

Abstract
The transcription factor C/EBPα is more potent than C/EBPβ in inducing granulocitic differentiation and inhibiting BCR/ABL-expressing cells. We took a “domain swapping” approach to assess biological effects, modulation of gene expression, and binding to C/EBPα-regulated promoters by wild-type and chimeric C/EBPα/C/EBPβ proteins. Wild-type and N-C/EBPα+ C/EBPβ-DBD induced transcription of the granulocyte-colony stimulating factor receptor (G-CSFR) gene, promoted differentiation, and suppressed proliferation of K562 cells vigorously; instead, wild-type C/EBPβ and N-C/EBPβ+C/EBPα-DBD had modest effects, although they bound the G-CSFR promoter like wild-type C/EBPα and N-C/EBPα+C/EBPβ-DBD. Chimeric proteins consisting of the TAD of VP16 and the DBD of C/EBPα or C/EBPβ inhibited proliferation and induced differentiation of K562 cells as effectively as wild-type C/EBPα. Gene expression profiles induced by C/EBPα resembled those modulated by N-C/EBPα+C/EBPβ-DBD, whereas C/EBPβ induced a pattern similar to that of N-C/EBPβ+C/EBPα-DBD. C/EBPα activation induced changes in the expression of more cell cycle- and apoptosis-related genes than the other proteins and enhanced Imatinib-induced apoptosis of K562 cells. Expression of FOXO3a, a novel C/EBPα-regulated gene, was required for apoptosis but not for differentiation induction or proliferation inhibition of K562 cells.
Keywords: Apoptosis, Differentiation, Gene Expression, Leukemia, Oncogene, Transcription Factors
Introduction
C/EBPα and C/EBPβ, two transcription factors of the CAAT/enhancer-Binding protein family, regulate the proliferation and differentiation of various cell types (1). In hematopoietic cells, C/EBPα is expressed by myeloid progenitors and precursors but not by monocytes (2, 3) and induces granulopoiesis and blocks monocytic differentiation upon expression in bipotential myeloid progenitors (4); consistent with this effect, loss of C/EBPα in vivo led to mice capable of producing monocytes but not mature granulocytes (5), and conditional knock-out of C/EBPα in hematopoietic stem cells/early progenitors increased the frequency of hematopoietic stem cells and blocked the transition of common myeloid progenitors into granulocyte-monocyte precursors (6).
The requirement of C/EBPβ in hematopoietic cells is less well defined; C/EBPβ knock-out mice exhibit a lymphoproliferative disorder and defective splenic macrophage activation but no apparent changes in granulopoiesis (7, 8). Nevertheless, the knock-in of the C/EBPβ gene at the C/EBPα locus restored granulocytic differentiation of C/EBPα knock-out mice but did not rescue their metabolic disorder, suggesting that C/EBPβ has overlapping but not entirely redundant functions with C/EBPα (9).
Although C/EBPα expression is finely regulated in normal hematopoietic cells, its expression/activity is consistently altered in myeloid leukemias (10), suggesting that genetic or functional inactivation of C/EBPα is an important event in leukemogenesis (10, 11). For example, transformation of myeloid precursor cells by p210BCR/ABL and chronic phase-to-blast crisis transition in chronic myelogenous leukemia (CML)8 is associated with decreased expression of C/EBPα by a mechanism that depends, in part, on inhibition of C/EBPα mRNA translation (12, 13). C/EBPα is mutated in ∼10% of acute myelogenous leukemia (AML) patients (14–16); the identified mutants have reduced DNA binding and/or transactivation activity, suggesting that they suppress differentiation of blast cells through a dominant-negative effect or that reduced levels of functional protein are insufficient to promote blast cell differentiation. Genetic evidence indicates that expression of p30 C/EBPα (an isoform lacking the N-terminal transactivation domain) or of a DNA binding-deficient C terminus mutant in the absence of wild-type C/EBPα promotes the development of acute myeloid leukemia in mice (17, 18). In AML patients with the t(8;21) translocation, the AML-1-ETO fusion protein appears to block differentiation by physically interacting with C/EBPα and suppressing its activity and by transcriptional repression of C/EBPα expression (19, 20). C/EBPα expression is also down-regulated via enhanced degradation in AML-overexpressing Trib2 (21) and via translation inhibition in AML with the inv16 fusion protein and the AML1-MDS1-EVI1 fusion gene (22, 23). In AML with mutant FLT3, C/EBPα expression is in some cases down-modulated, but its function may also be inhibited by mitogen-activated protein kinase-dependent phosphorylation in the N terminus (24, 25). Treatment of FLT3-ITD myeloid leukemia lines with a mitogen-activated protein kinase inhibitor induced partial differentiation and was associated with C/EBPα activation (25), and ectopic expression of C/EBPα in the t(8;21)-positive Kasumi cell line and in BCR/ABL-expressing cells induced granulocytic differentiation and suppressed proliferation in vitro and in leukemic mice (19, 26, 27), emphasizing the therapeutic potential of restoring expression of functional C/EBPα in leukemic cells.
Unlike C/EBPα, much less is known about potential mechanisms of structural or functional inactivation of C/EBPβ in AML and CML-BC cells. We found that expression of C/EBPβ was induced in Imatinib (IM)-treated BCR/ABL-expressing cells and that levels of full-length C/EBPβ were lower in CML-blast crisis than in CML-chronic phase or healthy individuals CD34+ progenitors (28). The effect of IM in myeloid precursor 32D-BCR/ABL-expressing cells is reminiscent of all-trans-retinoic acid in acute promyelocitic leukemia cells. In acute promyelocitic leukemia cells, C/EBPβ expression was induced by all-trans-retinoic acid treatment and was required for all-trans-retinoic acid-dependent differentiation (29).
Consistent with these findings, ectopic expression of C/EBPβ inhibited proliferation, induced differentiation, and suppressed leukemogenesis of myeloid precursor 32D-BCR/ABL cells (28). However, C/EBPβ was considerably less potent than C/EBPα (27), perhaps because C/EBPα exerts its biological functions not only via DNA binding and transcription activation of C/EBP-regulated genes but also via protein-protein interactions involving cell cycle regulators and chromatin remodeling proteins (30–33). In addition, the less potent effects of C/EBPβ may also reflect the subset of genes transcriptionally regulated by C/EBPα, C/EBPβ, or both. C/EBPα and C/EBPβ have a global homology at the protein level of 34%, but the homology in their TAD at the N terminus is only 24%, whereas it increases to 71% in the C terminus, where the DBD, the leucine-zipper domain, and the basic region reside (34).
To obtain additional insights on the mechanisms underlying the different potency of C/EBPα and C/EBPβ in p210BCR/ABL expressing cells, we assessed the biological effects of 4-hydroxytamoxifen (4-HT)-regulated proteins consisting of C/EBPα-C/EBPβ hybrids or of VP16-C/EBPα or C/EBPβ DBD chimera in K562 cells. We report here that the potent effect of C/EBPα in inhibiting proliferation and inducing differentiation of BCR/ABL-expressing cells depends on the strong activity of its TAD more than on different specificities of the DBDs; in addition to a subset of genes whose expression is modulated by C/EBPα, C/EBPβ, and the chimeric proteins, expression of most C/EBP-regulated genes is influenced by their respective TAD or appears to be C/EBPα- or C/EBPβ-specific. In particular, C/EBPα modulates the expression of a subset of apoptosis and cell cycle-regulatory genes that includes FOXO3a; expression of FOXO3a is essential for the apoptosis-enhancing effect of C/EBPα but not for its roles in cell proliferation and differentiation.
EXPERIMENTAL PROCEDURES
Plasmids
C/EBPα-ER and C/EBPβ-ER plasmids were previously described (26, 27). N-C/EBPα+C/EBPβ-DBD-ER and N-C/EBPβ+C/EBPα-DBD-ER plasmids were generated as follows; a NotI site was inserted in C/EBPβ-ER plasmid with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) at nucleotide 623 upstream of the DBD; a NotI site was eliminated in C/EBPα-ER plasmid at nucleotide 460, and a NotI site was inserted at nucleotide 810 upstream of the DBD. Each plasmid (sequenced to verify presence of mutations) was digested with NotI and BamHI to release the C/EBPα or C/EBPβ DBDs, leaving the MigRI plasmids with the N terminus of C/EBPα or C/EBPβ and the ER cassette. The C/EBPα DBD was directionally cloned into the NotI/BamHI-digested N terminus-C/EBPβ-ER-MigRI to obtain N-C/EBPβ+C/EBPα-DBD-ER plasmid, whereas the C/EBPβ DBD was directionally cloned into the NotI/BamHI-digested N terminus-C/EBPα-ER-MigRI to obtain N-C/EBPα+C/EBPβ-DBD-ER plasmid.
VP16+C/EBPα-DBD-ER, VP16+C/EBPβ-DBD-ER, and VP16+K298E-DBD-ER plasmids were generated as follows; the VP16 fragment was PCR-amplified from a pVP16-Nedd4 plasmid with 5′ and 3′ primers containing flapping XhoI and NotI sites, respectively; C/EBPα-DBD-ER, C/EBPβ-DBD-ER and K298E-DBD-ER fragments were generated by PCR from N-C/EBPβ+C/EBPα-DBD-ER, N-C/EBPα+C/EBPβ-DBD-ER, and K298E-ER (27) plasmids, respectively, with 5′ and 3′ primers containing flapping NotI and EcoRI sites, respectively; the VP16 and C/EBPα-DBD-ER, C/EBPβ-DBD-ER or K298E-DBD-ER products were directionally cloned into the XhoI/EcoRI-digested MigRI vector. FOXO3a A-B-C pshRNA and scramble vectors were previously described (35).
Cell Culture, Retroviral Infection, and shRNA Transfection
K562 and derivative cell lines were cultured in Iscove's modified Dulbecco's medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 0.1 mg/ml streptomycin, 2 mm l-glutamine. 293T and Phoenix cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 100 units/ml penicillin, 0.1 mg/ml streptomycin, 2 mm l-glutamine.
For retroviral infections, amphotropic Phoenix cells were transiently transfected with the indicated plasmids. The infectious supernatant was collected 48 h later and used to infect (a 48-h procedure) K562 cells. Twenty-four hours later, cells were sorted (EPICS Profile Analyzer; Coulter, Hialeah, FL) for green florescent protein (GFP) expression.
