in boxes.
Mutation in the NHR3 domain that attenuates PKA(RIIα) binding
Based on the complex structure, we generated mutations in NHR3(585-615) to disrupt its interaction with PKA (RIIα), with the ultimate goal of determining the importance of PKA (RIIα) association for AML1-ETO function. Residues in NHR3 that were shown to be in intimate contact with PKA (RIIα) were selected as possible energetic hot spots important to the protein-protein interaction (Figure 3C). We observed that W594 (which is conserved with the other mammalian ETO homologues MTG16 and MTGR1, but not with Drosophila nervy) interacts with both V18 and Q14 in the N-terminal helix of protomer B of PKA (RIIα), and with V589 in the N-terminal extended region of NHR3. Mutation of W594 to an alanine could potentially disrupt both binding to PKA (RIIα) as well as destabilize the end of the NHR3 helix. As stated above, the highly conserved residues V601 and V604 in NHR3 make multiple contacts deep inside the hydrophobic pocket of PKA (RIIα). V601 and V604 are positioned on either side of the NHR3 helix, and contact identical residues from each N-terminal helix of PKA (RIIα) making them strong candidates for point mutations that might attenuate binding (Figure 3A). The equivalents of both V601P and V601A mutations in the ETO homologue MTG16 were previously shown to disrupt PKA (RIIα) binding (Figure 3D) 32. We individually substituted all three of these amino acids (W594, V601, V604) with alanine (Figure 3C), and performed CD spectroscopy to examine the NHR3 domain’s secondary structure (Supplementary Figure 2). The binding affinities of the mutant NHR3 domains to PKA (RIIα) were measured by ITC and compared to that of the wild type NHR3 domain (Figure 1A and 4A–C). The V604A mutant is the only one that showed a significant change in the secondary structure, consisting of an increase in helical content (Supplementary Figure 2). However, this mutation did little to disrupt the binding affinity for PKA (RIIα) (Kd = 383 nM) (Figure 4B). On the other hand, CD spectra of the W594A and V601A mutants showed little change compared with the wild type protein, indicating that these mutations cause no significant change in secondary structure (Supplementary Figure 2). The binding of the W594A mutant to PKA (RIIα) was impaired only modestly, by approximately 4-fold (Kd = 909 nM) (Figure 4A). Binding of the V601A mutant, on the other hand, was impaired by approximately 55-fold (Kd = 14μM), making this a useful mutation for functional studies (Figure 4C).
Figure 4. NHR3 mutations impair PKA (RIIα) binding.

(A) ITC for binding of W594A Trx-NHR3 (585-615) to PKA(RIIα). Data numbers presented are averages with standard deviations from two independent runs. Eight μL injections were made at 25°C. Kd and stoichiometry (N) are indicated in the box.
(B) ITC for binding of V604A Trx-NHR3 (585-615) to PKA(RIIα), performed as described in B.
(C) ITC for binding of V601A Trx-NHR3 (585-615) to PKA(RIIα), performed as described in B.
Contribution of the NHR3 domain – PKA (RIIα) interaction to AML1-ETO’s ability to inhibit granulocyte differentiation
We introduced the V601A mutation into full-length AML1-ETO (Figure 5A), and assessed the protein’s ability to interact with PKA (RIIα) upon overexpression in Cos7 cells. The V601A mutation decreased the interaction of AML1-ETO with PKA (RIIα) by approximately 2 fold (Figure 5B), as measured in titration experiments (not shown). Since AML1-ETO can form mixed oligomers with wild type ETO and its family members MTG16 and MTGR1 10; 33, all of which have intact NHR3 domains, PKA (RIIα) could be bound by these mixed oligomers. We therefore performed co-immunoprecipitations with a monomeric form of AML1-ETO that has seven mutated residues in the NHR2 oligomerization domain (m7) 10. The V601A mutation in the context of monomeric AML1-ETO (m7 V601A) decreased the amount of co-immunoprecipitated protein 3–4 fold relative to the m7 mutant alone (Figure 5B). These relatively small perturbations in binding could be due to the presence of additional proteins in cellular extracts that might stabilize the interaction of PKA (RIIα) with the NHR3 domain.
Figure 5. AML1-ETO is unaffected by mutations that impair PKA (RIIα) binding.

(A) Schematic diagram of AML1-ETO and the location of mutations (asterisks) that disrupt oligomerization (m7) and the binding of PKA (RIIα) (V601A).
(B) Cos7 cells were co-transfected with AML1-ETO and its mutated derivatives along with FLAG-tagged PKA (RIIα). Top panel; cell lysates immunoprecipitated (IP) with anti-FLAG and blotted with antibody to the Runt domain (RD) in AML1-ETO. Middle panel; 10% of input lysate blotted with anti-RD to detect AML1-ETO proteins. Bottom panel; membranes from the top panel were reprobed with anti-FLAG antibodies.
(C) Effect of mutations on AML1-ETO’s (AE) repression of granulocyte differentiation of primary Lin− BM cells following seven days of culture in the presence of IL3, IL6, SCF, and G-CSF. Plotted are triplicate samples from two independent experiments (n=6). Error bars represent 95% confidence intervals. Significant differences from AE are shown (ANOVA and Dunnett’s multiple comparison test).
(D) Granulocyte differentiation as assessed by the percentage of Gr1+Mac1+ cells. Cells within the forward and side scatter gates were further gated for GFP expression, and GFP positive cells examined for Mac1 and Gr1 expression. The V601A mutation did not significantly attenuate either AE’s activity or that of the AE m7 mutant. The data are included in the summary graph in panel C.
(E) Representative flow of BrdU incorporation 48 hours after the transduction of Lin− BM cells with MigR1 expressing GFP, AE, or the AE V601A mutant. Forward/side scatter and GFP (not shown) were gated as described in D.
