

Noncovalent interactions between aromatic molecules are widely believed to be important contributing factors in the stabilization of organized structure in biological macromolecules.1,2 Among the most significant aromatic–aromatic interactions are those found in helical nucleic acid structures. Since the identity of the nearest neighbors to a given base pair is the best single predictor of thermodynamics in DNA duplexes,3 it is clear that aromatic π–π interactions are crucial to the stabilization of these structures.4 While there have been a considerable number of theoretical studies aimed at modeling the π–π interaction in DNA,5 there have been remarkably few experimental studies specifically addressing the thermodynamics of stacking (separate from base pairing) in DNA itself.6 For that reason we have undertaken a study of aromatic stacking in the context of duplex DNA, and we hope to begin to elucidate what are the important forces which stabilize this organized structure. We report here the first experimental comparison of the stacking abilities of natural DNA bases and of nonnatural aromatic analogs in double-stranded DNA.
To separate stacking from pairing (hydrogen-bonding) interactions in duplex DNA we placed the natural or nonnatural nucleotide of interest in a “dangling” position (without a pairing partner) at the end of a base-paired duplex (Figure 1).7 The resulting stabilization of the duplex by the dangling base can be measured by thermal denaturation experiments, with comparison to the duplex lacking the added nucleotide.
Figure 1.
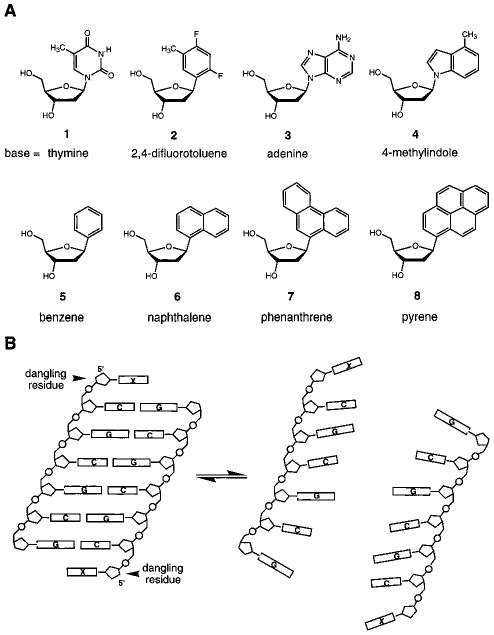
Structures and DNA sequences in this study. (A) Structures of natural (1, 3) and nonnatural (2, 4–8) nucleosides examined here. (B) Illustration of how placement of a single nucleotide residue at the 5′-end of a self-complementary strand brings it into an unpaired “dangling” position directly adjacent to the duplex; comparison with the unsubstituted 6-base-pair core duplex allows a measure of stacking interactions by the dangling bases.7
Electrostatic effects resulting from such localized charge have been implicated both in the stabilization and in the geometry of aromatic stacking.5 To examine such effects we compared not only natural DNA bases but also nonpolar molecules with similar shape and surface area. Thus, we compared the DNA base thymine (1) and adenine (3) with their respective nonpolar isosteres difluorotoluene (2) and 4-methylindole (4).9 We also compared the stacking of the aromatic hydrocarbons benzene (5), naphthalene (6), phenanthrene (7), and pyrene (8). The synthesis of these nucleoside analogs has been reported.10–13
Results of the thermodynamic measurements made at pH 7.0 and 1 M NaCl are presented in Table 1. We measured melting transitions (Tm) as a function of concentration for the duplexes and calculated thermodynamic parameters by plotting 1/Tm vs ln([oligonucleotide]). The linear fits were quite good (r2 ≥ 0.97), with error in free energies of approximately ±2%. The unsubstituted core duplex under these conditions has a Tm (5 μM) of 41.0(±0.5) °C and a free energy (37 °C) of −8.05(±0.16) kcal/mol.
Table 1.