For shRNAs transfection, K562 cells (100 μl at a density of 106/ml) were resuspended in nucleofector V (Amaxa, Gaithersburg, MD) solution and mixed with 5 μg of shRNA. The solution was added to Amaxa electrode cuvettes and electroporated in Amaxa Nucleofector II using program T-16. Then cells were diluted in 2 ml of Iscove's modified Dulbecco's medium and 24 h later seeded at 104/ml and treated with 0.2 μm IM. 12 h later, 250 nm 4-HT was added, and samples were collected after 24, 48, and 72 h for cell cycle and apoptosis analyses.
Cell Proliferation, Cell Cycle Analysis, Apoptosis, and Differentiation Assays
For proliferation and cell cycle analyses, K562 cells were washed with phosphate-buffered saline (PBS) and treated with 4-HT (250 nm; Sigma). Viable cells were counted by trypan blue exclusion.
For colony formation assays, cells were pretreated (1 h) with 250 nm 4-HT and plated in methylcellulose (5 × 102/plate) in the presence of 250 nm 4-HT. Colonies were counted 6 days later. Cell cycle distribution was determined by DNA content analysis of propidium iodide-stained nuclei as described (27).
Differentiation was monitored by May-Grunwald/Giemsa staining and by detection of CD11b and CD15 differentiation markers, as described (27). Images were visualized using an Olympus CK2 microscope with a 40×/0.65 numeric aperture objective and were photographed using an Olympus SC35 type 12 camera (Olympus, Melville, NY). JPEG images were viewed using Adobe Photoshop (Adobe Systems, San Jose, CA), and contrast adjustments were made. For induction of apoptosis, K562 cells were seeded at 104/ml and pretreated with 0.2 μm IM for 12 h, then 250 nm 4-HT was added to IM-treated cells, and the percentage of apoptotic cells was evaluated 24, 48, and 72 h after 4-HT addition.
Reverse Transcription-PCR Analysis and Real-time Quantitative PCR
For RT-PCR analysis, RNA of untreated or 4-HT-treated cells was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA) according to manufacturer's protocol. Extracted RNA was digested with DNase-RNase free (Roche Applied Science) for 1 h at 37 °C and deactivated 15 min at 65 °C. RT-PCR was performed with 200 ng of RNA using the ONE STEP RT-PCR kit (Qiagen) and oligodeoxynucleotides specific for granulocyte colony-stimulating factor (G-CSF) receptor and for FOXO3a cDNA (supplemental Table 1). cDNA samples were adjusted to yield relatively equal amplification of HPRT transcripts.
For real time quantitative-PCR, total RNA was isolated using the RNeasy Mini kit (Qiagen). Extracted RNA was digested with DNase-RNase free (Roche Applied Science) for 1 h at 37 °C and deactivated for 15 min at 65 °C. 2 μg of total mRNA were reverse-transcribed, and the resulting first-strand cDNA was used as the PCR template. All reactions were done in triplicate. Primer pairs for all analyzed genes (supplemental Table 1) were designed using the ABI Primer Express software. Real time quantitative-PCR was performed using iQ SYBR Green supermix (Bio-Rad) on a MyIQ thermocycler (Bio-Rad) and quantified using MyIQ software (Bio-Rad). HPRT, a housekeeping gene with constant expression, was used as an internal control to normalize input cDNA. For relative comparison of each gene, we used the MyIQ software that analyzes the Ct value of real-time PCR data with ΔΔCt method.
Luciferase Assay
293T cells were transiently transfected using ProFection Mammalian Transfection System-Calcium Phosphate (Promega, Madison, WI) with 3 μg of reporter plasmid pTK-G-CSFR-luciferase (which contains four C/EBPα binding sites from the G-CSF receptor promoter), 3 μg of the indicated C/EBP expression plasmid, and 1/50 Renilla luciferase plasmid to account for variation in transfection efficiencies. 24 h after co-transfection, 293T cells were treated with 250 nm 4-HT for 12 h. Firefly and renilla luciferase activity was recorded on a luminometer using the Dual-Luciferase Reporter Assay System (Promega). Results are expressed as -fold activation relative to empty vector after correction for Renilla luciferase activity and are representative of three independent experiments (performed in triplicate).
Microarray Analysis
RNA was isolated from 4-HT- or vehicle-treated C/EBP-ER K562 cells and purified with RNeasy Column (Qiagen). 5 μg of total RNA were used to generate biotin-labeled cRNA using an oligo T7 primer in a reverse transcription reaction followed by in vitro transcription reaction with biotin labeled UTP and CTP. Ten micrograms of cRNA were fragmented and hybridized to HGU133 2.0 Plus arrays (Affymetrix, Santa Clara, CA) representing nearly 50,000 RNA transcripts and variants. Hybridized arrays were stained according to the manufacturer's protocols on a Fluidics Station 450 and scanned on an Affymetrix scanner 3000. All array images were visually inspected for defects and quality. Signal values were determined using the Gene Chip Operating System 1.0 (GCOS, Affymetrix). For each array, all probe sets were normalized to a mean signal intensity value of 100. A qualifier was considered detectable if the mean expression was greater than 50 signal units and the percentage of samples with a Present call, as determined by GCOS default settings, was greater than or equal to 25%. A qualifier was considered to be regulated if the difference between 4-HT- and vehicle-treated samples met the following criteria; 1) the qualifier was detected in at least 25% of the samples in either the vehicle or 4-HT-treated samples, 2) the -fold change was at least 2, and 3) the p value based on an t test was ≤0.05. For identification of the gene associated with specific biological processes, we used EPLtool, an automatic gene clustering tool developed by Wyeth Bioinformatics for Affymetrix qualifiers.
Chromatin Immunoprecipitation
ChIP assays were performed using the EZ-ChIP assay kit (Upstate Biotechnology, Inc.). Briefly, 3 × 107 exponentially growing K562 cells (untreated and 4-HT-treated, 24 h) were cross-linked with 1% formaldehyde, incubated for 15 min, and treated with glycine at a final concentration of 125 mm for 5 min at room temperature. Cells were then washed with ice-cold PBS and resuspended in 1 ml of lysis buffer with a protease inhibitor containing mixture and sonicated at 24% power for 12 pulses of 10 s each in a Branson Sonifer 450 (Branson Ultrasonics, Danbury, CT). Chromatin was precleared with 50 μl of protein A-agarose beads for 60 min at 4 °C with rotation, and precleared lysates were immunoprecipitated with the anti-estrogen receptor α antibody (12 μg; SRA-1010 Stressgen) at 4 °C overnight with rotation. Immunoprecipitations without antibody (no antibody control) and an anti-rabbit IgG were included with each experiment. Immune complexes were collected with 50 μl of protein A-agarose beads for 60 min at 4 °C with rotation (except for 10 μl of supernatant of the no antibody control, saved as Input) and washed with the buffer recommended in the ChIP protocol. Immune complexes were next eluted using freshly prepared elution buffer (1% SDS and 0.1 m NaHCO3). Cross-links were reversed by heating at 65 °C overnight in the presence of 0.2 m NaCl. ChIP DNA (2 μl) was next used as a template for real time quantitative-PCR using the following primers: human FOXO3a promoter primers (−655/−453), which encompass the putative C/EBP binding site TATTTCCACA at nucleotides −608 to −599 identified through both the AliBaba2 and PATCH programs; human FOXO3a promoter primers (−960/−756), which do not contain putative C/EBP binding sites as the negative control (supplemental Table 1); human G-CSFR promoter primers, which include a canonical C/EBP binding site TGTTGCAATC at nucleotide −55 as the positive control (supplemental Table 1).
Recovered DNA was analyzed by real time quantitative-PCR with the primers in supplemental Table 1 to normalize ChIP-quantitative PCR data were analyzed with the Percent Input Method. Percent input was calculated by 100 × 2(CtInput − CtEnriched).
Statistical Analyses
Data (presented as the means ± S.D. of two or three experiments) were analyzed for statistical significance by the unpaired, 2-tailed Student's t test. p values of less than 0.05 were considered statistically significant.
RESULTS
Biological Effects of Wild-type and Chimeric C/EBPα/β Proteins in K562 Cells
Activation of C/EBPα-ER in K562 cells induces granulocytic differentiation rapidly and efficiently, whereas C/EBPβ-ER had no effects (33, 36). To assess the mechanisms underlying such distinct effects, we generated retroviruses for two chimeric proteins by swapping the C terminus of C/EBPα and C/EBPβ (Fig. 1A) and stably expressed wild-type and chimeric proteins into K562 cells (Fig. 1B). After treatment with 4-HT to activate ectopic C/EBP proteins, we first assessed the effects on K562 cell proliferation by cell counts and DNA content analyses. Compared with empty vector-transduced cells, activation of C/EBPα-ER or N-C/EBPα+C/EBPβ-DBD-ER led to statistically significant inhibition of proliferation (Fig. 1C). Likewise, DNA content analysis showed a statistically significant increase of G1 phase cells and a decrease of S and G2/M phase cells only after a 72-h activation of C/EBPα-ER or N-C/EBPα+C/EBPβ-DBD-ER (Fig. 1D).
FIGURE 1.

Effects of wild-type and chimeric C/EBP proteins on proliferation of K562 cells. A, shown is a schematic diagram of C/EBP-ER chimeric proteins. B, a Western blot shows levels of C/EBP-ER proteins in K562 cells detected by anti-estrogen receptor α monoclonal antibody (Stressgen); expression of GRB2, as loading control, was detected by anti-GRB2 monoclonal antibody (610112, BD Transduction Laboratories). Cell counts (C) and cell cycle distribution (D) at 72 h of 4-HT-treated MigRI- or C/EBP-ER-transduced K562 cells are shown; values represent the mean ± S.D. of three independent experiments. * p < 0.05 relative to MigRI transduced K562 cells. E, methylcellulose colony formation of 4-HT-treated MigRI- or C/EBP-ER-transduced K562 cells is shown. Colonies were scored 6 days after seeding 5 × 102 cells/plate; values (mean ± S.D. of two independent experiments performed in duplicate) are expressed as the percentage of colonies from 4-HT-treated cells compared with those derived from the corresponding untreated cells taken as 100%. p values indicate statistical significance of the difference in colony number of untreated MigRI- or C/EBP-ER-transduced K562 cells versus 4-HT treated calculated using unpaired, two-tailed Student's t test.
We also tested the effects of the C/EBPα/β chimeric proteins on colony formation of K562 cells. Consistent with the effects on cell proliferation, activation of C/EBPα-ER or N-C/EBPα+C/EBPβ-DBD-ER markedly suppressed (∼90% inhibition compared with controls) K562 colony formation (Fig. 1E); activation of C/EBPβ-ER or N-C/EBPβ+ C/EBPα-DBD-ER also suppressed K562 colony formation, but the effect was considerably less striking (40–50% inhibition compared with controls) (Fig. 1E).