(F) Percentage of GFP+ cells that had incorporated BrdU following a one-hour BrdU pulse. Data are from two experiments each with triplicate samples (error bars = 95% confidence intervals, significance determined by ANOVA and Dunnett’s multiple comparison test).
(G) Serial replating of BM cells. Graphs represent the average number of colonies from each round of replating in the presence of IL3, IL6, and SCF. Day 7 represents colony numbers per 103 cells plated and days 14, 21, and 28 from 1×104 plated cells. Numbers are averaged from three experiments, each containing triplicate samples. The numbers of colonies derived from AE V601A transduced cells were significantly lower than those from AE transduced cells at d14 and d28 (P < 0.01, ANOVA and Dunnett’s multiple comparison test).
We transduced primary, lineage depleted (Lin−) mouse BM cells with retroviruses expressing green fluorescent protein (GFP) alone (MigR1), AML1-ETO, or AML1-ETO with the V601A substitution in the NHR3 domain, and assessed granulocyte differentiation. The V601A mutation did not significantly affect AML1-ETO’s ability to repress granulocyte differentiation as measured by the percentage of Gr-1+ Mac-1+ cells after seven days of in vitro culture in the presence of IL-3, IL-6, SCF, and G-CSF (Figure 5C). The m7 mutation, which creates an AML1-ETO monomer, partially ameliorated AML1-ETO’s inhibition of granulocyte differentiation, as shown previously 10. When the V601A mutation was combined with m7, it did not significantly improve granulocyte differentiation as compared to the m7 mutation alone (Figure 5C,D). We conclude that an in vitro fifty five-fold reduction of PKA (RIIα) binding to the NHR3 domain does not significantly affect AML1-ETO’s ability to repress granulocyte differentiation.
Contribution of the NHR3 domain – PKA (RIIα) interaction to AML1-ETO’s ability to inhibit short-term proliferation and long-term self-renewal
AML1-ETO represses the short-term proliferation of human and mouse primary BM cells 11; 34. We introduced MigR1 expressing GFP and wild-type AML1-ETO or the V601A mutant into Lin− BM cells and measured proliferation 48 hours later by BrdU incorporation (Figure 5E). The V601A mutation did not release the proliferation block imposed by AML1-ETO (Figure 5E,F).
AML1-ETO increases the clonogenicity of primary BM cells, and as a result, cells transduced with AML1-ETO can be serially replated for several weeks in vitro 6; 10; 11; 34. The self-renewal activity of AML1-ETO correlates with its ability to promote leukemia 7. We transduced Lin− BM cells with retroviruses expressing wild-type AML1-ETO or AML1-ETO V601A, cultured them, and replated them weekly in the presence of IL3, IL6, and SCF. The V601A mutation did not eliminate AML1-ETO’s ability to confer increased self-renewal capacity on hematopoietic progenitors ex vivo (Figure 5G). The number of colonies regenerated in rounds 2 and 4 of replating was, however, significantly lower than that of cells expressing wild-type AML1-ETO, suggesting that the interaction with PKA (RIIα) (or another protein that requires V601 to bind the NHR3 domain) may contribute to a small amount of AML1-ETO’s activity.
Effect of the V601A mutation on AML1-ETO + TEL-PDGFβR induced leukemia
Full length AML1-ETO can cooperate with activated forms of PDGFβR (TEL-PDGFβR) and FLT3 to generate AML in mice 5; 7; 35. We retrovirally transduced BM cells from 5-FU treated mice with AML1-ETO and the V601A mutant plus TEL-PDGFβR to assess the contribution of PKA (RIIα) binding to leukemogenesis (Fig 6A). The retrovirus expressing AML1-ETO produced GFP from an internal ribosome entry site, and the TEL-PDGFβR virus produced hCD4. The percentages of doubly transduced cells (GFP+hCD4+) were similar for AML1-ETO and the V601A mutant at two days post spinfection (not shown). We transplanted lethally irradiated primary recipient animals with 1×106 retrovirally-transduced, unsorted cells and monitored the animals for disease. The AML that develops in mice transplanted with BM cells expressing both AML1-ETO and TEL-PDGFβR is marked clinically by splenomegaly, hepatomegaly, thrombocytopenia, and anemia with an accumulation of myeloid blasts in the BM and an infiltration thereof in the spleen and liver 5. TEL-PDGFβR will independently generate a chronic myeloproliferative disorder (CMPD) marked by splenomegaly, hepatomegaly, and neutrophilic leukocytosis following a longer latent period 36.
Figure 6. The V601A mutation in AML1-ETO does not impair its leukemogenic activity.

(A) Schematic of transplantation protocol. BM mononuclear cells harvested from 5-fluoruracil treated C57BL/6 mice were co-infected with MigR1 expressing AE (or AE V601A) and TEL-PDGFβR (TP). IRES-mediated expression of GFP marks AE expressing cells while hCD4 marks TP expressing cells. Retrovirally transduced cells (1 × 106) were transplanted along with 2 × 105 normal BM cells into lethally irradiated mice.
(B) Kaplan-Meier survival curve of mice after transplantation with retroviruses expressing AE or AE V601A plus TP. Control mice expressing MigR1 (+GFP) and TP (+hCD4) are shown. The study endpoint was 63 days. The number of mice in each group is indicated.
(C) Whole BM of diseased mice was gated based on forward and side scatter characteristics and subsequently analyzed for expression of hCD4 (TEL-PDGFβR) and GFP (AML1-ETO). GFP+hCD4+ cells were then analyzed for the expression of myeloid and progenitor markers. Plots are from a single animal that is representative of the experimental group. The dropout antibody controls were performed using normal BM. Although there appears to be a higher percentage of B220+ cells in the AE+TP versus the AE V601A+TP samples, this difference was not significant (not shown).