Comparison of Stacking Affinities of Natural DNA Bases and Simple Aromatic Analogs, As Measured by Dangling End Studies in a Self-Complementary DNA Duplex (Sequence d(XCGCGCG)2)a
| dangling moiety | Tm (°C)b | ΔTm | −ΔG°37 (kcal/mol)c | ΔΔG° stacking |
|---|---|---|---|---|
| (none) | 41.0 | 8.1 ± 0.2 | ||
| thymine | 48.1 | 7.1 | 9.2 ± 0.2 | 1.1 ± 0.2 |
| difluorotoluene | 54.4 | 13.4 | 10.7 ± 0.2 | 2.6 ± 0.3 |
| adenine | 51.6 | 10.6 | 10.1 ± 0.2 | 2.0 ± 0.3 |
| 4-methylindole | 54.6 | 13.6 | 11.1 ± 0.2 | 3.1 ± 0.3 |
| benzene | 48.3 | 7.3 | 9.4 ± 0.2 | 1.4 ± 0.2 |
| naphthalene | 56.2 | 15.2 | 10.9 ± 0.2 | 2.9 ± 0.3 |
| phenanthrene | 57.3 | 16.3 | 10.7 ± 0.2 | 2.6 ± 0.3 |
| pyrene | 64.1 | 23.1 | 11.4 ± 0.2 | 3.4 ± 0.3 |
Stacking parameters (ΔTm, ΔΔG°) are obtained by subtracting data for the core hexamer duplex from that for duplexes with 1–8 added at the unpaired (X) position.
Conditions: 1 M NaCl, 10 mM Na·phosphate pH 7.0, 5.0 μM DNA strand concentration.
Values obtained by plotting 1/Tm vs ln(CT) with data from at least five concentrations.
Measurement of the duplexes with dangling thymine and adenine residues shows, perhaps not surprisingly, that the purine stacks on the duplex more strongly than the smaller pyrimidine base. The two unpaired deoxyadenosines add 2.0 kcal of stabilizing interaction to the self-complementary sequence, and thymines add 1.1 kcal to the duplex stability. This relative stacking ability is as predicted from nearest-neighbor parameters3 and is consistent with dangling-end studies carried out in RNA.7
Interestingly, the data show that the nonpolar DNA base mimics stack considerably more strongly than their natural counterparts. Difluorotoluene raises the Tm of the duplex by 13.4 °C, about twice the effect of thymine, although the two have the same size, shape, and surface area.9 Similarly, the isosteric analog 4-methylindole increases the Tm by more than does the natural base adenine. The effects on free energies of the duplexes mirror these results (Table 1).
These results show clearly that surface area alone is not a good predictor of stacking affinity. It seems likely that the increased hydrophobicity of these two nonpolar analogs contributes favorably to their enhanced stacking. An alternative explanation might be differences in polarizability, which would influence van der Waals forces. However, methylindole is calculated to be only slightly more polarizable than adenine, and difluorotoluene is equally as polarizable as thymine.14 Thus it appears that hydrophobicity differences exert the largest influence here. The superior stacking of the two purine-sized compounds relative to the pyrimidine-sized cases might be explained by increased surface area, which would allow for stronger interaction with the neighboring C–G base pair.
To investigate further the effects of size and hydrophobicity on stacking interactions, we then examined the series of four nucleotides in which the “base” moiety is benzene, naphthalene, phenanthrene, and pyrene (Figure 1A). The results show (Table 1) that these compounds stack relatively strongly, and the interaction generally becomes more favorable with increasing size. The benzene nucleotide stacks with slightly greater affinity than thymine. The naphthalene compound shows a large Tm increase over the unsubstituted duplex of 15.2 °C and a total stacking energy of 2.9 kcal/mol, or twice that of benzene.
Interestingly, the phenanthrene nucleoside, although larger, has a stacking propensity essentially the same as that of naphthalene (compare seventh and eighth entries in Table 1). Models indicate that the third ring in phenanthrene (which is absent in naphthalene) does not contact the adjacent bases and instead is likely to protrude toward the major groove of the DNA. We surmise that lack of added contact with the nearest neighbor precludes added favorable interactions. Since phenanthrene is significantly more polarizable than naphthalene14 but does not stack more strongly, the results are most easily explained by solvophobic effects rather than dipole-induced dipole effects. The amount of nonpolar surface area excluded from solvent by stacking is expected to be similar for naphthalene and phenanthrene in these structures.