The effects on differentiation were assessed by morphology and expression of granulocytic markers. Activation of C/EBPα-ER or N-C/EBPα+C/EBPβ-DBD-ER promoted granulocytic differentiation of K562 cells, whereas activation of C/EBPβ-ER or N-C/EBPβ+C/EBPα-DBD-ER induced the early appearance of metamyelocytes, as indicated by the presence of cells with “doughnut”-shaped nuclei, but the vast majority of these cells failed to undergo terminal differentiation (Fig. 2A, Table 1).
FIGURE 2.

Effects of wild-type and chimeric C/EBP proteins on differentiation of K562 cells. A, morphology is shown. Light microscopy images of May-Grünwald-stained, untreated or 4-HT-treated K562 cells are shown; original magnification ×40. Counts of differentiated cells, summarized in Table 1, were performed in at least 10 fields. B, CD11b positivity is shown. C, CD15 positivity is shown. Values represent the mean ± S.D. of three independent experiments. * p < 0.05 relative to MigRI transduced K562 cells. GCSF-R mRNA expression was assessed by real time-Q-PCR (D) or semiquantitative RT-PCR (E) in 4-HT-treated (24 h in panel D; 24, 48, and 72 h in panel E) K562 cells retrovirally transduced with MigRI or cDNAs encoding wild-type or chimeric C/EBP proteins. HPRT expression was used as internal control in both PCR. In panel D results are reported as normalized -fold variation of the expression of G-CSFR mRNA; values are expressed as -fold variation relative to expression in the 4-HT-treated empty vector-transduced sample taken as 1. HPRT expression was used as the internal loading control. Error bars denote S.D. of normalized means of two independent experiments performed in triplicate.
TABLE 1.
Frequency of undifferentiated and differentiated myeloid cells in 4HT-treated C/EBP ER-transduced K562 cells
Numbers in parentheses are the number of cells.
| C/EBP-ER-transduced K562 cells | Percent differentiated cells |
|||
|---|---|---|---|---|
| Time 0 | 24 h | 48 h | 72 h | |
| % | ||||
| C/EBPα−ER | ||||
| Blasts | 75.7 (318) | |||
| Promyelocytes | 15.7 (66) | 13.9 (36) | ||
| Myelocytes | 8.5 (36) | 30.3 (78) | 12 (25) | |
| Metamyelocytes | 25.6 (66) | 26 (54) | 20.3 (66) | |
| Bands | 21.3 (55) | 37.5 (78) | 45.5 (148) | |
| Segments | 8.9 (23) | 24.5 (51) | 34.2 (111) | |
| Total no. of cells | 100 (420) | 100 (258) | 100 (208) | 100 (325) |
| C/EBPβ−ER | ||||
| Blasts | 73.2 (238) | 14.3 (49) | 14.2 (39) | 34.3 (141) |
| Promyelocytes | 16.6 (54) | 46.5 (159) | 30.4 (90) | 44 (181) |
| Myelocytes | 9.6 (31) | 28.4 (97) | 39 (117) | 12.9 (53) |
| Metamyelocytes | 0.6 (2) | 8.8 (30) | 14 (43) | 8.8 (36) |
| Bands | 2 (7) | 1.7 (5) | ||
| Segments | 0.7 (2) | |||
| Total no. of cells | 100 (325) | 100 (342) | 100 (296) | 100 (411) |
| N-C/EBPα+C/EBPβ-DBD-ER | ||||
| Blasts | 69.2 (275) | 0.3 (1) | ||
| Promyelocytes | 17.9 (71) | 5.3 (15) | 0.3 (1) | 0.9 (2) |
| Myelocytes | 11.9 (47) | 16.4 (47) | 5.5 (16) | 0.4 (1) |
| Metamyelocytes | 1 (4) | 30.6 (88) | 45.3 (132) | 10 (23) |
| Bands | 35.9 (103) | 31.8 (93) | 55.7 (128) | |
| Segments | 11.8 (34) | 16.8 (49) | 33 (76) | |
| Total no. of cells | 100 (397) | 100 (287) | 100 (292) | 100 (230) |
| N-C/EBPβ+C/EBPα-DBD-ER | ||||
| Blasts | 62.4 (164) | 16.8 (55) | 28.8 (79) | 38.4 (119) |
| Promyelocytes | 25.8 (68) | 32.4 (106) | 35 (96) | 46.3 (144) |
| Myelocytes | 10.7 (28) | 34.6 (113) | 26.6 (73) | 9.7 (30) |
| Metamyelocytes | 1.1 (3) | 12.5(41) | 8.4 (23) | 5.6 (17) |
| Bands | 3.7 (12) | 1.2 (3) | ||
| Segments | ||||
| Total no. of cells | 100 (263) | 100 (327) | 100 (274) | 100 (310) |
Analysis of granulocytic differentiation markers confirmed these morphological changes; compared with empty vector-transduced cells, activation of C/EBPα-ER or N-C/EBPα+C/EBPβ-DBD-ER led to a statistically significant induction of CD11b and CD15, whereas expression of these two markers did not increase in cells expressing wild-type C/EBPβ-ER or N-C/EBPβ+C/EBPα-DBD-ER (Fig. 2, B and C, respectively). Moreover, G-CSFR mRNA transcripts were readily detected by real-time or by semiquantitative RT-PCR upon activation of wild-type C/EBPα-ER or the N-C/EBPα+C/EBPβ-DBD-ER chimera (Fig. 2, D and E, respectively).
Effects of Wild-type and Chimeric C/EBPα/β Proteins on the GCSF-R Promoter
To further assess the effects of wild-type and chimeric C/EBPα and C/EBPβ proteins, we performed luciferase assays on the C/EBPα-responsive G-CSFR promoter. After co-transfection of 293T cells with the G-CSFR promoter-luciferase reporter (G-CSFR/Luc) plasmid, the Renilla luciferase control plasmid, to account for variation in transfection efficiencies and C/EBP plasmids or the MigRI empty vector, cells were treated with 4-HT for 12 h, and luciferase activity was measured thereafter. Results are expressed as -fold activation relative to vector alone after normalization for Renilla luciferase activity (Fig. 3A). C/EBPα and N-C/EBPα+C/EBPβ-DBD transactivated the G-CSFR/Luc plasmid more effectively than C/EBPβ or the N-C/EBPβ+C/EBPα-DBD chimera; however, the results of these assays do not allow us to exclude differences in DNA binding that could account for the distinct effects of the chimeric proteins. Thus, quantitative ChIP assays were performed upon activation of C/EBP-ER proteins in K562 cells. As shown in Fig. 3B, quantitative PCR showed comparable binding of C/EBPα, C/EBPβ, and the chimeric proteins to a segment of the G-CSFR promoter containing a canonical C/EBPα binding site, in sharp contrast with the inability of C/EBPβ N terminus containing proteins to activate G-CSFR expression (Fig. 2, D and E).
FIGURE 3.

Effect of wild-type and chimeric C/EBP proteins on the G-CSFR promoter. A, a histogram shows luciferase activity in 293T cells co-transfected with pTK-G-CSFR-luciferase and C/EBP plasmids or the MigRI empty vector after treatment (12 h) with 4-HT. Results (three independent experiments) are expressed as -fold activation over that in empty vector-transfected cells after normalization for Renilla luciferase activity and are reported as the means of triplicate determinations; error bars represent the S.D. of the mean; p values indicate statistical significance of the difference in transactivation activity of C/EBPα-ER versus C/EBPβ-ER or N-C/EBPβ-C/EBPα-DBD-ER calculated using unpaired, two-tailed Student's t test. B, quantitative ChIP assays show interaction of C/EBPα, C/EBPβ, and C/EBPα-C/EBPβ chimeric proteins with a segment of the G-CSFR promoter (nucleotides −147 to + 25) containing a functional C/EBPα binding site detected by real time Q-PCR. Error bars denote S.D. of the means of one representative experiment (of two) performed in triplicate. A Western blot shows total and immunoprecipitated (IP) C/EBPα, C/EBPβ, and chimeric proteins from K562 cell lysates used for ChIP assays. Chimeric proteins were immunoprecipitated and detected by Western blot with the anti-estrogen receptor α monoclonal antibody (Ab) (Stressgen).
Effects of VP16-C/EBPα/β-DBD-ER Chimeric Proteins on the GCSF-R Promoter and the Proliferation and Differentiation of K562 Cells
To further address the possibility that the more potent biologic effects of C/EBPα depend on the stronger activity of its TAD more than on different specificities of the DBD, we generated K562 cell lines expressing chimeric proteins consisting of the VP16 TAD juxtaposed to the DBD of C/EBPα or C/EBPβ fused to the tamoxifen-responsive estrogen receptor ligand binding domain (ERTAM) (Fig. 4, A and B).
FIGURE 4.

Effect of VP16-C/EBP-DBD chimeric proteins on the G-CSFR promoter activity. A, shown is a schematic representation of the VP16-C/EBP-DBD-ER chimeric proteins. B, expression of VP16-C/EBP-DBD-ER chimeric proteins in retrovirally transduced K562 cells is shown. Chimeric proteins were detected by Western blot with the anti-estrogen receptor α monoclonal antibody (Stressgen). Expression of GRB2 was detected by anti-GRB2 monoclonal antibody (610112, BD Transduction Laboratories). C, shown is luciferase activity of the GCSF-R promoter in 293T cells transfected with the VP16-C/EBP-DBD-ER expression plasmids and the GCSF-R-LUC reporter after treatment with 4-HT (12 h, 250 nm). The histogram shows -fold activation of luciferase activity over that in cells transfected with the MigRI empty vector after normalization for Renilla; error bars represent the S.D. of the mean of three independent experiments performed in triplicate. NS indicates that the difference in transactivation between N-VP16-C/EBPβ-DBD-ER and N-VP16-C/EBPα-DBD-ER is not significant; statistical significance was calculated using unpaired, two-tailed Student's t test.
The herpes simplex virus type 1 (HSV-1) tegument protein VP16 is a structural protein of HSV-1 that activates transcription of the immediate early promoters of the virus (37, 38). Although VP16 specifically activates promoters containing the so-called TAATGARAT element, specificity is conferred by the cellular DNA-binding protein(s) fused to the N-terminal domain of VP16 (39, 40).