(D) The percentage of GFP+ hCD4+ BM cells that were positive for both c-kit and Sca-1. Error bars indicate 95% confidence intervals. AE+TP, n=9 mice; V601A + TP, n = 8, MigR1+TP, n=7. Differences relative to AE+TP were determined using Dunnett’s test and ANOVA.
(E) Differences in the percentage of GFP+hCD4+ BM cells that were positive for both Gr-1 and Mac-1, analyzed as in D.
Mice transplanted with BM cells expressing both AML1-ETO and TEL-PDGFβR developed a rapid, completely penetrant lethal leukemia, as reported previously 5; 7 and succumbed to their disease within seven weeks (Fig 6B). Diseased mice presented with anemia, splenomegaly, and pale livers (not shown). Mice transplanted with cells expressing TEL-PDGFβR alone, on the other hand, developed a CMPD after a significantly longer latent period. GFP+hCD4+ BM cells from mice transplanted with cells expressing AML1-ETO plus TEL-PDGFβR contained significantly higher percentages of c-kit+ Sca-1+ cells than those transplanted with cells expressing TEL-PDGFβR alone (Fig 6C,D). The percentage of Gr-1+Mac-1+ cells in the GFP+hCD4+ BM population and the intensity of Gr-1 staining was correspondingly lower in the AML1-ETO plus TEL-PDGFβR group, reflective of impaired granulocyte differentiation and consistent with a diagnosis of AML (Fig 6C,E). The AML1-ETO V601 mutant (plus TEL-PDGFβR) caused AML with a similar latent period as AML1-ETO. There was a moderate decrease in the percentage of c-kit+ Sca-1+ cells, but cell surface expression of Gr-1 and Mac-1 was not significantly different (Figure 6C–E). Along with the serial replating data, these results suggest that the V601 mutation only very modestly affects AML1-ETO’s activity.
In summary, cells expressing AML1-ETO + TEL-PDGFβR gave rise to a rapid AML as reported previously 5; 7, and this was not ameliorated by the V601A mutation.
Discussion
Previous studies identified PKA(RIIα) as an AML1-ETO interacting protein that binds the NHR3 domain 4. We solved the structure of the NHR3 domain in a complex with PKA(RIIα). We show that the NHR3 domain forms an amphipathic α-helix when bound to PKA(RIIα), in agreement with other studies of AKAP binding 27. NHR3 makes extensive contacts to PKA(RIIα) through residues at positions A597, A600, V601, and V604, and makes additional contacts through K605 A608, M609, and L612. Other AKAPs contain hydrophobic residues at similar positions that are important for PKA(RIIα) interaction (Figure 3D) 27. In addition, K605 forms a hydrogen bond with residue T10 on protomer B of PKA(RIIα). We also observe interactions of NHR3 residues I593, W594, K596 and Q613 with PKA(RIIα). These residues lengthen the NHR3 helix several turns beyond the AKAP structures observed in other studies, which suggests that the AKAP constructs used in other studies were not long enough and caused the helical fraying at the C-terminus observed in those structures 27. Residues in the N-terminal extended region also make multiple contacts to PKA(RIIα) through hydrophobic interactions (V589) and the formation of hydrogen bonds (G587 and Y588 to R22 of protomer B) suggesting that this region may also contribute to binding.
While the AKAPs do not share a conserved amino acid sequence, they do have a consensus amphipathic helical structure (Figure 3D) 19; 37. This may explain why certain AKAPs can interact with the different isoforms of PKA, and also seem to bind PKAs in different structural conformations29. The amphipathic helical structure is obviously the main driver of these interactions, however the amino acid sequence, along with the strategic placement of certain branched hydrophobic residues, seems to dictate PKA specificity as well as subtle differences in the structure of the PKA/AKAP complex29. This is especially evident in the case of the D-AKAP2, a dual AKAP, which can interact with either the PKA(RIIα) or PKA(RIα) isoforms. Interestingly, D-AKAP2 interacts with both isoforms, as stated above, but there are distinct differences in the complex structures, not only due to the structural differences of the PKA isomers, but because of the change in the helical register of the AKAP29; 31. It was observed that during its interaction with PKA(RIIα) that D-AKAP2 indeed fits tightly into the hydrophobic groove, but in contrast to the complex structure of Ht31 bound to PKA(RIIα), only one of the flexible N-terminal regions of the A protomer is shown to interact with the AKAPs, while the other N-terminal region from the second protomer remains flexible, disordered, and ultimately does not participate in the interaction29; 31. This may explain how PKA(RIIα) is able to recognize different AKAP sequence motifs while maintaining high affinity interactions, which is consistent with the ubiquitous expression of PKA(RIIα) and its importance in regulation of many cellular processes during growth and development 19; 20; 27; 38.
In contrast to the D-AKAP2/PKA(RIIα) complex, we observe that the interaction between NHR3 and PKA(RIIα) resembles the interaction between PKA(RIIα) and Ht31 in that both flexible N-terminal regions of PKA(RIIα) make contact with the NHR3 helix27. As stated above, residues I3 and I5 from both N-terminal regions make contacts to the NHR3 helix. While I3 and I5 from protomer B do make contacts to the NHR3 helix and show partial ordering, the observed NOEs are relatively weak, a possible result of the protonation of the N-terminal histidines that occurs at the pH 4.0 conditions employed for solubility27. We observe extensive ordering of I5 from protomer A as well as partial ordering of I3. While our experimental conditions may contribute to the partial disordering of this flexible region, it is clear that both N-terminal regions make contacts to the NHR3 helix and contribute to binding, which is a distinction from the D-AKAP2 complex.