The pyrene compound interacts the most strongly of all the natural and nonnatural bases studied. The duplex substituted by this species has a melting transition 23.1 °C higher than the unsubstituted duplex and 12.5 °C higher than for the case with adenine. Addition of the two pyrene residues to the self-complementary duplex adds a quite large 3.4 kcal / mol of stabilization to the helix. Models indicate that a large fraction of one face of this molecule can stack with the neighboring base pair, and so the relatively high solvophobicity is apparently put to good effect.
The overall order of stacking ability of these molecules is thus found to be thymine < benzene < adenine < difluorotoluene ≤ naphthalene ∼ phenanthrene ≤ methylindole < pyrene, at least in this sequence context. It seems significant that solvophobic effects appear to be the most important factor in determining this order. When molecules of the same size are compared, we find that the less polar structure stacks more strongly than one of higher polarity. When nonpolar molecules of different sizes are compared, we find that stacking propensity appears to correlate with the surface area excluded from solvent.
A number of studies aimed at measuring aromatic–aromatic interactions in smaller model systems have been reported previously.15,16 A few of these have examined interactions of DNA bases;16 and mono- and dinucleotides have been studied as models for stacking interactions.17 The present results agree with the previously known superiority of purines over pyrimidines at stacking, which was also observed by Leonard et al. using nucleobases bridged by simple trimethylene linkers.16a In a comparison of adenines and naphthalenes linked by three-carbon bridges, Gellman found evidence that naphthalene–adenine interactions were qualitatively stronger than adenine–adenine interactions,16c a result in accord with our finding of superior stacking of naphthalene over adenine. In a simple receptor system, Rebek et al. found increasing interaction of adenine with aromatics of increasing size,16b also in agreement with the present results.
Our results indicate that the natural DNA bases are not particularly effective at stacking, at least in a global comparison of aromatic structures. Evolution may not have favored bases which stack too strongly, since helix unwinding is a requirement for DNA replication. For designed nucleic acid structures, where thermodynamic stability is often desirable, our results indicate that one may gain stability simply by placing large nonpolar aromatics at the ends of helical structure. It will of course be useful to study this series of compounds in additional sequence contexts to explore the generality of the conclusions. It is worth noting that in a considerably different structure (a hairpin loop) we have noted similar stabilization by nonpolar DNA base analogs.11 Since thermodynamic rather than structural evidence of stacking is presented here, it will be of significant future interest to obtain structural information18 about dangling nucleotides to better evaluate the specific interactions involved.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM52956) for support.
Footnotes
Supporting Information Available: Plots of thermodynamic data, sample thermal melting profiles, and proton NMR spectra (3 pages). See any current masthead page for ordering and Internet access instructions.
References
- 1.See, for example: Yu MH, Weissman JS, Kim PS. J Mol Biol. 1996;249:388–397. doi: 10.1006/jmbi.1995.0304.
- 2.Werner MH, Gronenborn AM, Clore GM. Science. 1996;271:778–784. doi: 10.1126/science.271.5250.778. [DOI] [PubMed] [Google Scholar]
- 3.See, for example: Breslauer KJ, Frank R, Blocker H, Marky LA. Proc Natl Acad Sci USA. 1986;83:3746–3750. doi: 10.1073/pnas.83.11.3746.SantaLucia J, Allawi HT, Seneviratne PA. Biochemistry. 1996;35:3555–3562. doi: 10.1021/bi951907q.
- 4.Petersheim M, Turner DH. Biochemistry. 1983;22:256–263. doi: 10.1021/bi00271a004. [DOI] [PubMed] [Google Scholar]
- 5.(a) Jernigan RL, Sarai A, Ting KL, Nussinov R. J Biomol Struct Dyn. 1986;4:41–48. doi: 10.1080/07391102.1986.10507645. [DOI] [PubMed] [Google Scholar]; (b) Hunter CA. J Mol Biol. 1993;230:1025–1054. doi: 10.1006/jmbi.1993.1217. [DOI] [PubMed] [Google Scholar]; (c) Maroun RC, Olson WK. Biopolymers. 1988;27:561–584. doi: 10.1002/bip.360270403. [DOI] [PubMed] [Google Scholar]; (d) Ornstein RL, Rein R, Breen DL, Macelroy RD. Biopolymers. 1978;17:2341–2360. doi: 10.1002/bip.1978.360171005. [DOI] [PubMed] [Google Scholar]; (e) Friedman RA, Honig B. Biopolymers. 1992;32:145–159. doi: 10.1002/bip.360320205. [DOI] [PubMed] [Google Scholar]; (f) Maroun RC, Olson WK. Biopolymers. 1988;27:585–603. doi: 10.1002/bip.360270404. [DOI] [PubMed] [Google Scholar]; (g) Sarai A, Mazur J, Nussinov R, Jernigan RL. Biochemistry. 1988;27:8498–8502. doi: 10.1021/bi00422a030. [DOI] [PubMed] [Google Scholar]
- 6.For a review of helical structure (stacking) in single-stranded, heterogeneous polymeric DNA, see: Felsenfeld G, Miles HT. Annu Rev Biochem. 1967;36:407. doi: 10.1146/annurev.bi.36.070167.002203.