To assess the transactivation ability of the two VP16-C/EBPs-DBD-ER chimeric proteins, we performed luciferase assays on the C/EBPα-responsive G-CSFR promoter. After co-transfection of 293T cells with the G-CSFR/Luc plasmid, the Renilla luciferase control plasmid to account for variation in transfection efficiency, VP16-C/EBPs-DBD-ER expression vectors, or the MigRI empty vector, cells were treated with 4-HT for 12 h, and luciferase activity was measured thereafter. Results are expressed as -fold activation relative to vector only-transfected cells after normalization for Renilla luciferase activity. The assays show that the two chimeric proteins are essentially undistinguishable in the ability to transactivate the C/EBPα-responsive G-CSFR promoter (Fig. 4C).
To analyze the biological effects of these chimeric proteins, differentiation and proliferation were analyzed upon 4-HT treatment of VP16-C/EBPα/β-DBD- ER retrovirally transduced K562 cells (Fig. 4B). Upon 4-HT activation, both chimeric proteins rapidly induced granulocytic differentiation, as indicated by morphology (Fig. 5A), expression of CD15 and CD11b differentiation markers (Fig. 5B), and G-CSFR mRNA levels (Fig. 5C).
FIGURE 5.

Effect of VP16-C/EBP-DBD chimeric proteins on proliferation and differentiation of K562 cells. A, morphology is shown. Light microscopy images of May-Grünwald-stained untreated or 4-HT-treated MigRI-transduced or VP16-C/EBP-DBD-expressing K562 cells are shown; original magnification −40. B, CD11b (upper panel) and CD15 (lower panel) expression was detected by flow cytometry with phycoerythrin-conjugated monoclonal antibodies (Pharmingen). Values represent the mean ± S.D. of three independent experiments. *, p < 0.03 relative to MigRI transduced K562 cells. C, GCSF-R expression was assessed by semiquantitative RT-PCR. HPRT expression was used as internal loading control. Results are representative of two independent experiments. D, cell cycle distribution (DNA content analysis of propidium iodide-stained nuclei) of 4-HT-treated (72 h) K562 cells is shown. Values represent the mean ± S.D. of three independent experiments. E, shown is a colony formation assay of 4-HT-treated MigRI- or VP16-C/EBP-DBD-ER-transduced K562 cells. Values (mean ± S.D. of two independent experiments performed in duplicate) are expressed as the percent of colonies of 4-HT-treated versus untreated cells taken as 100%. p values indicate statistical significance of the difference in colony number of untreated MigRI- or VP16-C/EBP-DBD-ER-transduced K562 cells versus 4-HT treated calculated using unpaired, two-tailed Student's t test.
We also assessed cell cycle distribution and methylcellulose colony formation of untreated and 4-HT-treated VP16-C/EBPα/β-DBD-ER/K562 cells. After a 72-h treatment with 4-HT, either chimeric protein induced a marked increase in the number of G1 cells and a decrease in the fraction of S and G2/M phase cells (Fig. 5D); likewise, both proteins markedly suppressed methylcellulose colony formation of K562 cells (Fig. 5E). Of note, the effects of the VP16-C/EBPα/β-DBD-ER proteins appear to be undistinguishable from those of wild-type C/EBPα-ER and N-C/EBPα+C/EBPβ-DBD-ER (compare Fig. 1 and Fig. 5).
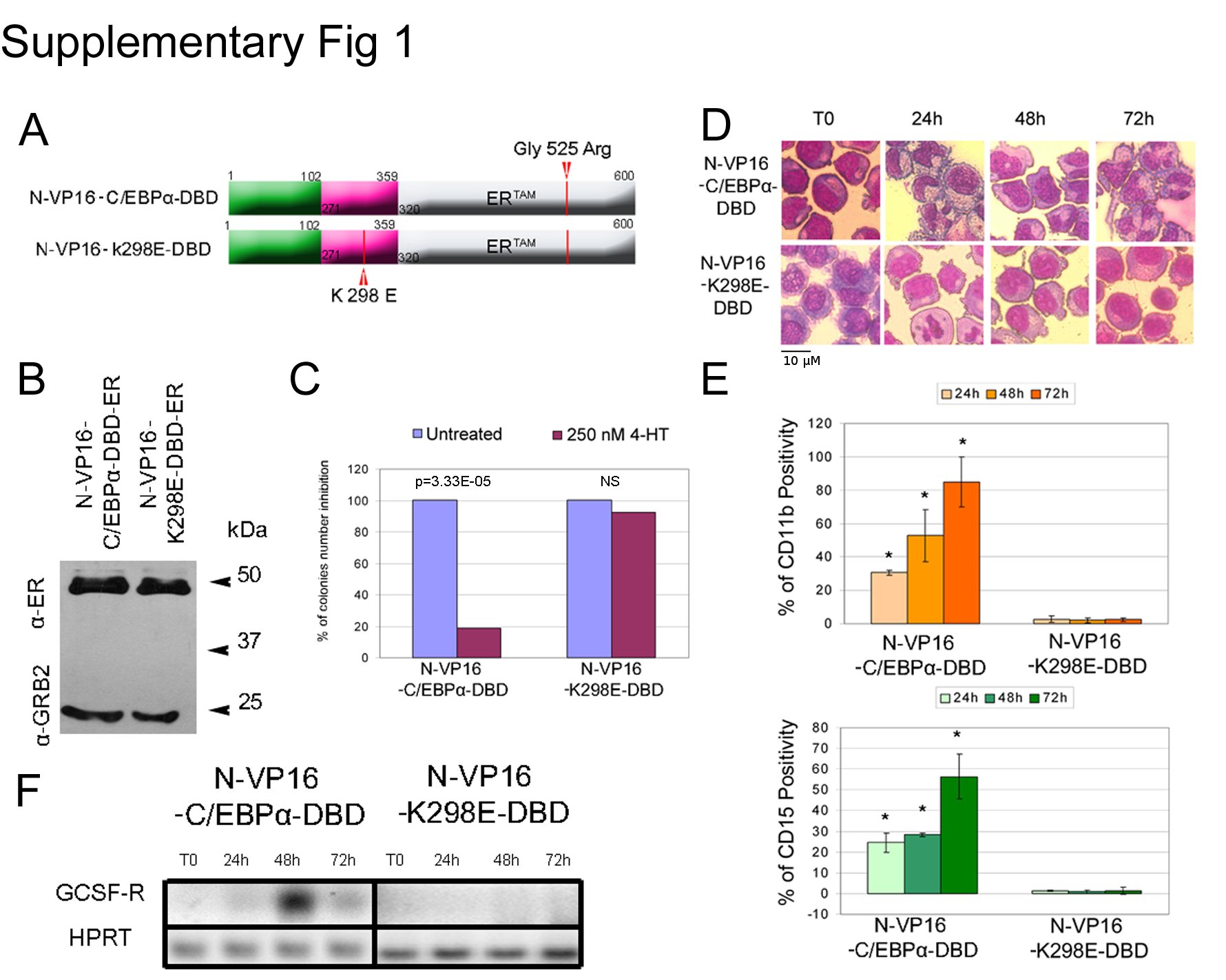
As control, we also generated a VP16-C/EBPα K298E-DBD-ER expression plasmid in which the K298E mutation prevents DNA binding (27) to assess whether the effects of the chimeric VP16-C/EBP DBD-ER proteins were DNA binding-dependent. As shown in supplemental Fig. 1, activation of this chimeric protein failed to induce granulocytic differentiation or to suppress colony formation of K562 cells. Together, these studies further confirm that the markedly different biological effects of C/EBPα and C/EBPβ in K562 cells depend on the potency of their transactivation domain.
Gene Expression Profiles Regulated by Wild-type and Chimeric C/EBPα/β Proteins
The different transactivation activities of wild-type and chimeric C/EBPα/β proteins should be reflected in the modulation of distinct patterns of gene expression; thus, we performed microarray hybridization assays using RNA of untreated and 4-HT-treated (12 h) C/EBP-ER K562 cells to assess overall patterns of gene expression and specific gene subsets regulated by wild-type and chimeric C/EBPα/β proteins.
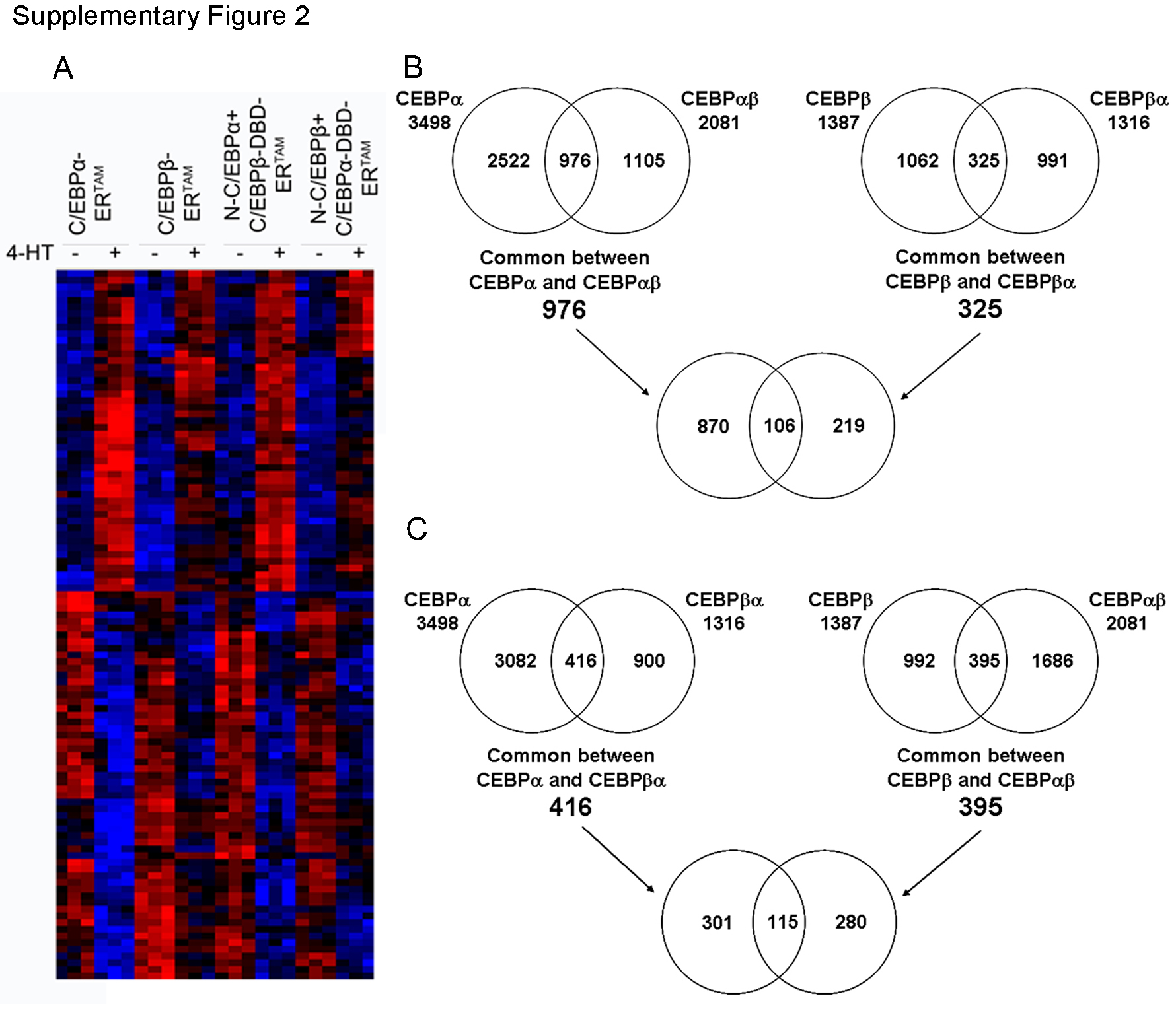
An overview of the genes differentially regulated by wild-type and chimeric C/EBPα/β proteins in K562 cells is shown in supplemental Fig. 2, panel A. Upon 4-HT treatment, C/EBPα-ER and N-C/EBPα-C/EBPβ-DBD-ER regulated the expression of a larger number of genes, 3498 (1644↓;1854↑) and 2081 (1018↓;1063↑), respectively, than C/EBPβ-ER and N-C/EBPβ-C/EBPα-DBD-ER, 1387 (744↓;643↑), and 1316 (663↓;653↑), respectively.