A main difference between our structure and the Ht31/PKA(RIIα) structure is the residues identified as essential to the binding interaction. In the previous study, alignments showed that residues at equivalent positions 604 and 605 of NHR3 were identified as essential to the AKAP interaction27. The results from our mutagenesis study clearly show that the valine at position 604 does not contribute significantly to binding PKA(RIIα). Additionally, the lysine at position 605 is not completely buried in the hydrophobic pocket. Small contacts to the hydrophobic core of PKA (RIIα) were observed through the aliphatic portion of the side chain, but the main contacts were observed between the side chain and the N-terminal region of protomer B, and through a hydrogen bond formed with T10 in protomer B of PKA(RIIα). K605 and the hydrogen bond may be important to PKA isomer specificity, as alignments with other AKAPs generally contain a hydrophobic residue at this position. Our results show that the valine at position 601 is required for high affinity binding to PKA(RIIα). This position represents a shift in the helical registry of a full turn when compared to AKAP sequence alignments that show the equivalent of position 604 and 605 as necessary to the PKA interaction. These results agree with other studies that show AKAPs have the ability to bind at different positions along the helical registry, a fact illustrated by D-AKAP2’s ability to interact with two PKA isoforms at different helical registries29.
Using the structural information, we showed that a fifty-five fold disruption of binding between the isolated NHR3 domain and PKA(RIIα) in vitro did not affect AML1-ETO’s in vivo leukemogenic activity. The V601A mutation we introduced into AML1-ETO only modestly disrupted PKA(RIIα) binding (3–4 fold) in cell extracts, potentially raising the concern that we had not sufficiently impaired binding to cause an in vivo effect. This scenario is well-illustrated by recent studies that examined the importance of CBFβ binding to AML1-ETO, in which mutations that decreased CBFβ binding by approximately 50 fold had little effect on AML1-ETO’s leukemogenic activity, whereas a >400 fold decrease in affinity severely impeded leukemogenesis 6; 7; 39. Another possible concern is that AML1-ETO can recruit PKA(RIα) through the RI Specifier Region (RISR) motif, as this was observed to be the case in the MTG16b homologue of ETO40; 41. A sequence alignment of AML1-ETO with other proteins containing the RISR motif shows that AML1-ETO does in fact contain a RISR motif in the NHR3 domain just C-terminal to the AKAP site (Figure 7). This could allow AML1-ETO to recruit a different PKA R isoform even though the V601A mutant interrupts binding to PKA(RIIα). It is also possible that the AKAP site in NHR3 could bind both isoforms of PKA R subunits, as is the case with D-AKAP229; 31. Therefore, the V601A mutation could inhibit the interaction with the RIIα isomer, while still allowing the interaction with RIα to occur. Point mutations of certain branched hydrophobic residues induced changes in binding affinity of one PKA R isomer while having little effect, or the opposite effect, on the other29; 31. This is unlikely, as alignments of the NHR3 AKAP site to D-AKAP2 do not meet the criteria for dual specificity established by Sarma et al. as two charged residues align with positions reserved for hydrophobic residues. Regardless, previous studies showed that deletion of the entire NHR3 domain did not impair the serial replating ability conferred by AML1-ETO on primary BM cells8. The ability to confer serial replating ex vivo is a useful surrogate for the in vivo self-renewal and leukemogenic activity conferred by oncogenic nuclear proteins 6; 7; 42; 43; 44; 45. Thus we can reasonably conclude, as have others8, that proteins that interact with NHR3 contribute very little to AML1-ETO’s leukemogenic activity.
Figure 7. Alignment of RISR sequences and NHR3 domain.

Amino acids 618-647, directly C-terminal to NHR3(585-615), shows homology to other RI specifier regions that have been identified. Residues in red have been shown to be functionally conserved.
Several studies showed that a truncation that removes both the NHR3 and NHR4 domains augments AML1-ETO’s leukemogenic activity, enabling it to cause disease in the absence of an experimentally induced secondary mutation 17; 18. NHR4 appears to be largely responsible for this dampening of AML1-ETO’s leukemogenic activity, as a mutation that unfolds the domain had the same effect as removing NHR3 and NHR4 16. We did not directly assess whether the V601A mutation in NHR3 augmented AML1-ETO’s leukemogenic activity in the absence of an experimentally-induced second hit, thus this remains an open question. However, given the observation that the AML1-ETO V601A mutation significantly decreased the percentage of c-kit+ cells in the BM of leukemic mice, we would predict that the interaction with PKA (RIIα) is not actively blocking AML1-ETO’s activity.
It has been proposed that ETO and the related MTG16 proteins are AKAPs based on their interaction with PKA(RIIα) 4; 32; 46. We show that the NHR3 domain indeed directly binds PKA (RIIα) with high affinity. AKAPs are located in several subcellular compartments, and participate in multiple signaling pathways 22. Although we did not demonstrate a role for the NHR3 – PKA (RIIα) interaction for AML1-ETO mediated leukemogenesis, it may be important for normal ETO function. It was recently shown, for example, that mammalian MTG16b, an ETO homologue and an AKAP found in immune cells, interacts with Plexins A1 and A3 as well as with both type I and II PKAs thus forming a signaling complex 46. This study observed that phosphorylation of Plexin A3 by type I PKA increased the interaction with MTG16b. This is significant because it suggests the possibility that the interaction of MTG16b and PKA is involved in cAMP targeting and integration of semaphorin signaling in immune cells. In addition, it was reported that the Drosophila ETO homologue nervy interacts at the plasma membrane with Plexin A, a receptor for semaphorin 47. Nervy also interacts with PKA, and both nervy and PKA antagonize repulsion mediated by the interaction between semiphorin and Plexin A. Nervy with a mutation equivalent to V601P, which would break the NHR3 α-helix and presumably disrupt the domain’s structure, could not rescue a nervy loss of function phenotype, indicating that the NHR3 domain is essential for nervy function. The equivalent of a V601A mutation in other AKAPs would represent a useful reagent to more precisely investigate the interactions of AKAPs with PKA (RIIα) and their functional significance.
