

Abstract
Recent findings have implicated tight junction (TJ) protein Occludin (OCLN) as an essential factor for Hepatitis C Virus (HCV) to enter human hepatocytes. To gain insights into OCLN-mediated HCV entry, we created a panel of OCLN deletion mutants and found that without impairing OCLN’s cell surface localization, removal of the extracellular loop 2 (EL2) from OCLN abolished both its ability to mediate HIV-HCV pseudotypes’ (HCVpp) entry as well as its ability to coprecipitate HCV glycoprotein E2. Recombinant OCLN EL2, however, failed to robustly bind soluble E2 (sE2) in pull-down assays. Subsequent studies revealed that OCLN formed complex with Dynamin II, an important GTPase for endocytosis, in an EL2-dependent fashion. HCVpp, as well as cell culture grown HCV (HCVcc), was sensitive to Dynamin knockdown or inhibition. We conclude that OCLN EL2 dictates the Dynamin-dependent HCV entry. Furthermore, OCLN could function to bridge virions to Dynamin -dependent endocytic machineries.
Keywords: Hepatitis C Virus, Entry, Tight junction, Occludin, Dynamin
Introduction
Hepatitis C virus (HCV) exacts a heavy toll on global health by causing acute and chronic hepatitis, cirrhosis, as well as hepatocellular carcinoma. Infection of this single-stranded RNA virus is almost exclusively restricted to human hepatocytes, a phenomenon that is at least partially attributed to the specific viral entry into these cells. Accumulated evidence suggests that HCV entry into a cell is a complex multi-step process: infectious virions first attach to cell surface via low-affinity binding and then associate with cellular receptors including CD81 (Pileri et al., 1998) and SR-BI (Scarselli et al., 2002) at higher affinity; in the presence of tight junction (TJ) proteins Claudin-1 (CLDN1) (Evans et al., 2007) and Occludin (OCLN) (Liu et al., 2009; Ploss, 2009), bound virions undergo clathrin-mediated endocytosis (Blanchard et al., 2006; Codran et al., 2006; Meertens, Bertaux, and Dragic, 2006) and traffic to endosomal compartments where the viral envelope proteins undergo a conformational change and then fuse with cellular membrane for entry (Hsu et al., 2003). Among the four required cellular factors, CD81 and SR-BI are believed to mediate virus attachment to the cell by interacting with viral glycoproteins (gps) E1 and E2 (reviewed in (Bartosch and Cosset, 2006)). By contrast, the specific roles of CLDN1 and OCLN in HCV entry remain elusive.
Similar to CLDN1, OCLN is also a four-transmembrane protein present in the TJ of polarized cells, where it functions to regulate paracellular permeability and to confer cell adhesiveness (Van Itallie and Anderson, 1997). The predicted OCLN topology consists of a 66 amino acid (aa)- long N-terminus intracellular domain that does not exist in CLDN1, two extracellular loops (EL1 and EL2) of similar size, a short intracellular loop linking transmembrane domain 2 and 3, and a 259 aa long cytoplasmic tail (significantly longer than that of CLDN1). Both ELs of OCLN are enriched with tyrosine residues, and more than half of the residues found in EL1 are tyrosines and glycines (~60% in human, mouse and dog). EL2, in addition, was shown to mediate the homophilic interaction between OCLNs expressed on two adjacent cells (Van Itallie and Anderson, 1997; Wang et al., 2005; Wong and Gumbiner, 1997). The distal C-terminal cytoplasmic domain of OCLN is highly homologous to an important functional domain in the C terminus of the ELL family of RNA polymerase II transcription factors; this region was reported to dictate protein targeting and endocytosis (Matter and Balda, 1998). In addition, the C terminus can also bind to the scaffolding proteins ZO-1, ZO-2, ZO-3; cingulin; the membrane trafficking protein VAP33; and the cytoskeletal protein F-actin (Feldman, Mullin, and Ryan, 2005).
The observation that the OCLN ELs failed to substitute CLDN1 ELs highlights the different roles that CLDN1 and OCLN play in mediating HCV entry (Liu et al., 2009). We have previously shown that the endogenous OCLN co-precipitated with HCV E2 in HCVcc-infected hepatoma cells (Liu et al., 2009). However, it is unclear whether the OCLN-E2 complex is derived from an intracellular pool or it represents the interaction between HCV particles and cell surface-anchored OCLN. In the present study, we attempt to further investigate the interaction between OCLN and HCV glycoproteins using OCLN deletion mutants and recombinant proteins. To our surprise, although co-transfected OCLN and HCV E2 readily co-precipitated in a EL2-dependent manner, the direct binding between purified OCLN ELs and E2 was nearly undetectable when measured by in vitro pull-down assays. Subsequently, we observed that OCLN could form complex with Dynamin II, the large GTPase that is critical for multiple endocytic pathways. Silencing Dynamin II by RNA interference (RNAi) or blocking Dynamin function inhibited HCVpp and HCVcc entry. Altogether, HCV entry appears to be Dynamin-dependent and requires intact OCLN EL2 domain.