Analysis of the gene expression profiles modulated by the proteins that share the same TAD shows that C/EBPα and N-C/EBPα+C/EBPβ-DBD regulated 976 (413↓;563↑) common genes, whereas C/EBPβ and N-C/EBPβ+C/EBPα-DBD regulated only 325 (182↓;143↑) common genes. 106 (58↓;48↑) of these genes qualified as differentially expressed in all four cell lines (supplemental Fig. 2, panel B).
On analysis of the gene expression profiles modulated by the proteins that share the same DBD, C/EBPα, and N-C/EBPβ+C/EBPα-DBD regulated 461 (240↓;221↑) common genes, whereas C/EBPβ and N-C/EBPα+C/EBPβ-DBD regulated 395 (211↓;184↑) common genes. 115 of these genes qualified as differentially expressed in all four cell lines (supplemental Fig. 2, panel C). Although a subset of genes is regulated by both C/EBPα and C/EBPβ, it is clear that C/EBPα is a more effective gene expression modulator than C/EBPβ and that this higher activity is probably due to a more potent TAD, as the C/EBPα- and N-C/EBPα+C/EBPβ-DBD-regulated gene expression profiles are very similar.
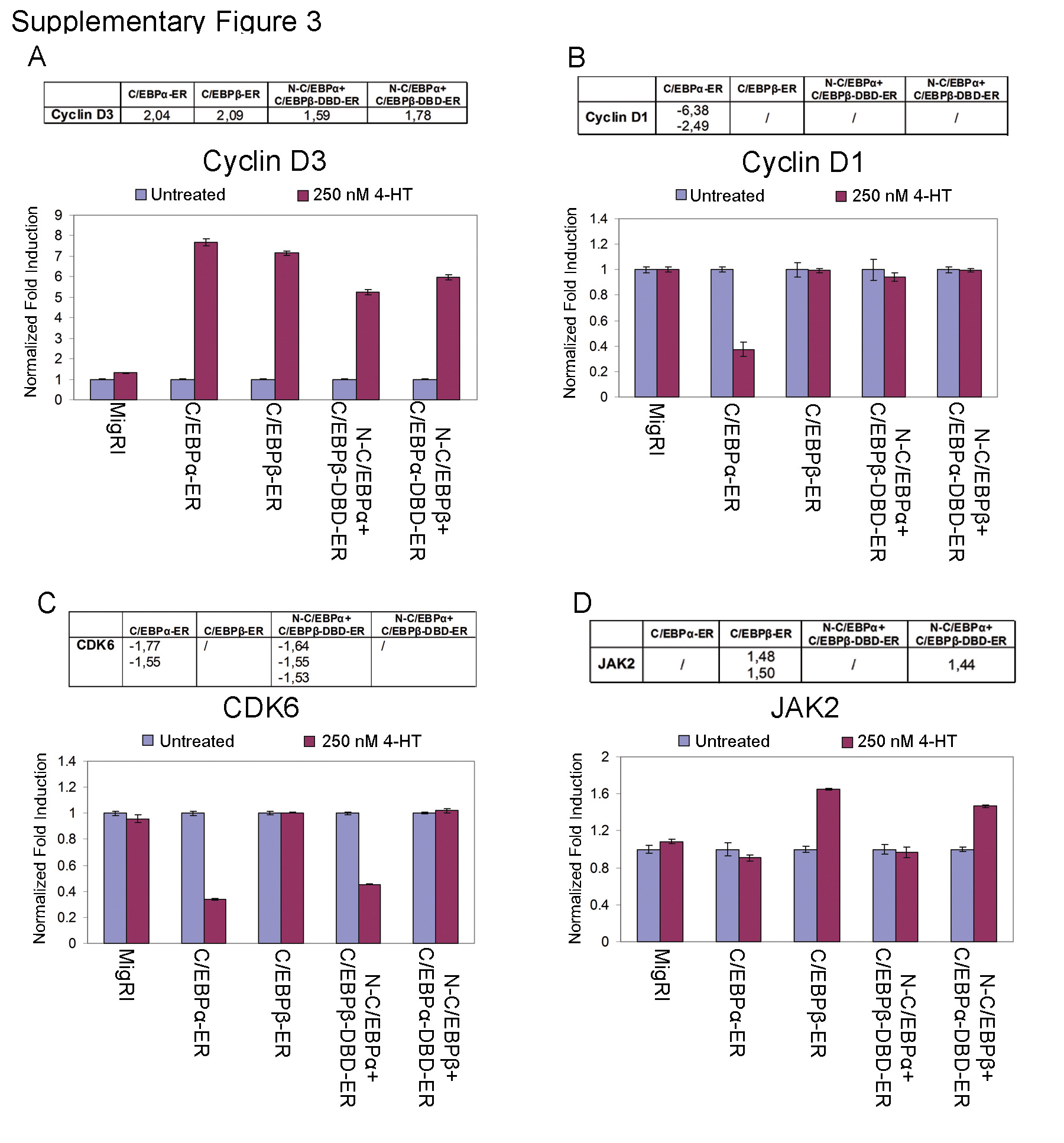
To validate the microarray hybridization data, expression of some genes differentially regulated by wild-type and chimeric C/EBPα/β proteins in K562 cells was assessed by real time quantitative PCR; as shown in supplemental Fig. 3, levels of cyclin D1, cyclin D3, CDK6, and JAK2 mRNA were differentially modulated by the wild-type and chimeric C/EBP proteins, in agreement with the results of the microarray hybridization data.
We then asked to which functional groups the C/EBP-regulated genes belonged in order to correlate gene expression profiles and biological effects induced by wild-type and chimeric C/EBPα/β proteins. Within the group of cell cycle-related genes, C/EBPα and N-C/EBPα+C/EBPβ-DBD modulated the expression of 114 and 81 cell cycle-related genes, respectively; of these, 58 were unique for C/EBPα and 35 for N-C/EBPα+C/EBPβ-DBD chimeric protein, whereas 26 were modulated by both proteins. C/EBPβ and N-C/EBPβ+C/EBPα-DBD regulated 56 and 61 cell cycle-related genes, respectively; of these, 20 were unique for C/EBPβ and 37 for the N-C/EBPβ+C/EBPα-DBD chimeric protein, whereas 5 were regulated by both proteins, suggesting that the more potent inhibitory effects of C/EBPα and the N-C/EBPα+C/EBPβ-DBD chimeric protein in proliferation and colony formation of K562 cells probably depend on a more complex pattern of cell cycle-related gene regulation.
The same analysis was performed on genes involved in apoptosis. C/EBPα regulated 80 apoptosis-related genes, 40 of which were unique, whereas C/EBPβ, N-C/EBPα+C/EBPβ-DBD, and N-C/EBPβ+C/EBPα-DBD modulated 40, 51, and 40 apoptosis-related genes, respectively; of these, 10, 17, and 18 were unique.
The Effect of C/EBPα in Apoptosis Is, in Part, Dependent on FOXO3a Expression
Although a pro-apoptotic effect was not discerned upon C/EBPα activation in BCR/ABL-expressing cells in vitro and in leukemic mice (27), the more complex subset of apoptosis-related genes regulated by C/EBPα raised the possibility that, under certain circumstances, C/EBPα activation could also inhibit cell survival, possibly enhancing its anti-leukemia effects.