Our long-term goal is to identify protein-protein interactions that are essential for AML1-ETO’s activity, and then to develop inhibitors that block those interactions 48. Of the six protein-protein (or protein-DNA) interactions thus far analyzed, only three would appear to be potential targets for small molecule or peptide inhibitors: the interaction of the Runt domain with DNA and CBFβ, and oligomerization through the NHR2 domain. Higher order protein complexes formed on AML1-ETO, or proteins that bind to non-conserved domains may also present therapeutic opportunities, but they first require identification and/or validation.
Materials and Methods
Protein Expression and Purification
Nervy Homology Region 3 (NHR3) residues A585-N654 were cloned into both pHis-parallel and pET-32a expression vectors (Novagen, Madison WI) between the BamHI and XhoI sites using standard PCR cloning methods 49. Truncated versions of NHR3 A585-A615 and K623-D648 were also cloned into the pET-32a expression vector. Point mutations were introduced into NHR3 constructs using the Quick Change Site-Directed Mutagenesis kit (Stratagene, La Jolla CA). Wild type PKA (RIIα) was cloned between the BamHI and XhoI sites of the pHIS-parallel vector. All NHR3 constructs as well as PKA (RIIα) were expressed in Rosetta (DE3) competent cells. Cells were induced at 30 °C using 0.8 mM IPTG at O.D.600 nm 0.6–0.8. NHR3 constructs and PKA (RIIα) were purified separately using the same protocols. Cells were incubated at 4°C in the presence of EDTA-free protease inhibitors (Roche, Indianapolis IN) for 30 min., lysed by three passes through a French press, and clarified by centrifugation. The supernatant was applied to a Ni-NTA column (Qiagen, Valencia CA), washed with 50 mM Tris-HCl (pH=7.2), 300 mM NaCl, 30 mM imidazole and 1 mM Pefabloc, and eluted with 50 mM Tris-HCl (pH=7.2), 300 mM NaCl, 250 mM imidazole and 1mM Pefabloc. The protein was dialyzed against 50 mM Tris-HCl (pH=7.2), 25 mM NaCl, 1mM EDTA and 1 mM DTT to remove imidazole while cleaving the 6xHis-tag or Trx fused His-tag with Ac-TEV (Invitrogen, Carlsbad CA). The cleaved protein was passed through the Ni-NTA column to remove the His-tag or Trx fused His-tag. The non-adherent fraction was further purified using Q-sepharose (GE Healthcare, Piscataway NJ). For formation of the complex, NHR3 and PKA (RIIα) were dialyzed against 25 mM KPi (pH 6.5), 150 mM KCl, and 1 mM EDTA for three hours and then mixed together in a 2:1 ratio of PKA RIIα to NHR3. The mixture was incubated at room temperature for one hour and then the pH of the solution was rapidly dropped to pH 4.0 and dialyzed against 20 mM NaPi (pH 4.0) and 1 mM EDTA. The protein complex was applied to an S-100 size exclusion chromatography column (GE Healthcare, Piscataway NJ) to remove any unbound protein.
Isothermal titration Calorimetry (ITC)
ITC experiments were carried out on a VP-ITC MicroCalorimeter system (MicroCal, Inc., Northampton MA) at 25 °C. Samples were dialyzed against 25 mM KPi (pH=6.5), 150 mM KCl, 1mM EDTA, and 1mM DTT, centrifuged to remove any particulates, and degassed. A solution of 18 μM wild-type PKA (RIIα) was titrated with 129 μM wild-type NHR3(585-654). 36 μM wild-type PKA (RIIα) was titrated with 124 μM Trx-NHR3(585-654). 27 μM of PKA (RIIα) was titrated with 100 μM Trx-NHR3(585-615), 100 μM Trx-NHR3(585-615) W594A, 100 μM Trx-NHR3(585-615) V601A, and 100 μM Trx-NHR3(585-615) V604A. Data were corrected for dilution enthalpy and then analyzed using Origin 7.0 (Origin Lab, Northampton MA).
NMR spectroscopy
All NMR experiments were performed at 30 °C on a Varian Inova 500 MHz spectrometer or a 600 MHz spectrometer (Palo Alto CA) equipped with a cryoprobe. All NMR samples were prepared in 20 mM NaPi (pH 4.0), 1 mM EDTA, and 5% (v/v) D20. U-[15N, 13C]-labeled NHR3 domain complexed with unlabeled PKA (RIIα) was prepared for conventional triple resonance experiments and peak assignments for the NHR3 domain. U-[15N, 13C]-labeled PKA (RIIα) complexed with unlabeled NHR3 domain was prepared for conventional triple resonance experiments and peak assignments for the PKA (RIIα). A series of half-filtered 13C-edited-12C-filtered/13C-filtered and 15N-edited-14N-filtered/15N-filtered NOESY experiments were performed to determine intermolecular NOEs between the two proteins. In addition, a sample consisting of 50% unlabeled PKA(RIIα) and 50% U-[15N, 13C] was complexed to unlabeled NHR3 domain to determine interdimer NOEs, as described previously for studies of PKA(RIIα) complexes with Ht31 and AKAP79 27. U-[15N, 13C]-labeled PKA (RIIα)/unlabeled NHR3 domain and U-[15N, 13C]-labeled NHR3 domain/unlabeled PKA (RIIα) were each soaked into a 6% compressed positively charged and a 4% compressed zwitterionic polyacrylamide gel for collecting residual dipolar coupling data 50. Four types of RDCs, 1DHN, 1DNC′, 1DC′Cα, and 2DHNC′ were measured using HNCO-based experiments 51.