- 7.Such studies have been carried out by Turner et al. with the natural bases in RNA (ref 4 and Turner DH, Sugimoto N. Annu Rev Biophys Biophys Chem. 1988;17:167–192. doi: 10.1146/annurev.bb.17.060188.001123.), but there are no such single-base stacking studies reported in DNA.
- 8.Senior M, Jones RA, Breslauer KJ. Biochemistry. 1988;27:3879–3885. doi: 10.1021/bi00410a053. [DOI] [PubMed] [Google Scholar]
- 9.Schweitzer BA, Kool ET. J Org Chem. 1994;59:7238–7242. doi: 10.1021/jo00103a013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moran S, Ren RXF, Sheils CJ, Kool ET. Nucleic Acids Res. 1996;24:2044–2052. doi: 10.1093/nar/24.11.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren XF, Schweitzer BA, Sheils CJ, Kool ET. Angew Chem. 1996;108:834–837. doi: 10.1002/anie.199607431. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem, Int Ed Engl. 1996;35:743–746. [Google Scholar]
- 12.Chaudhuri NC, Ren RXF, Kool ET. Synlett. in press. [Google Scholar]
- 13.Millican TA, Mock GA, Chauncey MA, Patel TP, Eaton MAW, Gunning J, Cutbush SD, Neidle S, Mann J. Nucleic Acids Res. 1984;12:7435. doi: 10.1093/nar/12.19.7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molecular polarizabilities calculated using SPARTAN were for thymine, 10.6 Å3; difluorotoluene, 11.0 Å3; adenine, 12.3 Å3; 4-methylindole, 14.0 Å3; naphthalene, 14.2 Å3; phenanthrene, 18.3 Å3.
- 15.(a) Petti MA, Sheppodd TJ, Barrans RE, Jr, Dougherty DA. J Am Chem Soc. 1988;110:6825. [Google Scholar]; (b) Cowart MD, Sucholeiki I, Bukownik RR, Wilcox CS. J Am Chem Soc. 1988;110:6204. doi: 10.1021/ja00226a040. [DOI] [PubMed] [Google Scholar]; (c) Ferguson SB, Sanford EM, Seward EM, Diederich F. J Am Chem Soc. 1991;113:5410–5419. [Google Scholar]; (d) Smithrud DB, Wyman TB, Diederich F. J Am Chem Soc. 1991;113:5420–5426. [Google Scholar]
- 16.(a) Leonard NJ. Acc Chem Res. 1979;12:423–429. [Google Scholar]; (b) Rotello VM, Viani EA, Deslongchamps G, Murray BA, Rebek J. J Am Chem Soc. 1993;115:797–798. [Google Scholar]; (c) Newcomb LF, Gellman SH. J Am Chem Soc. 1994;116:4993–4994. [Google Scholar]
- 17.(a) Warshaw MM, Tinoco I., Jr J Mol Biol. 1966;20:29–38. doi: 10.1016/0022-2836(66)90115-x. [DOI] [PubMed] [Google Scholar]; (b) Ts'o POP, Melvin JS, Olson AC. J Am Chem Soc. 1963;85:1289. [Google Scholar]
- 18.Proton NMR spectra of oligonucleotide trimers with 6–8 at the center position show strong upfield shifts in neighboring bases (see supporting information) and thus provide evidence of stacking with nearest neighbors even in single-stranded DNA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.