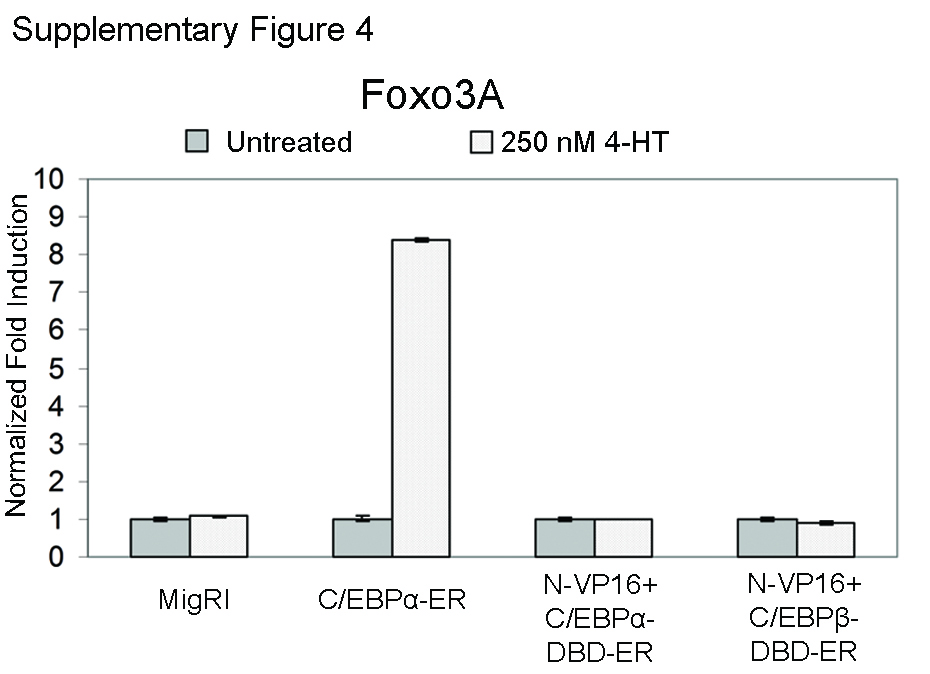
To address this possibility, K562 cells expressing C/EBPα, C/EBPβ, or the chimeric proteins were pretreated for 12 h with a suboptimal concentration of IM (0.2 μm) followed by incubation with 250 nm 4-HT, and apoptosis was assessed by propidium iodide staining to detect cells with hypodiploid DNA content. As shown in Fig. 6A, only activation of C/EBPα led to a statistically significant increase in the number of apoptotic cells. Among apoptosis-related genes modulated by C/EBPα, it seemed especially interesting to further assess the role of FOXO3a, a transcription factor of the FoxO family that appears to play an important role in cell cycle arrest and apoptosis of normal and BCR/ABL-expressing hematopoietic cells (41–43). From the microarray data, FOXO 3a was differentially regulated by C/EBPα, C/EBPβ, and N-C/EBPα+C/EBPβ-DBD, but only C/EBPα was able to enhance the expression of five different FOXO3a qualifiers, one of which showed a 2.5-fold increase. Indeed, upon a 12-h 4-HT treatment of K562 cells expressing C/EBPα, C/EBPβ, or the chimeric proteins, expression of FOXO3a mRNA was readily induced only by C/EBPα (Fig. 6B); the increased expression of FOXO3a was confirmed by anti-FOXO3a Western blotting (Fig. 6C). Of interest, both VP16-C/EBPα/β-DBD-ER chimeric proteins failed to activate FOXO3a mRNA expression (supplemental Fig. 4).
FIGURE 6.

Activation of C/EBPα enhances IM-induced apoptosis of K562 cells. A, shown are the number of apoptotic cells (mean plus S.D., three experiments) in MigRI-transduced and C/EBP-ER-expressing K562 cells after treatment with 0.2 μm IM alone or with 4-HT; * p < 0.05 relative to corresponding 4-HT-untreated sample. B, the histogram shows FOXO3a mRNA levels, assessed by real time Q-PCR, in 4-HT-treated C/EBP-ER-transduced K562 cells. HPRT expression was used as internal control. Error bars denote S.D. of the normalized means of one representative (of two) experiment performed in triplicate. C, shown is expression of FOXO3a in 4-HT-treated C/EBPα-ER K562 cells; expression of FOXO3a was detected by anti-FKHRL/FOXO3a rabbit polyclonal antibody (07-702, UBI). D, FOXO3A mRNA levels, assessed by semiquantitative RT-PCR, in C/EBPα-ER-expressing K562 cells after treatment with 4-HT alone or in the presence of cycloheximide (CHX) are shown. HPRT expression was used as internal loading control. Results are representative of two independent experiments. E, quantitative ChIP assays show binding of C/EBPα, C/EBPβ, and C/EBPα-C/EBPβ chimeric proteins to a segment (nucleotides −655 to −453) of the FOXO3a promoter containing a putative C/EBP binding site (nucleotides −608 to −599) detected by real time Q-PCR. Error bars denote S.D. of the means of one representative experiment (of two) performed in triplicate.
To assess whether FOXO3a is directly regulated by C/EBPα, FOXO3a mRNA levels were measured in K562 cells treated with 250 nm 4-HT (to activate C/EBPα) in the presence of the protein synthesis inhibitor cycloheximide. In control experiments using K562 cells transduced with the MigRI empty vector, co-treatment with 4-HT and cycloheximide had no effect on FOXO3a mRNA levels (not shown); by contrast, expression of FOXO3a mRNA was rapidly induced after C/EBPα-activation, and this early increase was not prevented by a 4-h treatment with cycloheximide (CHX, Fig. 6D), consistent with FOXO3a being a direct target of C/EBPα.
Additional proof in support of FOXO3a being a transcriptional target of C/EBPα was obtained by ChIP assays; a search for C/EBP binding sites in the 5′-flanking region of the human FOXO3a gene identified a putative binding site at nucleotides −608 to −599 (underlined in Fig. 6E). PCR amplification of a segment including this site after anti-ER ChIP from 4-HT-treated K562 cells expressing wild-type or chimeric C/EBPα-C/EBPβ proteins demonstrated that each of these proteins was able to interact with the segment (nucleotides −655 to −453) of the FOXO3a promoter (Fig. 6E), although only C/EBPα was able to activate FOXO3a mRNA expression (Fig. 6B). C/EBP proteins did not interact with a more distal segment of the FOXO3a promoter lacking putative C/EBP binding sites (not shown).
To assess if the enhanced propensity of IM-treated C/EBPα-expressing K562 cells to undergo apoptosis was associated with the ability of C/EBPα to induce FOXO3a expression, levels of FOXO3A were down-regulated by RNAi in empty vector-transduced or C/EBPα-ER-expressing K562 cells (Fig. 7A), and the frequency of apoptotic cells was assessed after treatment with a suboptimal concentration of IM (0.2 μm). Down-regulation of FOXO3a expression suppressed the increase in apoptosis induced by co-treatment of C/EBPα-ER-expressing K562 cells with 4-HT and IM (Fig. 7B). Thus, under certain conditions (i.e. IM or chemotherapy activation of apoptotic pathways), C/EBPα may enhance apoptosis through FOXO3a-dependent mechanisms.
FIGURE 7.

Suppression of FOXO3a expression blocks the apoptosis-inducing effect of C/EBPα. A, shown are FOXO3A mRNA levels, assessed by real time Q-PCR, in 4-HT-treated scramble or FOXO3A shRNA-transfected K562 cells (MigRI-transduced or C/EBPα-ER-expressing). HPRT expression was used as internal control. Error bars denote S.D. of normalized means of one representative experiment (of three) performed in triplicate. B, shown is frequency of apoptotic cells (mean plus S.D., three experiments) in scrambled or shRNAFOXO3A-transfected K562 cells (MigRI-transduced or C/EBPα-ER-expressing) after treatment with 0.2 μm IM alone or with 4-HT (250 nm) assessed by ipodiploid percentage after propidium iodide staining. * p < 0.05 relative to corresponding 4-HT-untreated sample.
FOXO3a Expression Is Not Required for C/EBPα-dependent Granulocytic Differentiation and Proliferation Inhibition in K562 Cells
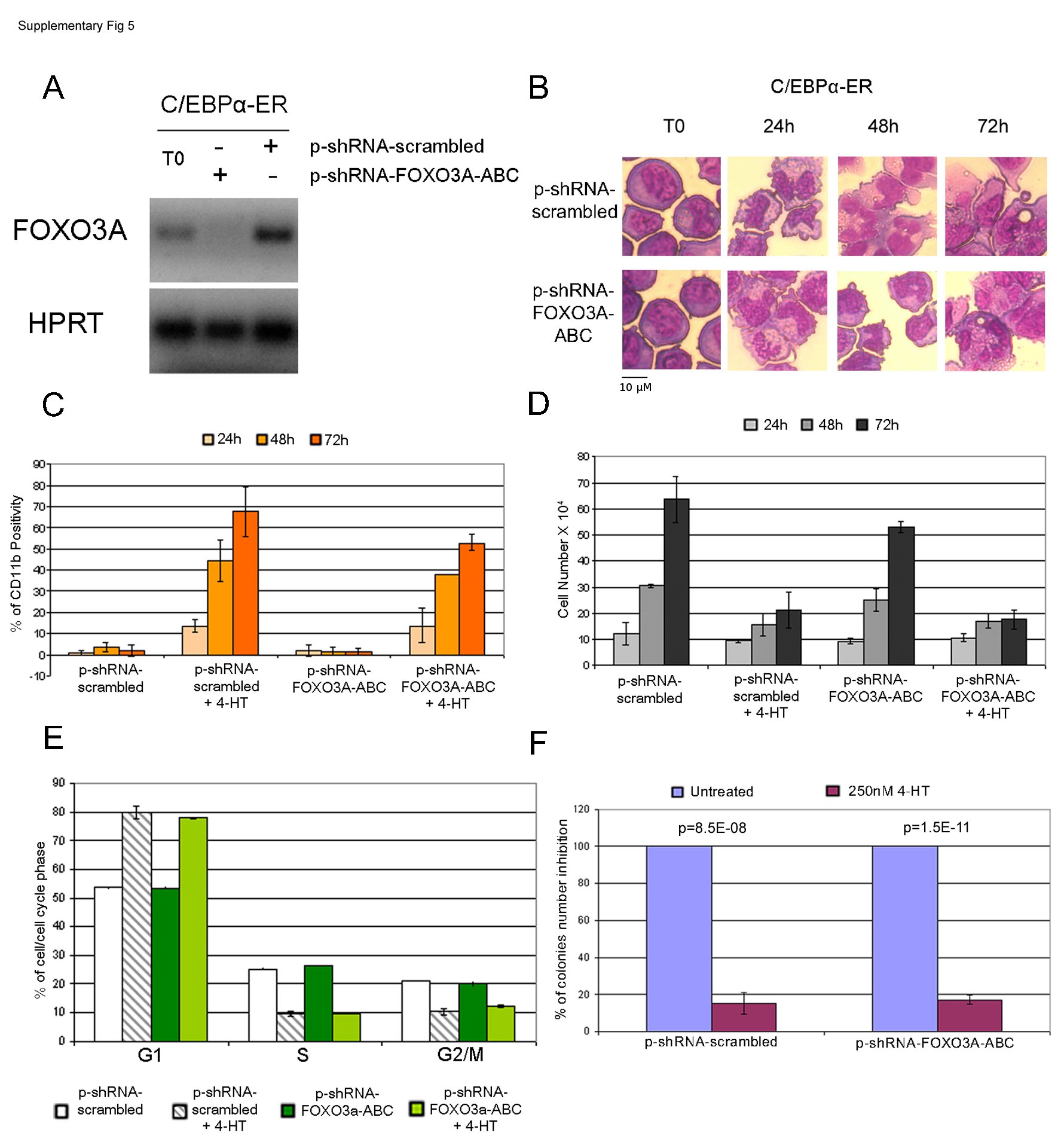
Because expression of constitutively active FOXO3a induces erythroid differentiation of K562 cells (44), we also assessed the role of C/EBPα-dependent FOXO3a expression in C/EBPα-induced granulocytic differentiation of K562 cells. Thus, FOXO3a expression was down-regulated by RNAi in C/EBPα-ER-expressing K562 cells (supplemental Fig. 5A), and the induction of granulocytic differentiation was assessed upon 4-HT treatment. K562 cells transfected with scramble or FOXO3a-specific shRNAs underwent granulocytic differentiation as indicated by morphology (supplemental Fig. 5B) and expression of the myeloid differentiation marker CD11b (supplemental Fig. 5C).