Results
OCLN EL2 is necessary to confer cellular susceptibility to HCVpp
To gain insights into the OCLN-mediated HCV entry, we first sought to determine which domain of OCLN, a four-transmembrane protein with a relatively long C-terminus tail, is required for its function as a HCV entry cofactor. To this end, we created a panel of deletional constructs expressing various regions of human OCLN (Fig. 1A). Following lentiviral transduction, all of the mutants were expressed as N-terminal Flag tagged proteins in 786-O cell line which was recently shown to be deficient in endogenous OCLN (Ploss, 2009) (Fig. 1A). To our surprise, the N-terminus deletant (OCLN/ΔN) was not detected well by the anti-Flag antibody (Fig 1A upper panel), but could be easily identified using an OCLN monoclonal antibody (Fig. 1A lower panel). This observation is consistent with that in a previous report (Bamforth et al., 1999). Notably, the expression level of deletants was comparable to that of the full-length OCLN. Besides the predicted ~62kd band (full-length OCLN), several bands with smaller size were also noticed, which could be truncated forms of OCLN, as a number of host enzymes reportedly cleave OCLN at multiple sites in different cell types (Feldman, Mullin, and Ryan, 2005).
Fig. 1.

OCLN EL2 is essential to mediate HCVpp entry. (A) 5×105 786-O cells were transduced with lentiviruses to express Flag-tagged hOCLN, OCLN/ΔN, OCLN/ΔC, OCLN/ΔEL1, and OCLN/ΔEL2. pTrip-GFP virus (expressing the green fluorescent protein) was included to control for transduction. Cell lysates were made at 48 hr p.i. and immunoblotted with an anti-Flag antibody (top panel) or a monoclonal anti-OCLN antibody (bottom panel). OCLN/ΔC (indicated with a star sign in top panel) could not be recognized by the anti-OCLN antibody as this antibody was raised against the C-terminus of OCLN. (B) All of the OCLN constructs except OCLN/ΔEL2 rendered 786-O cells susceptible to HCVpp infection. Data are representative of at least three independent experiments and are presented as mean ± standard deviation.
Subsequent analyses revealed that overexpression of full-length OCLN in 786-O cells significantly increased HCVpp entry (Fig. 1B). Importantly, removing EL2 completely abolished OCLN’s ability to mediate HCVpp entry (Fig. 1B). By contrast, deletion of N-terminal, C-terminal cytoplasmic domain or OCLN EL1 had no impact on OCLN-dependent HCVpp entry (Fig 1B). We also examined the infection of 786-O cells by VSV-G pseudotyped virus (VSV-Gpp) and found that expressing OCLN mutants had no effect on VSV-Gpp infection at all (Fig. 1B). Together, these results indicate that the OCLN EL2 is specifically required for HCVpp entry.
Next, the cellular distribution of OCLN deletants was characterized to gain clues as to whether any of the deletions impairs the membrane targeting of OCLN. Initial attempts to image 786-O cells, however, were unsuccessful, we therefore transfected 293T cells with GFP-tagged OCLN deletants and found that all OCLN deletants were able to locate to the plasma membrane, except that OCLN/ΔC also displayed pronounced cytoplasmic retention (Fig. 2). Consistently, the GFP-OCLN/ΔC was not as effective in rendering 786-O cells susceptible to HCVpp as the Flag-OCLN/ΔC did (data not shown). Perhaps due to a larger size, addition of GFP to OCLN/ΔC altered the protein folding to a greater extent than did the Flag tag. Nevertheless, OCLN without EL2 clearly preserved the cell surface localization pattern. To validate this part of the observation, we performed cell surface biotinylation assay to label plasma membrane-bound Flag-tagged OCLN and deletants. Biotinylated cell surface proteins were enriched with streptavidin-conjugated agarose beads. Precipitated proteins were then analyzed by western blotting with the anti-OCLN or anti-Flag antibody. Again, we found that all of the mutants were transported to the cell surface (Fig. 3). Collectively, the observed inability for OCLN/ΔEL2 to mediate HCV entry was unlikely caused by failed protein targeting to cell surface.
Fig. 2.

Cellular localization of OCLN mutants. Lenti-X 293T cells expressing indicated GFP-tagged OCLN deletants were fixed and imaged by laser scanning confocal microscopy. Nuclei were stained with Draq5 (blue). Orthogonal views were presented here.
Fig 3.