Structure calculation
Because the symmetry between the two protomers of the PKA (RIIα) dimer is partially broken upon binding with NHR3(585-615), peak splitting in the spectra of the labeled PKA (RIIα) was observed for ~50% of the residues. Since symmetry for the other 50% of the residues remains, it was impossible to assign the two protomers of the PKA (RIIα) unambiguously. Therefore, it was necessary to ambiguously assign all of the residues in the PKA (RIIα) spectra using an assignment nomenclature that included different numbering systems for each protomer of PKA (RIIα) and for the NHR3(585-615) monomer. The assignment nomenclature was as follows: amino acids 1/101-50/150 designate protomer A of PKA (RIIα), and amino acids 101/1-150/50 designate protomer B of PKA (RIIα). Residues 1/101-7/107 and 101/1-107/7 are residual from the expression vector, therefore residues 8/108-50/150, or 108/8-150/50, correspond to residues 2-44 of PKA (RIIα). The Sparky program allowed for the ambiguous assignment of all NOEs generated by the PKA (RIIα) dimer. Therefore, any NOE generated by the PKA (RIIα) dimer was assigned as being generated by protomer A or protomer B (ex. Y22/Y122N-Q21/Q121CA-Y22/Y122HN). Assignments for the residues in the NHR3(585-615) portion of the complex were designated 201-237 to further distinguish the different subunits in this trimer. Residues 201-207 are residual from the expression vector, therefore residues 208-238 correspond to residues 585-615 of NHR3. While intramolecular NOEs of the NHR3(585-615) were all unambiguously assigned, intermolecular long range NOEs to PKA (RIIα) were all ambiguously assigned to both protomers of PKA (RIIα).
The structure of the NHR3-PKA RIIα complex was calculated using CNS, employing the simulated annealing protocol 52. NOE restraints were obtained from manual assignment and classified into 3 groups: 1.8-1.1, 1.8–2.5, and 1.8–3.7 Å. ϕ and ψ dihedral angle restraints were generated by TALOS prediction based on Cα, Cβ, C′, and N chemical shifts 53. Eight sets of RDCs (1DHN, 1DNC′, 1DC′Cα, and 2DHNC′ from each gel condition) were used in the structure calculations. Initial values of the Da and R of the alignment tensor were estimated from a histogram of RDC distribution 54. The 15 lowest energy structures out of 200 calculated structures were selected to represent the structure.
Retroviral transduction, in vitro and in vivo assays
Mutated AML1-ETO proteins were prepared using the QuickChange Site Directed Mutagenesis kit and coding sequences transferred from pBluescript into the MigR1 vector. Retroviruses were prepared and transduced into C57BL/6 mouse BM cells as described previously 7; 9; 10; 13. Granulocyte differentiation assays, serial replating assays, cell cycle kinetics, and leukemogenesis assays were performed as described previously 7; 9; 10; 13. Mice were monitored for development of leukemia by observation of physical symptoms characterized by lethargy and splenomegaly. Sick mice were euthanized and all remaining mice were sacrificed at 63 days. The diagnosis of chronic myeloproliferative disease and acute myeloid leukemia was based on criteria described previously 7. All animal work was approved by the Institute Animal Care and Use Committee at Dartmouth.
Flow Cytometry
Samples were analyzed either on a FACSCalibur or LSRII (BD Biosciences, San Jose CA). Antibodies used were: APC-Cy7 or PE conjugated anti-Mac-1 (clone M1/70, BD Pharmingen), PerCP-Cy5.5 or APC conjugated anti-Gr-1 (clone RB6-8C5, BD Pharmingen), PE-Cy7 conjugated anti B220 (clone RA3-6B2, eBioscience), APC conjugated anti CD3 (clone 145-2C11, BD Pharmingen), PE-Cy7-conjugated anti-Sca1 (clone D7, BD Pharmingen), APC-conjugated anti-CD117 (clone 2B8, eBioscience), and PE-conjugated anti-human CD4 (clone L3T4, eBioscience).
Co-Immunoprecipitations
Immunoprecipitation of FLAG-tagged PKA RIIα from Cos7 cell extracts and immunoblotting using anti-FLAG or monoclonal anti-Runt domain antibodies was performed as described previously 9.
Supplementary Material
Acknowledgments
The authors thank Gary Gilliland for the TEL-PDGFBR cDNA and Warren Pear for the MigR1 vector.