The role of FOXO3a expression for the proliferation inhibitory effects of C/EBPα were tested by assessing cell counts, cell cycle distribution, and methylcellulose colony formation of untreated and 4-HT-treated K562 cells transfected with scramble or FOXO3a specific shRNAs. Activation of C/EBPα suppressed proliferation (supplemental Fig. 5D) and induced a marked increase in the number of G1 cells and a decrease in the fraction of S and G2/M phase cells (supplemental Fig. 5E) as effectively in scrambled and in FOXO3a shRNA-transfected cells; likewise, down-regulation of FOXO3a expression did not rescue the inhibition of methylcellulose colony formation induced by expression of functional C/EBPα in K562 cells (supplemental Fig. 5F).
DISCUSSION
Expression of C/EBPα and C/EBPβ is reduced in BCR/ABL-transformed cell lines and in blast cells from patients with CML-blast crisis (27, 28), suggesting that loss of C/EBP activity is necessary to suppress the differentiation potential of CML-blast crisis progenitors. Consistent with this hypothesis, ectopic expression of C/EBPα or C/EBPβ induced granulocytic differentiation and inhibited proliferation of BCR/ABL-expressing cells in vitro and in mice, but the effects of C/EBPβ were considerably less potent than those of C/EBPα (27, 28). This contrasts with the apparent complete rescue of granulocytic differentiation in mice in which the C/EBPα gene was replaced by C/EBPβ (9) and is probably due to the severe block of differentiation induced by BCR/ABL. Because C/EBPα and C/EBPβ are highly homologous in their C terminus, where basic region, DNA binding, and leucine zipper domain reside, but are highly divergent in the N terminus, where the TAD is located, we took a domain-swapping approach to evaluate biological effects and gene expression profiles in K562 cells expressing 4-HT-regulated C/EBPα-C/EBPβ chimeric proteins.
Activation of C/EBPα or of the chimeric protein N-C/EBPα+C/EBPβ-DBD that shares the TAD of wild-type C/EBPα, suppressed proliferation and promoted an increase in the number of G1 phase cells, whereas C/EBPβ or N-C/EBPβ+C/EBPα-DBD-ER that shares the TAD of wild-type C/EBPβ did not. Likewise, activation of C/EBPα and N-C/EBPα+C/EBPβ-DBD led to efficient granulocytic differentiation of K562 cells, whereas C/EBPβ and N-C/EBPβ+C/EBPα-DBD-ER did not. These effects correlated with those on the transactivation of the G-CSFR promoter. However, ChIP assays did not reveal any difference in the ability of C/EBPα, C/EBPβ, or the chimeric proteins to interact with the G-CSFR promoter, suggesting that the TAD of C/EBPα is more potent of that of C/EBPβ, perhaps through the recruitment of interacting proteins capable of enhancing its transcription activation function.
In an attempt to distinguish between the role of C/EBPα N terminus-specific interactions versus TAD strength, we the assessed biological effects (induction of differentiation and inhibition of proliferation) and transcription activation of chimeric proteins consisting of the same N terminus (the VP16 TAD) fused to the C/EBPα or C/EBPβ DBD. The effects of these two chimeric proteins were undistinguishable, confirming that the DBD does not confer specificity in modulating the transcription-dependent program required for proliferation inhibition and differentiation induction in K562 cells. Of equal importance, the effects of the VP16-C/EBPα/β-DBD chimeric proteins were identical to those of wild-type C/EBPα- or N-C/EBPα+C/EBPβ-DBD, indicating that the gene expression program responsible for the ability of C/EBPα to induce granulocytic differentiation and to inhibit proliferation of K562 cells can be faithfully recapitulated by a chimeric protein in which the C/EBP DBD (which determines specificity of gene targets) is under the control of a strong TAD. Of interest, mutation of the DNA binding domain suppressed differentiation induction and colony formation inhibition by the VP16-C/EBP-DBD chimera, suggesting that C/EBPα can suppress proliferation through both transcriptional mechanisms and the interaction with cell cycle regulatory proteins (30–33). The differences in the biological and biochemical effects of C/EBPα, C/EBPβ, and chimeric proteins were reflected in the activation of distinct gene expression profiles, although a subset of genes was modulated by each protein.
The gene expression profile modulated by C/EBPα and N-C/EBPα+C/EBPβ-DBD was more complex than that regulated by C/EBPβ and N-C/EBPβ+C/EBPα-DBD, most likely reflecting the higher transactivation activity of C/EBPα and N-C/EBPα+C/EBPβ-DBD; however, the number of C/EBPα-regulated genes was higher of that modulated by N-C/EBPα+C/EBPβ-DBD, raising the possibility that certain changes in gene expression may be due to a DBD-specific effect or to loss of C/EBPα N terminus protein-protein interactions in the N-C/EBPα+C/EBPβ-DBD chimeric protein.
Because gene expression profiles modulated by C/EBP proteins were analyzed 12 h post-4-HT treatment, the difference in the number of C/EBPα- and N-C/EBPα+C/EBPβ-DBD-regulated genes might have been magnified by secondary changes possibly due to protein-protein interactions in the N terminus or DBD target specificity, although this would be inconsequential for the ability of N-C/EBPα+C/EBPβ-DBD to inhibit proliferation and induce differentiation of K562 cells as effectively as wild-type C/EBPα.
Functional analysis of the gene subsets regulated by C/EBPα, C/EBPβ, and the two chimeric proteins revealed that genes in the apoptosis pathway were especially modulated by C/EBPα; this finding was of interest because activation of C/EBPα, per se, does not promote apoptosis of BCR/ABL-expressing cells (27), but it enhanced IM-induced cell death of K562 cells. One of the genes regulated by C/EBPα and potentially required for its biological effects is FOXO3a, a transcription factor of the FoxO family with crucial roles in regulating the proliferation and survival of hematopoietic stem cells (41).
Of interest, expression of FOXO3a was not activated by N-C/EBPα-C/EBPβ-DBD or by the VP16-C/EBP-DBD chimeric proteins; this suggests that the transcription of some C/EBPα targets depends not only on the potency of the transactivation domain but also on specific protein interactions that may not be limited to the N-terminal region, although it cannot be excluded that changes in conformation of the chimeric proteins may have affected the interaction with cofactors necessary for FOXO3a transcription.
Although we did not demonstrate that C/EBPα transactivates the FOXO3a promoter, a direct role of C/EBPα in enhancing FOXO3a expression is suggested by ChIP assays demonstrating interaction with a C/EBP binding site in the human FOXO3a promoter and by the finding that C/EBPα induced FOXO3a mRNA expression in cells treated with cycloheximide to block de novo protein synthesis.
Recent studies have revealed that BCR-ABL inhibits FOXO3a activity via Akt-dependent phosphorylation, which promotes interaction with 14-3-3 protein and sequestration of FOXO3a into the cytoplasm (42, 43), leading to lower expression of pro-apoptotic genes such TRAIL and BIM (44); furthermore, FOXO3A inhibition by BCR-ABL also affects the expression of cyclin D1 and D2 (45, 46).
Consistent with the pro-apoptotic effects of FOXO3a, down-regulation of FOXO3A expression by RNAi blocked the increase in apoptosis induced by C/EBPα activation in IM-treated K562 cells. Based on the finding that expression of a constitutively active mutant of FOXO3a induced erythroid differentiation of K562 cells (47) and that proliferation regulatory genes are among the FOXO3A targets (45, 46), we suspected that FOXO3a was also involved in C/EBPα-regulated granulocytic differentiation and proliferation of K562 cells; however, FOXO3a silencing did not rescue either effect, indicating that distinct C/EBPα-regulated genes function as essential downstream effectors to enhance apoptosis susceptibility, promote differentiation, and inhibit proliferation. It remains to be established whether differentiation induction and proliferation inhibition are regulated by common C/EBPα target genes or are under the control of separate effectors.
In summary, we have used a C/EBPα-C/EBPβ domain swapping approach to demonstrate that (i) a strong TAD is responsible for the biological effects of C/EBPα in K562 cells, and (ii) the effects correlate with C/EBPα-dependent activation of gene expression profiles, which include many cell cycle and apoptosis regulatory genes.
Although activation of C/EBPα is insufficient to induce apoptosis of BCR/ABL-expressing cells, it enhanced IM-induced cell death via expression of FOXO3a, further emphasizing the therapeutic potential of restoring functional C/EBPα alone or together with other anti-cancer agents in CML-blast crisis and perhaps other types of acute myeloid leukemia.
Supplementary Material
Acknowledgments
We thank Dr. A. Morrione (Thomas Jefferson University, Philadelphia) for the gift of plasmid pVP16-Nedd4 and Dr. D. G. Tenen (Harvard Institutes of Medicine, Cambridge, MA) for the gift of plasmid pTK-G-CSFR-luciferase.
This work was supported, in whole or in part, by National Institutes of Health Grant CA95111 (NCI; to B. C.). This study was supported by grants from Fondazione Cassa di Risparmio di Modena, Programmi di Ricerca di Rilevante Interesse Nazionale (PRIN2007), Fondazione Cassa di Risparmio di Vignola (to B. C.) and by the Associazione Italiana Ricerca sul Cancro (to C. G.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–5 and Table 1.
- CML
- chronic myelogenous leukemia
- DBD
- DNA binding domain
- TAD
- transactivation domain
- AML
- acute myelogenous leukemia
- 4-HT
- 4-hydroxytamoxifen
- HPRT
- hypoxanthine-guanine phosphoribosyltransferase
- ER
- estrogen receptor
- IM
- Imatinib
- G-CSFR
- granulocyte-colony stimulating factor (G-CSF) receptor.