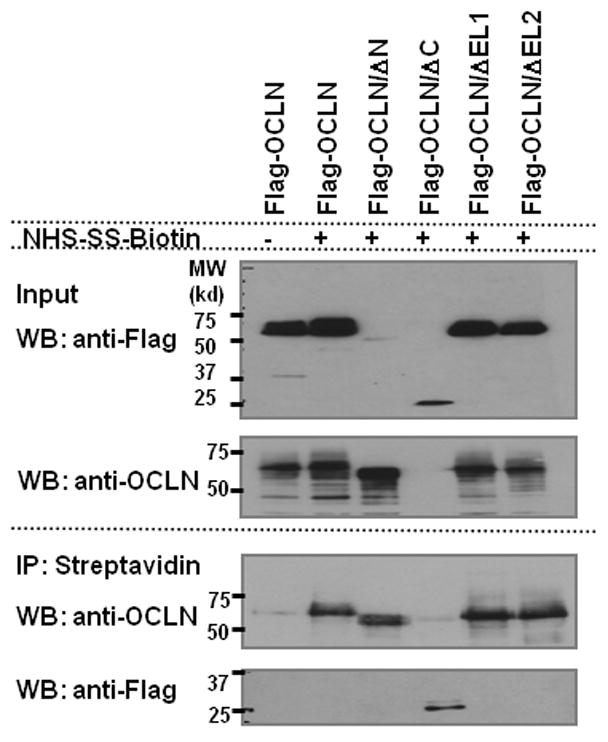
OCLN deletant that abrogates HCV entry can locate to cell surface. Lenti-X 293T cells expressing indicated Flag-tagged OCLN deletants were surface biotinylated or mock treated and assessed as described in Materials and Methods. All deletants were able to reach cell surface. Lysate from mock treated Flag-OCLN expressing cells was included as negative control to demonstrate the specific pull-down of biotinylated proteins.
Interestingly, several isoforms of OCLN are present in Huh 7.5.1 cells (Supplementary Fig. 1). A recent report described the existence of up to eight different splicing isoforms of OCLN in normal human liver and other tissues (Kohaar et al., 2010). Since a construct lacking both the N terminus and extracellular domains of OCLN reportedly exerted a dominant negative effect on tight junction integrity (Bamforth et al., 1999), we explored whether our OCLN deletants would have similar effects on HCV entry in naturally permissive cell types. It was observed that expressing these deletants in Huh7.5.1 cells did not have appreciable effect on HCVpp entry (Supplementary Fig. 2). Hence, the shorter forms of OCLN do not appear to interfere with the function of the full-length protein as a key mediator of virus entry.
Deletion of EL2 abolished OCLN-E2 association
We have previously shown that the viral glycoprotein E2 precipitated with endogenous OCLN in HCVcc-infected hepatoma cells (Liu et al., 2009). HCV E1, however, did not appear to form complex with OCLN (Figure 4A). To locate the portion of OCLN that is required for its association with E2, we transfected OCLN deletional constructs together with HCV E2 expression plasmid into 293T cells and performed co-immunoprecipitation (Co-IP) studies. All OCLN constructs except the EL2 deletant efficiently precipitated with HCV E2 (Fig. 4B), indicating that the OCLN EL2 region is essential for OCLN-E2 association. Consistent with what we observed earlier, although the Flag-OCLN/ΔN was not detected well using anti-Flag antibody, the OCLN monoclonal antibody efficiently recognized the exogenous protein (Fig. 4B).
Fig. 4.

OCLN EL2 is important for its association with HCV E2 in co-immunoprecipitation study. (A) 293T cells were co-transfected with indicated combinations of DNA constructs. Cell lysates were immunoprecipitated with anti-Flag M2 affinity resin at 48 hr p.i. Input cell lysates and immunoprecipitates were blotted with antibodies against HCV E1, E2, OCLN, CLDN1 as well as β-actin. Flag-OCLN co-precipitated with HCV E2 but not with E1 alone. (B) Lysates of 293T cells transfected with indicated OCLN deletant together with an E2 expressing plasmid (CMV-E1E2, H77) were immunoprecipitated with anti-Flag affinity resin. Inputs as well as immunoprecipitates were immunoblotted with indicated antibodies. The positions of Flag-OCLN/ΔN and ΔC were marked by a star sign.
Analysis of E2-OCLN direct binding using recombinant proteins
Results from the above study suggest that OCLN EL2 may harbor an interacting interface with HCV glycoprotein. To test this possibility, we generated a panel of recombinant proteins consisting of individual domains of OCLN or CLDN1 fused with human Fc. Fusion proteins were expressed in 293T cells and then secreted into the media, which could be further purified through affinity columns. The expressions and secretions of represented proteins are shown in Figure 5A.
Fig. 5.

OCLN does not bind soluble HCV E2 (sE2) directly. (A) Examples of production of human Fc (hFc) fusion proteins in 293T cells using a secretion system. Fusion proteins could be readily detected by western blotting using an anti-human secondary antibody in either cell lysates or supernatants. Notably, the OCLN-EL1-hFc expression is relatively low. To ensure the specific detection, a stop codon was introduced between the EL and hFc, which results in the production of proteins without hFc tag. (B) Purified Fc fusion proteins were bound to protein G beads first and then incubated with sE2. After wash, bound protein was eluted and immunoblotted with anti-E2 mAb, striped, and reprobed with anti-hFc. Only the hCD81 large extracellular loop (hCD81-LEL-hFc) could bind sE2. CLDN1-EL1(E48K)-hFc harbors an E→K mutation at residue 48.