Abbreviations
- NMR
nuclear magnetic resonance
- ITC
isothermal titration calorimetry
- CD
circular dichroism
- NHR3
nervy homology region three
- AML1
acute myeloid leukemia
- ETO
eight-twenty one
- PKA
protein kinase-A
- AKAP
A-kinase anchoring proteins
- CBF
core binding factor
Footnotes
Accession numbers: BMRB ID: 16954; PDB ID: 2kyg
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci U S A. 1991;88:10431–4. doi: 10.1073/pnas.88.23.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hug BA, Lazar MA. ETO interacting proteins. Oncogene. 2004;23:4270–4. doi: 10.1038/sj.onc.1207674. [DOI] [PubMed] [Google Scholar]
- 3.Davis JN, McGhee L, Meyers S. The ETO (MTG8) gene family. Gene. 2003;303:1–10. doi: 10.1016/s0378-1119(02)01172-1. [DOI] [PubMed] [Google Scholar]
- 4.Fukuyama T, Sueoka E, Sugio Y, Otsuka T, Niho Y, Akagi K, Kozu T. MTG8 proto-oncoprotein interacts with the regulatory subunit of type II cyclic AMP-dependent protein kinase in lymphocytes. Oncogene. 2001;20:6225–32. doi: 10.1038/sj.onc.1204794. [DOI] [PubMed] [Google Scholar]
- 5.Grisolano JL, O’Neal J, Cain J, Tomasson MH. An activated receptor tyrosine kinase, TEL/PDGFbetaR, cooperates with AML1/ETO to induce acute myeloid leukemia in mice. Proc Natl Acad Sci U S A. 2003;100:9506–11. doi: 10.1073/pnas.1531730100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwok C, Zeisig BB, Qiu J, Dong S, So CW. Transforming activity of AML1-ETO is independent of CBFbeta and ETO interaction but requires formation of homo-oligomeric complexes. Proc Natl Acad Sci U S A. 2009;106:2853–8. doi: 10.1073/pnas.0810558106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roudaia L, Cheney MD, Manuylova E, Chen W, Morrow M, Park S, Lee CT, Kaur P, Williams O, Bushweller JH, Speck NA. CBFbeta is critical for AML1-ETO and TEL-AML1 activity. Blood. 2009;113:3070–9. doi: 10.1182/blood-2008-03-147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan M, Ahn EY, Hiebert SW, Zhang DE. RUNX1/AML1 DNA binding domain and ETO/MTG8 NHR2 dimerization domain are critical to AML1-ETO9a leukemogenesis. Blood. 2009 doi: 10.1182/blood-2008-04-153742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park S, Chen W, Cierpicki T, Tonelli M, Cai X, Speck NA, Bushweller JH. Structure of the AML1-ETO eTAFH domain-HEB peptide complex and its contribution to AML1-ETO activity. Blood. 2009;113:3558–67. doi: 10.1182/blood-2008-06-161307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Cheney MD, Gaudet JJ, Chruszcz M, Lukasik SM, Sugiyama D, Lary J, Cole J, Dauter Z, Minor W, Speck NA, Bushweller JH. The tetramer structure of the Nervy homology two domain, NHR2, is critical for AML1/ETO’s activity. Cancer Cell. 2006;9:249–60. doi: 10.1016/j.ccr.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Hug BA, Lee SY, Kinsler EL, Zhang J, Lazar MA. Cooperative function of Aml1-ETO corepressor recruitment domains in the expansion of primary bone marrow cells. Cancer Res. 2002;62:2906–12. [PubMed] [Google Scholar]
- 12.Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol. 1998;18:7185–91. doi: 10.1128/mcb.18.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Chen W, Gaudet J, Cheney MD, Roudaia L, Cierpicki T, Klet RC, Hartman K, Laue TM, Speck NA, Bushweller JH. Structural Basis for Recognition of SMRT/N-CoR by the MYND Domain and Its Contribution to AML1/ETO’s Activity. Cancer Cell. 2007;11:483–97. doi: 10.1016/j.ccr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR, Huynh KD, Bardwell VJ, Lavinsky RM, Rosenfeld MG, Glass C, Seto E, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol. 1998;18:7176–84. doi: 10.1128/mcb.18.12.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci U S A. 1998;95:10860–5. doi: 10.1073/pnas.95.18.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahn EY, Yan M, Malakhova OA, Lo MC, Boyapati A, Ommen HB, Hines R, Hokland P, Zhang DE. Disruption of the NHR4 domain structure in AML1-ETO abrogates SON binding and promotes leukemogenesis. Proc Natl Acad Sci U S A. 2008;105:17103–8. doi: 10.1073/pnas.0802696105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan M, Burel SA, Peterson LF, Kanbe E, Iwasaki H, Boyapati A, Hines R, Akashi K, Zhang DE. Deletion of an AML1-ETO C-terminal NcoR/SMRT-interacting region strongly induces leukemia development. Proc Natl Acad Sci U S A. 2004;101:17186–91. doi: 10.1073/pnas.0406702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, Chen IM, Chen Z, Rowley JD, Willman CL, Zhang DE. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006;12:945–9. doi: 10.1038/nm1443. [DOI] [PubMed] [Google Scholar]
- 19.Carnegie GK, Means CK, Scott JD. A-kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB Life. 2009;61:394–406. doi: 10.1002/iub.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beene DL, Scott JD. A-kinase anchoring proteins take shape. Curr Opin Cell Biol. 2007;19:192–8. doi: 10.1016/j.ceb.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 2008;40:651–62. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dessauer CW. Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–41. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 24.Rubin CS. Characterization and comparison of membrane-associated and cytosolic cAMP-dependent protein kinases. Studies on human erythrocyte protein kinases. J Biol Chem. 1979;254:12439–49. [PubMed] [Google Scholar]
- 25.Luo Z, Shafit-Zagardo B, Erlichman J. Identification of the MAP2- and P75-binding domain in the regulatory subunit (RII beta) of type II cAMP-dependent protein kinase. Cloning and expression of the cDNA for bovine brain RII beta. J Biol Chem. 1990;265:21804–10. [PubMed] [Google Scholar]
- 26.Scott JD, Stofko RE, McDonald JR, Comer JD, Vitalis EA, Mangili JA. Type II regulatory subunit dimerization determines the subcellular localization of the cAMP-dependent protein kinase. J Biol Chem. 1990;265:21561–6. [PubMed] [Google Scholar]