REFERENCES
- 1.Ramji D. P., Foka P. (2002) Biochem. J. 365, 561–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott L. M., Civin C. I., Rorth P., Friedman A. D. (1992) Blood 80, 1725–1735 [PubMed] [Google Scholar]
- 3.Cheng T., Shen H., Giokas D., Gere J., Tenen D. G., Scadden D. T. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 13158–13163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radomska H. S., Huettner C. S., Zhang P., Cheng T., Scadden D. T., Tenen D. G. (1998) Mol. Cell. Biol. 18, 4301–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang N. D., Finegold M. J., Bradley A., Ou C. N., Abdelsayed S. V., Wilde M. D., Taylor L. R., Wilson D. R., Darlington G. J. (1998) Science 269, 1108–1112 [DOI] [PubMed] [Google Scholar]
- 6.Zhang P., Iwasaki-Arai J., Iwasaki H., Fenyus M. L., Dayaram T., Owens B. M., Shigematsu H., Levantini E., Huettner C. S., Lekstrom-Himes J. A., Akashi K., Tenen D. G. (2004) Immunity 21, 853–863 [DOI] [PubMed] [Google Scholar]
- 7.Screpanti I., Romani L., Musiani P., Modesti A., Fattori E., Lazzaro D., Sellitto C., Scarpa S., Bellavia D., Lattanzio G. (1995) EMBO J. 14, 1932–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka T., Akira S., Yoshida K., Umemoto M., Yoneda Y., Shirafuji N., Fujiwara H., Suematsu S., Yoshida N., Kishimoto T. (1995) Cell 80, 353–361 [DOI] [PubMed] [Google Scholar]
- 9.Jones L. C., Lin M. L., Chen S. S., Krug U., Hofmann W. K., Lee S., Lee Y. H., Koeffler H. P. (2002) Blood 99, 2032–2036 [DOI] [PubMed] [Google Scholar]
- 10.Tenen D. G. (2003) Nat. Rev. Cancer 3, 89–101 [DOI] [PubMed] [Google Scholar]
- 11.Nerlov C. (2004) Nat. Rev. Cancer 4, 394–400 [DOI] [PubMed] [Google Scholar]
- 12.Perrotti D., Cesi V., Trotta R., Guerzoni C., Santilli G., Campbell K., Iervolino A., Condorelli F., Gambacorti-Passerini C., Caligiuri M. A., Calabretta B. (2002) Nat. Genet. 30, 48–58 [DOI] [PubMed] [Google Scholar]
- 13.Chang J. S., Santhanam R., Trotta R., Neviani P., Eiring A. M., Briercheck E., Ronchetti M., Roy D. C., Calabretta B., Caligiuri M. A., Perrotti D. (2007) Blood 110, 994–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pabst T., Mueller B. U., Zhang P., Radomska H. S., Narravula S., Schnittger S., Behre G., Hiddemann W., Tenen D. G. (2001) Nat. Genet. 27, 263–270 [DOI] [PubMed] [Google Scholar]
- 15.Gombart A. F., Hofmann W. K., Kawano S., Takeuchi S., Krug U., Kwok S. H., Larsen R. J., Asou H., Miller C. W., Hoelzer D., Koeffler H. P. (2002) Blood 99, 1332–1340 [DOI] [PubMed] [Google Scholar]
- 16.Leroy H., Roumier C., Huyghe P., Biggio V., Fenaux P., Preudhomme C. (2005) Leukemia 19, 329–334 [DOI] [PubMed] [Google Scholar]
- 17.Kirstetter P., Schuster M. B., Bereshchenko O., Moore S., Dvinge H., Kurz E., Theilgaard-Mönch K., Månsson R., Pedersen T. A., Pabst T., Schrock E., Porse B. T., Jacobsen S. E., Bertone P., Tenen D. G., Nerlov C. (2008) Cancer Cell 13, 299–310 [DOI] [PubMed] [Google Scholar]
- 18.Bereshchenko O., Mancini E., Moore S., Bilbao D., Månsson R., Luc S., Grover A., Jacobsen S. E., Bryder D., Nerlov C. (2009) Cancer Cell 16, 390–400 [DOI] [PubMed] [Google Scholar]
- 19.Pabst T., Mueller B. U., Harakawa N., Schoch C., Haferlach T., Behre G., Hiddemann W., Zhang D. E., Tenen D. G. (2001) Nat. Med. 7, 444–451 [DOI] [PubMed] [Google Scholar]
- 20.Westendorf J. J., Yamamoto C. M., Lenny N., Downing J. R., Selsted M. E., Hiebert S. W. (1998) Mol. Cell. Biol. 18, 322–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keeshan K., He Y., Wouters B. J., Shestova O., Xu L., Sai H., Rodriguez C. G., Maillard I., Tobias J. W., Valk P., Carroll M., Aster J. C., Delwel R., Pear W. S. (2006) Cancer Cell 10, 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heibling D., Mueller B. U., Timchenko N. A., Hagemeijer A., Jotlerand M., Meyer-Monard S., Lister A., Rowley J. D., Huegli B., Fey M. F., Pabst T. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 13312–13317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helbling D., Mueller B. U., Timchenko N. A., Schardt J., Eyer M., Betts D. R., Jotterand M., Meyer-Monard S., Fey M. F., Pabst T. (2005) Blood 106, 1369–1375 [DOI] [PubMed] [Google Scholar]
- 24.Zheng R., Friedman A. D., Levis M., Li L., Weir E. G., Small D. (2004) Blood 103, 1883–1890 [DOI] [PubMed] [Google Scholar]
- 25.Radomska H. S., Bassères D. S., Zheng R., Zhang P., Dayaram T., Yamamoto Y., Sternberg D. W., Lokker N., Giese N. A., Bohlander S. K., Schnittger S., Delmotte M. H., Davis R. J., Small D., Hiddemann W., Gilliland D. G., Tenen D. G. (2006) J. Exp. Med. 203, 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tavor S., Park D. J., Gery S., Vuong P. T., Gombart A. F., Koeffler H. P. (2003) J. Biol. Chem. 278, 52651–52659 [DOI] [PubMed] [Google Scholar]
- 27.Ferrari-Amorotti G., Keeshan K., Zattoni M., Guerzoni C., Iotti G., Cattelani S., Donato N. J., Calabretta B. (2006) Blood 108, 1353–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guerzoni C., Bardini M., Mariani S. A., Ferrari-Amorotti G., Neviani P., Panno M. L., Zhang Y., Martinez R., Perrotti D., Calabretta B. (2006) Blood 107, 4080–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duprez E., Wagner K., Koch H., Tenen D. G. (2003) EMBO J. 22, 5806–5816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H., Iakova P., Wilde M., Welm A., Goode T., Roesler W. J., Timchenko N. A. (2001) Mol. Cell 8, 817–828 [DOI] [PubMed] [Google Scholar]
- 31.Porse B. T., Pedersen T. A., Xu X., Lindberg B., Wewer U. M., Friis-Hansen L., Nerlov C. (2001) Cell 107, 247–258 [DOI] [PubMed] [Google Scholar]
- 32.Müller C., Calkhoven C. F., Sha X., Leutz A. (2004) J. Biol. Chem. 279, 7353–7358 [DOI] [PubMed] [Google Scholar]
- 33.D'Alo' F., Johansen L. M., Nelson E. A., Radomska H. S., Evans E. K., Zhang P., Nerlov C., Tenen D. G. (2003) Blood 102, 3163–3171 [DOI] [PubMed] [Google Scholar]
- 34.Guerzoni C., Ferrari-Amorotti G., Bardini M., Mariani S. A., Calabretta B. (2006) Cell Cycle 5, 1254–1257 [DOI] [PubMed] [Google Scholar]
- 35.Sunters A., Fernández de Mattos S., Stahl M., Brosens J. J., Zoumpoulidou G., Saunders C. A., Coffer P. J., Medema R..H., Coombes R. C., Lam E. W. (2003) J. Biol. Chem. 278, 49795–49805 [DOI] [PubMed] [Google Scholar]
- 36.Hirai H., Zhang P., Dayaram T., Hetherington C. J., Mizuno S., Imanishi J., Akashi K., Tenen D. G. (2006) Nat. Immunol. 7, 732–739 [DOI] [PubMed] [Google Scholar]
- 37.Sadowski I., Ma J., Triezenberg S., Ptashne M. (1988) Nature 335, 563–564 [DOI] [PubMed] [Google Scholar]
- 38.Triezenberg S. J., LaMarco K. L., McKnight S. L. (1988) Genes Dev. 2, 730–742 [DOI] [PubMed] [Google Scholar]
- 39.McKnight J. L., Kristie T. M., Roizman B. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 7061–7065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Preston C. M., Frame M. C., Campbell M. E. (1988) Cell 52, 425–434 [DOI] [PubMed] [Google Scholar]
- 41.Miyamoto K., Araki K. Y., Naka K., Arai F., Takubo K., Yamazaki S., Matsuoka S., Miyamoto T., Ito K., Ohmura M., Chen C., Hosokawa K., Nakauchi H., Nakayama K., Nakayama K. I., Harada M., Motoyama N., Suda T., Hirao A. (2007) Cell Stem Cell 1, 101–112 [DOI] [PubMed] [Google Scholar]
- 42.Ghaffari S., Jagani Z., Kitidis C., Lodish H. F., Khosravi-Far R. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 6523–6528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Komatsu N., Watanabe T., Uchida M., Mori M., Kirito K., Kikuchi S., Liu Q., Tauchi T., Miyazawa K., Endo H., Nagai T., Ozawa K. (2003) J. Biol. Chem. 278, 6411–6419 [DOI] [PubMed] [Google Scholar]
- 44.Essafi A., Fernández de Mattos S., Hassen Y. A., Soeiro I., Mufti G. J., Thomas N. S., Medema R. H., Lam E. W. (2005) Oncogene 24, 2317–2329 [DOI] [PubMed] [Google Scholar]
- 45.Fernández de Mattos S., Essafi A., Soeiro I., Pietersen A. M., Birkenkamp K. U., Edwards C. S., Martino A., Nelson B. H., Francis J. M., Jones M. C., Brosens J. J., Coffer P. J., Lam E. W. (2004) Mol. Cell. Biol. 24, 10058–10071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt M., Fernandez de Mattos S., van der Horst A., Klompmaker R., Kops G. J., Lam E. W., Burgering B. M., Medema R. H. (2002) Mol. Cell. Biol. 22, 7842–7852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Birkenkamp K. U., Essafi A., van der Vos K. E., da Costa M., Hui R. C., Holstege F., Koenderman L., Lam E. W., Coffer P. J. (2007) J. Biol. Chem. 282, 2211–2220 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}