To measure the direct binding between OCLN or CLDN1 and HCV E2, we performed pull-down assays using purified recombinant Fc fusion proteins and the soluble HCV E2 (sE2 aa 384–661). Strikingly, only CD81-LEL displayed robust binding to sE2, consistent with many previous observations (Fig. 5B). By contrast, none of other purified proteins bound to sE2 (Fig. 5B). The same observation could be made when the experiment was performed at pH of 5.7, suggesting that lowering pH alone does not induce direct binding of OCLN and E2 (data not shown). Because intracellular forms of E2(661) were found to bind CD81 with greater affinity than the extracellular forms (Flint et al., 2000), and because it was shown that E1E2 complexes efficiently bind to CD81 whereas truncated E2 is a weak binder (Cocquerel et al., 1999; Cocquerel et al., 2003), we repeated this part of the study using either intracellularly expressed or secreted full-length E2 or E1E2 and found the observations were reproducible (data not shown). Together, our data do not support a robust direct binding between OCLN and HCV glycoproteins. Rather, it appears that OCLN and E2 perhaps co-localized to an intracellular environment when co-expressed. In support of this notion, Benedicto et al. recently reported that HCV envelope components alter localization of hepatocyte TJ-associated proteins and promote OCLN retention in the endoplasmic reticulum (ER) (Benedicto et al., 2008).
OCLN associates with Dynamin II
One possible role of OCLN in virus entry is that it directly or indirectly mediates the internalization of virions as reported in group B coxsackieviruses (CVB) entry (Coyne et al., 2007). We previously found that treatment of Huh7 cells with a Dynamin inhibitor, Dynasore, significantly inhibited HCVpp entry (Liu et al., 2009). Dynamins are known GTPases that are essential for the internalization of clathrin-coated vesicles (Praefcke and McMahon, 2004). Dynamin I is primarily expressed in neurons, whereas Dynamin II is ubiquitously expressed (Praefcke and McMahon, 2004). To define the role of Dynamin in HCV entry, we expanded the study by using four different treatment protocols and found that addition of Dynasore prior or at the same time of virus infection suppressed HCVpp, HCVcc, as well as VSV-Gpp entry (Fig. 6), which is known to be Dynamin-dependent in certain cell types (Cureton et al., 2009). For HCVcc, addition of Dynasore post-infection even had an inhibitory effect, implying Dynamin may be important for post-entry events as well. Further experiments also demonstrated that Dynasore pre-treatment as short as two hours was sufficient to exert its inhibitory effect (Supplementary Fig. 3). Of note, Dynasore treatment could not completely block the HCVpp infection, which may be either due to incomplete blockage of Dynamin activity, or the existence of a Dynamin-independent entry route. To validate these observations, we knocked down the endogenous Dynamin II in Huh7.5.1 cells by short-hairpin-based RNAi (shRNA). As shown in Figure 7, lentivirus-mediated shRNA targeting Dynamin II specifically reduced the level of Dynamin II. Consistently, HCVpp and HCVcc infection decreased by 90% in Dynamin II knockdown cells. Reduction of endogenous Dynamin II also lowered the VSV-Gpp entry by 50%. By contrast, infection of cells by HIV pseudovirions bearing HTLV-1 envelop protein (HTLVpp) was not affected. Together, these data confirmed that HCV entry is a Dynamin-dependent event.
Fig. 6.

Dynasore inhibits both HCVpp and HCVcc entry. (A) Schematic drawing of the experimental setup. Huh7.5.1 cells were treated with Dynasore or DMSO under four different protocols, differing in duration of the treatment and the time when added. A series of concentrations were tested and the selected Dynasore concentration (80 μM) was tested not to exert noticeable effect on cell growth. (B) Treated Huh7.5.1 cells were analyzed for relative susceptibility to HCVpp, HCVcc-luc, and VSV-Gpp. All results are expressed as percent infection to the DMSO treated cells. Data are presented as mean ± standard deviation. *** p<0.0005, ** p<0.005, * p<0.05.
Fig. 7.
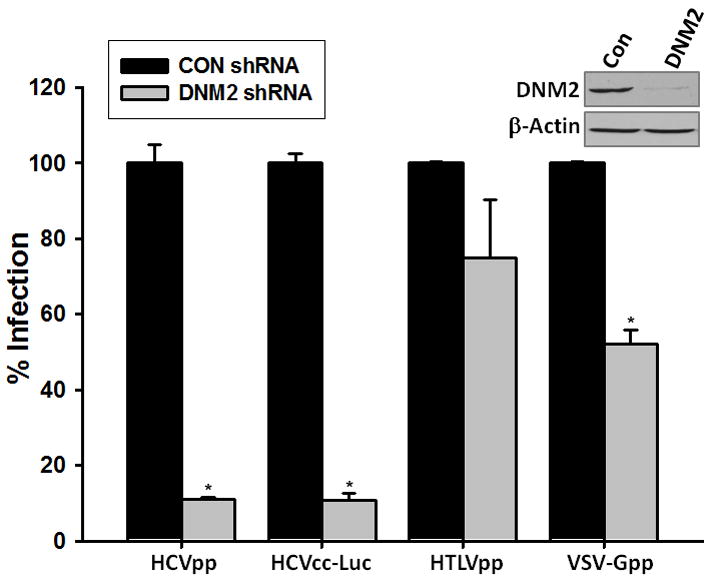
Silencing Dynamin II ablates HCV infection. Huh7.5.1 cells were transduced by lentivirus carrying Dynamin II (DNM2) or a scrambled (CON) shRNA. After 48 hours incubation, cells were infected by HCVpp, VSV-Gpp, HCVcc-Luc, and HTLVpp. Luciferase activity was measured at 24 hour post-infection. Data are presented as mean ± standard deviation. * p<0.05. (Inset) Western blot analysis of cells 48hr after transduction with CON or DNM2 shRNA. The specific knockdown of endogenous DNM2 by DNM2 shRNA was confirmed.