- 27.Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. Embo J. 2001;20:1651–62. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newlon MG, Roy M, Morikis D, Hausken ZE, Coghlan V, Scott JD, Jennings PA. The molecular basis for protein kinase A anchoring revealed by solution NMR. Nat Struct Biol. 1999;6:222–7. doi: 10.1038/6663. [DOI] [PubMed] [Google Scholar]
- 29.Sarma GN, Kinderman FS, Kim C, von Daake S, Chen L, Wang BC, Taylor SS. Structure of D-AKAP2:PKA RI complex: insights into AKAP specificity and selectivity. Structure. 18:155–66. doi: 10.1016/j.str.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burns LL, Canaves JM, Pennypacker JK, Blumenthal DK, Taylor SS. Isoform specific differences in binding of a dual-specificity A-kinase anchoring protein to type I and type II regulatory subunits of PKA. Biochemistry. 2003;42:5754–63. doi: 10.1021/bi0265729. [DOI] [PubMed] [Google Scholar]
- 31.Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schillace RV, Andrews SF, Liberty GA, Davey MP, Carr DW. Identification and characterization of myeloid translocation gene 16b as a novel a kinase anchoring protein in T lymphocytes. J Immunol. 2002;168:1590–9. doi: 10.4049/jimmunol.168.4.1590. [DOI] [PubMed] [Google Scholar]
- 33.Kitabayashi I, Ida K, Morohoshi F, Yokoyama A, Mitsuhashi N, Shimizu K, Nomura N, Hayashi Y, Ohki M. The AML1-MTG8 leukemic fusion protein forms a complex with a novel member of the MTG8(ETO/CDR) family, MTGR1. Mol Cell Biol. 1998;18:846–58. doi: 10.1128/mcb.18.2.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mulloy JC, Cammenga J, MacKenzie KL, Berguido FJ, Moore MA, Nimer SD. The AML1-ETO fusion protein promotes the expansion of human hematopoietic stem cells. Blood. 2002;99:15–23. doi: 10.1182/blood.v99.1.15. [DOI] [PubMed] [Google Scholar]
- 35.Schessl C, Rawat VP, Cusan M, Deshpande A, Kohl TM, Rosten PM, Spiekermann K, Humphries RK, Schnittger S, Kern W, Hiddemann W, Quintanilla-Martinez L, Bohlander SK, Feuring-Buske M, Buske C. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J Clin Invest. 2005;115:2159–68. doi: 10.1172/JCI24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomasson MH, Sternberg DW, Williams IR, Carroll M, Cain D, Aster JC, Ilaria RL, Jr, Van Etten RA, Gilliland DG. Fatal myeloproliferation, induced in mice by TEL/PDGFbetaR expression, depends on PDGFbetaR tyrosines 579/581. J Clin Invest. 2000;105:423–32. doi: 10.1172/JCI8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carr DW, Stofko-Hahn RE, Fraser ID, Bishop SM, Acott TS, Brennan RG, Scott JD. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem. 1991;266:14188–92. [PubMed] [Google Scholar]
- 38.Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G. PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259–69. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 39.Park S, Speck NA, Bushweller JH. The role of CBFbeta in AML1-ETO’s activity. Blood. 2009;114:2849–50. doi: 10.1182/blood-2009-07-231233. [DOI] [PubMed] [Google Scholar]
- 40.Jarnaess E, Ruppelt A, Stokka AJ, Lygren B, Scott JD, Tasken K. Dual specificity A-kinase anchoring proteins (AKAPs) contain an additional binding region that enhances targeting of protein kinase A type I. J Biol Chem. 2008;283:33708–18. doi: 10.1074/jbc.M804807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiedler SE, Schillace RV, Daniels CJ, Andrews SF, Carr DW. Myeloid translocation gene 16b is a dual A-kinase anchoring protein that interacts selectively with plexins in a phospho-regulated manner. FEBS Lett. 584:873–7. doi: 10.1016/j.febslet.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–35. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.So CW, Karsunky H, Passegue E, Cozzio A, Weissman IL, Cleary ML. MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell. 2003;3:161–71. doi: 10.1016/s1535-6108(03)00019-9. [DOI] [PubMed] [Google Scholar]
- 44.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, Akashi K, Gilliland DG. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6:587–96. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 45.Kwok C, Zeisig BB, Dong S, So CW. Forced homo-oligomerization of RARalpha leads to transformation of primary hematopoietic cells. Cancer Cell. 2006;9:95–108. doi: 10.1016/j.ccr.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 46.Fiedler SE, Schillace RV, Daniels CJ, Andrews SF, Carr DW. Myeloid translocation gene 16b is a dual A-kinase anchoring protein that interacts selectively with plexins in a phospho-regulated manner. FEBS Lett. doi: 10.1016/j.febslet.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 47.Terman JR, Kolodkin AL. Nervy links protein kinase a to plexin-mediated semaphorin repulsion. Science. 2004;303:1204–7. doi: 10.1126/science.1092121. [DOI] [PubMed] [Google Scholar]
- 48.Gorczynski MJ, Grembecka J, Zhou Y, Kong Y, Roudaia L, Douvas MG, Newman M, Bielnicka I, Baber G, Corpora T, Shi J, Sridharan M, Lilien R, Donald BR, Speck NA, Brown ML, Bushweller JH. Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins Runx1 and CBFbeta. Chem Biol. 2007;14:1186–97. doi: 10.1016/j.chembiol.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 49.Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr Purif. 1999;15:34–9. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- 50.Cierpicki T, Bushweller JH. Charged gels as orienting media for measurement of residual dipolar couplings in soluble and integral membrane proteins. J Am Chem Soc. 2004;126:16259–66. doi: 10.1021/ja046054g. [DOI] [PubMed] [Google Scholar]
- 51.Yang DW, Venters RA, Mueller GA, Choy WY, Kay LE. TROSY-based HNCO pulse sequences for the measurement of (HN)-H-1-N-15, N-15-(CO)-C-13, (HN)-H-1-(CO)-C-13, (CO)-C-13-C-13(alpha) and (HN)-H-1-C-13(alpha) dipolar couplings in N-15, C-13, H-2-labeled proteins. Journal of Biomolecular Nmr. 1999;14:333–343. [Google Scholar]
- 52.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54 ( Pt 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 53.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 54.Clore GM, Gronenborn AM, Bax A. A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J Magn Reson. 1998;133:216–21. doi: 10.1006/jmre.1998.1419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.