To investigate whether OCLN associates with Dynamin II, we co-expressed GFP-Dynamin and Flag tagged OCLN in 293T cells. Shown in Figure 8A, OCLN readily precipitated with Dynamin II (DNM2) and the dominant negative (DN) form of Dynamin II (DNM2 K44A). Importantly, such an association disappeared in OCLN/ΔEL2 transfected cells (Fig. 8A), but not in cells that were transfected with OCLN/ΔEL1, ΔN, and ΔC mutants. Interestingly, Flag-CLDN1 also precipitated with DNM2, but Flag-CD81 failed to do so. Therefore, the interactions are unlikely due to overexpression of recombinant proteins or to the tag added for the pull-down (Fig 8B).
Fig. 8.

Overexpressed OCLN precipitates with Dynamin II. (A) Flag-OCLN or deletants were co-transfected with GFP-Dynamin II (GFP-DNM2) or GFP-Dynamin II K44A (GFP-DNM2K44A) into 293T cells. Same amounts of cell lysates were immunoprecipitated with anti-Flag affinity resin and blotted for Dynamin II or Flag-tagged proteins. The positions of Flag-OCLN/ΔN and ΔC were indicated with star signs. (B) GFP-DNM2 was co-transfected with Flag-CD81, CLDN1, OCLN, OCLN/ΔEL1, respectively, into 293T cells. Immunoprecipitation and immunoblotting were carried out as described in (A).
Dynamin is required for HCV entry into polarized PHHs
Because those commonly used hepatoma cell lines are phenotypically distinct from hepatocytes in vivo, we repeated the above studies using polarized primary human hepatocytes (PHHs) in order to rule out the possibility that the observation is merely cell line specific. When seeded in collagen-coated or Matrigel overlaid plates, PHHs can develop into complex polarized cell monolayer, exhibiting visible bile canaliculi between adjacent hepatocytes (Fig 9A and Supplementary Fig. 4). Confocal microscopic characterization of the cells revealed polarized distribution of TJ protein CLDN1, OCLN, and the apical surface marker, CD26 (Fig. 9B, C, F). CD81, on the other hand, displayed a basolateral localization pattern (Fig. 9D). The staining of adherens junction protein, E-cadherin, was seen in lateral surface and concentrated at AJ-like structures (Fig. 9E). Altogether, such complex orientation of polarity is in sharp contrast to the simple (columnar) apical-basolateral polarity observed in intestinal epithelial cells and in other commonly used hepatoma cell lines (Brazzoli et al., 2008; Coyne, Kim, and Bergelson, 2007; Mee et al., 2008; Yang et al., 2008). Finally, treatment of PHHs with Dynasore significantly inhibited HCVpp entry (Fig. 9G), confirming the important role of Dynamin in virus entry. To ensure that the inhibitory effect was not a result of Dynasore-associated cytotoxicity, we determined the cell viability using the CytoVial Glo system. Shown in Figure 10, Dynasore at 80 μM did not exert any adverse effect on Huh7.5.1 cell viability even after 24hrs incubation.
Fig. 9.

Dynasore treatment inhibits HCVpp entry into polarized primary human hepatocytes (PHHs). (A) Phase contrast image of PHHs (48 hours after culturing on collagen-coated plates). Bile canaliculi were visible as widened gaps between cells (white arrows). For confocal images, cell were fixed in methanol and stained for CLDN1 (B), OCLN (C), CD81 (D), E-cadherin (E-cad) (E), and CD26 (F). In (F), both OCLN and CD26 were stained to show the relative location of TJ (OCLN, green) and apical membrane (CD26, red). (G) Pretreatment of PHHs with Dynasore (12 hrs, 80 μM) strongly inhibited HCVpp and VSV-Gpp entry. Data are shown as mean ± standard deviation. ** p<0.005, * p<0.05.
Fig. 10.
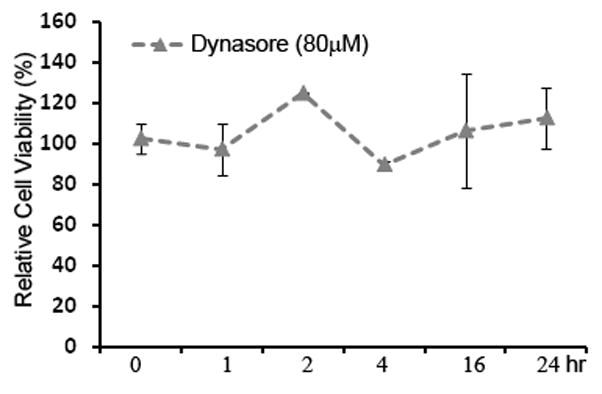
Dynasore has no adverse effect on cell viability. PHHs were treated with Dynasore (80μM) for 0, 1, 2, 4, 16, 24 hours. After removal of Dynasore, cells were incubated for additional 48 hours prior to the CellTiter- Glo cell viability assay (see Materials and Methods). Data are shown as mean ± standard deviation.
Discussion
Current model suggests that HCV entry into host cells is a multi-step event requiring four essential host factors including SR-BI, CD81, CLDN1, and OCLN. The question that exactly how CLDN1 and OCLN are involved in this stepwise entry process remains unanswered, despite some evidence pointing to a late step (Benedicto et al., 2009; Evans et al., 2007). Published data from several groups including us have indicated that a fraction of surface CLDN1 associates with CD81 (Brazzoli et al., 2008; Harris et al., 2010; Harris et al., 2008; Kovalenko, Yang, and Hemler, 2007; Yang et al., 2008) in multiple cell types. Most recently, it was reported that CLDN1 association with CD81 defines HCV entry (Harris et al., 2010). Yet, when Flag-hCD81 and OCLN or CLDN1 were co-expressed in 293T cells, CD81 selectively precipitated with CLDN1, but not OCLN (Supplementary Fig. 5), implying CLDN1 and OCLN play distinct roles in HCV entry. In the present study, we present two novel findings with regard to the specific role of OCLN in mediating HCV entry. First, although OCLN precipitates with HCV E2 in transfected cells, recombinant sE2 and OCLN ELs do not bind to each other. Second, OCLN forms complex with Dynamin, and HCV infection is sensitive to Dynamin inhibition. These findings reminisce of CVB entry, where CVB entry across epithelial TJ requires OCLN but OCLN does not interact directly with the virus, and CVB-induced macropinocytosis of OCLN appears to be a separate event from CVB-CAR interaction (Coyne et al., 2007).
Our study indicate that the EL2 domain plays a pivotal role in OCLN-dependent HCV entry. Ploss et al. previously demonstrated that OCLN EL2 harbors species-specific determinant for HCV entry (Ploss, 2009). Here, we show that OCLN EL2 is both required to confer HCVpp infectivity and its ability to precipitate with HCV E2. Unexpectedly, removal of C-terminus of OCLN did not impair its ability to mediate HCVpp infection of 786-O cells. We therefore infer that the C-terminus-dependent protein-protein interactions are not essential for HCVpp entry. It must be noted, however, that even though all OCLN deletants successfully located to plasma membrane, addition of different tags to OCLN does seem to have subtle effects on the topology of derived proteins. As shown in Figures 1, Flag-tagged OCLN/ΔN was not detected well by anti-Flag antibody, whereas GFP-tagged OCLN/ΔC displayed pronounced cytoplasmic distribution (Fig. 2) and was less effective in mediating HCVpp entry. Thus, it is possible that the topology of the mutants has altered in ways that are immeasurable in our study. Moreover, 786-O cells do express very low levels of endogenous OCLN (Supplementary Fig. 6), which could facilitate the deletant’s assembly to cell surface by compensating for the loss of C-terminus in the exogenous counterpart.
Both our previous report (Liu et al., 2009) and the current study show that OCLN precipitates with HCV E2 in infected or transfected cells. However, the observation that individually purified Fc-tagged OCLN ELs failed to robustly bind sE2 in the in vitro pull-down assay disfavors a direct interaction between HCV glycoproteins and surface OCLN. The discrepancy may reflect the nature of the multi-step HCV entry process. It is possible that HCV virions need to bind another surface molecule(s) prior to association with junctional OCLN. Nonetheless, addition of recombinant CD81 into the system failed to foster the OCLN-E2 direct binding either (data not shown). Owing to the relatively large size, addition of Fc to a protein may alter its conformation. Moreover, taking EL1 or EL2 out of the context of the full-length OCLN may result in the loss of its native conformation. As a result of this, recombinant proteins produced in the current study may not possess the proper conformation required for the direct binding. In spite of these caveats, purified Fc-tagged CD81-LEL strongly bound to sE2 in our system, confirming those previous reports (Cocquerel et al., 2003; Flint et al., 1999). Collectively, these results suggest that either OCLN is indirectly involved in HCV entry, or its association with HCV particles only occurs at some point during viral entry, which cannot be measured in the current study system.
As an integral TJ protein, OCLN was originally thought to be necessary but later found to be dispensable for the formation of TJ (Furuse et al., 1993; Saitou et al., 2000; Schulzke et al., 2005). The observation that OCLN internalization is required for and accompanied to CVB entry hints that OCLN may be a critical protein linking virions to endocytic pathways (Coyne et al., 2007). Indeed, TJs comprise a network of cytoplasmic scaffolding proteins that provide links to the cytoskeleton and to intracellular signaling molecules (Balda and Matter, 2008; Tsukita et al., 2008). Three modes of internalization have been reported to occur to TJ proteins: clathrin-mediated endocytosis, caveolar-mediated pathway, and macropinocytosis (Bruewer et al., 2005; Capaldo and Nusrat, 2009; Fujita et al., 2000; Ivanov, Nusrat, and Parkos, 2004). Notably, no known endocytic motif can be found in OCLN. Here, we provided evidence that OCLN precipitated with Dynamin II and blocking Dynamin activity inhibited HCV entry. The association between Dynamin II and OCLN has been previously reported at the blood-testis barrier in adult rat testes (Lie et al., 2006). Interestingly, we found that the EL2 of OCLN is needed for its association with Dynamin II. Whereas it is difficult to envision how EL2 could physically interact with Dynamin, because Dynamin II locates to the cytoplasmic side of cell membrane and EL2 is topologically outside of plasma membrane, perhaps the association between Occludin and Dynamin requires the presence of a third molecule that spans the two cellular compartments (extracellular-space, membrane/cytosol). The observation that Flag-CLDN1, but not Flag-CD81, could also precipitated Dynamin in a Co-IP experiment (Fig. 8B) further suggests a close association between TJ network and Dynamin.
Dynamin is a well-known regulator of endocytosis and actin cytoskeleton (Praefcke and McMahon, 2004). By maintaining a balance of membrane protein endocytosis and intracellular membrane traffic, Dynamin II was recently shown to play an important role in orchestrating epithelial cell polarity (Chua, Rikhy, and Lippincott-Schwartz, 2009). Depletion of Dynamin II, the only Dynamin in epithelial cells, prevents junctional formation and induces abnormal actin assembly (Chua, Rikhy, and Lippincott-Schwartz, 2009). Interestingly, it was recently reported that TNF-induced OCLN endocytosis is also a Dynamin-dependent process (Marchiando et al.). Perhaps, through its association with Dynamin, OCLN could be an important bridge between surface protein complex and endocytic machinery, even though it remains to be tested whether the association of OCLN and Dynamin is absolutely necessary for HCV entry. More analysis is needed to address whether surface OCLN is physically engaged with incoming HCV particles during the sequential entry process and/or facilitate virus internalization.
Materials and methods
Cell lines and Reagents
The human kidney epithelial cell line Lenti-X 293T was purchased from Clontech. The human renal carcinoma cell line 786-O (CRL-1932), human breast cancer cell line BT-549 (HTB-122), and human colorectal adenocarcinoma cell line HT-29 (HTB-38) were purchased from American type cell culture (ATCC). The human liver cell line Huh7 and its derivative clone Huh 7.5 were obtained from Apath, Inc. with permission from Dr. Charles Rice (Rockefeller University). The Huh7.5.1 line generated from a cured HCV replicon cell line was provided by Dr. Francis Chisari (Scripps Research Institute) (Zhong et al., 2005). All cell lines were maintained in DMEM supplemented with 5% Penicillin and streptomycin, 1% NEAA, and 10% fetal bovine serum (FBS) (Atlanta Biologicals). Primary human hepatocytes (PHHs) were isolated from healthy donor livers that were not used for liver transplantation. The detailed isolation methods have been described previously (Strom et al., 1996) with modifications as outlined in (Komoroski et al., 2004). PHHs were maintained in hepatocyte maintenance medium supplemented with 10−7 M insulin and dexamethasone (Lonza).
Antibodies were obtained from Dr. Jane McKeating (E2, clone 3/11); Dr. Harry Greenberg (anti-E1, A4); Zymed (anti-CLDN1, clones 2H10D10 and JAY.8; anti-OCLN, Cat. No. 71–1500); Santa Cruz (anti-CD81, clone 5A6); BD Biosciences (anti-CD81, JS-81 clone; anti-CD26); Cell Signaling (anti-E-cad, product #4065; anti-Dynamin, product #2342); BD transduction (anti-OCLN, mAB); and Sigma (anti-Flag M2 and β-actin). Secondary antibodies are purchased from Santa Cruz, Jackson ImmunoResearch Laboratories, Inc, and Molecular Probes (Invitrogen). The specific Dynamin inhibitor, Dynasore, was purchased from Sigma.
DNA constructs
pTrip-eGFP and pCMV-dR8.2 were kindly provided by Dr. Guangxia Gao (Institute of Biophysics, Chinese Academy of Sciences). Trip-luc was constructed by replacing eGFP with the firefly luciferase gene between BamHI and XhoI sites. The coding sequence of firefly luciferase was PCR amplified from the pGL4-Luc vector (Promega) with primers: 5′-AGAGGATCCACCGGTCGCCACCATGGAAGATGCCAAAAAC-3′ (sense) and 5′-ATAGCTCGAGTTAGACGTTGATCCTGGCGC-3′ (anti-sense). Flag-hCD81 and Flag-hCLDN1 were previously constructed in the lab containing double Flag tags and a calmodulin binding peptide (results in ~5kd increase in size). pTrip Flag-OCLN, pTrip Flag-OCLN/ΔN, pTrip Flag-OCLN/ΔC, pTrip Flag-OCLN/ΔEL1, and pTrip Flag-OCLN/ΔEL2 express Flag-tagged full-length human OCLN (hOCLN), OCLN/ΔN(deletion of aa 1–63), OCLN/ΔC (deletion of aa 267–522), OCLN/ΔEL1(deletion of aa 94–128), and OCLN/ΔEL2 (deletion of aa 200–238), respectively. The full length OCLN fragment was originally amplified from the ViraPort human liver cDNA library (Stratagene) and cloned into a modified Trip vector between EcoRV and XhoI sites. Truncated fragments were subsequently amplified by PCR using the following primers with the restriction sites in italics:
OCLN-F: 5′-GGCGATATCTCATCCAGGCCTCTTGAA-3′
OCLN/ΔN-F: 5′-GGCGATATCGTGATTCGGATCCTGTCT-3′
OCLN/ΔC-R: 5′-ATATAGCTCGAGTTATTTCACAGCAAAGAAAAT-3′
OCLN/ΔEL1-F: 5′-GCAGAGGCTATGGAGGCTATACAGACCCAAGA-3′
OCLN/ΔEL1-R: 5′-TGGGTCTGTATAGCCTCCATAGCCTCTGTCCCA-3′
OCLN/ΔEL2-F: 5′-ATGGGAGTGAACCCAGTGGATCCCCAGGAGGCC-3′
OCLN/ΔEL2-R: 5′-CTCCTGGGGATCCACTGGGTTCACTCCCATTAT-3′
OCLN-R: 5′-GACTCTCGAGCTATGTTT TCTGTCTATCATAGTCTC – 3′
For those fluorescent OCLN constructs, Flag sequence was replaced with the enhanced GFP.
Production of pseudoviral particles and HCVcc
In order to produce the pseudovirus, Lenti-X 293T cells were seeded one day before transfection at 6 × 105 cells in a 6 cm plate in 4 ml of DMEM containing 10% FBS. The next day cells were transfected using polyethylenimine (PEI, MW 25Kd, PolySciences, Inc.) (Rothwangl et al., 2008) or lipofectamine 2000 reagent (Invitrogen). HCVpp and VSV-Gpp were packaged by transfecting with DNA mixture (0.5 ml) comprising 2 μg of pTrip-Luc, 2 μg pCMV-dR8.2, and 2 μg of phCMV-HCV E1E2 (a generous gift from Dr. F. Cosset, INSERM, France, (Bartosch, Dubuisson, and Cosset, 2003)) or 1 μg of pHEF-VSV-G (expressing VSV-G Env protein). 16 hours after transfection, media were replaced, and supernatants containing HCVpp were typically harvested 36–48 h after transfection and then filtered through 0.45 μm syringe filter. Pseudotyped virus bearing the human T-cell leukemia virus type I envelope protein (HTLVpp) was packaged by the two-plasmid system consisting of 2 μg of pNL4.3-Luc-E-R- construct and 2 μg of HTLV Env (obtained from Dr. Fanning-Heidecker, NCI). In our study, non-enveloped lentivirus particles (Bald virus) were also made as the negative control. VSV-G pseudotyped lentiviruses carrying shRNA against human Dynamin II (Sigma, target sequence: 5′-AGTCCTACATCAACACGAA-3′) and the scramble shRNA (Addgene plasmid 1864) were similarly produced. Production procedure of HCVcc expressing luciferase that was inserted between NS5A and NS5B (sequence available upon request) (JFH1 strain) was described elsewhere (Liu et al., 2009; Yang, 2008; Yang et al., 2008).
HCVpp Infection assay
To conduct the infection assay, Huh 7.5.1 or other cell lines were seeded in a 48-well plate at the density of 5×104/well the day before transduction. The next day, 200 μl of supernatant containing HCVpp or Bald virus was added into each well in the presence of 4 μg/ml of polybrene and further incubated for 3–6 hours followed by media change. Approximately 36 hours after transduction, cells were lysed in 100 μl of passive lysis buffer, and 20–50 μl of lysates was incubated with 50 μl of luciferase assay buffer and luciferase activity was determined by a Veritas luminometer according to the manufacturer’s instruction (Promega luciferase assay kit). We typically obtain counts ranging from 10,000–900,000, whereas the background signal from a Bald virus infected sample is usually below 100.
Western blotting and Immunoprecipitation
For cell lysate preparation, monolayer cells were lysed with lysis buffer (50 mM Tris-HCl, pH7.5, 150mM NaCl, 1% Nonidet P40, 50 mM NaF, 1 mM Na3VO4, 5 mM β-glycerophosphate, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride) supplemented with protease inhibitor cocktail (Sigma or Pierce) on ice. The lysate was cleared by centrifuging at 14,000×g for 20 minutes. Boiled samples in 2×SDS loading buffer were loaded onto a 9–12% polyacrylamide gel. After electrophoresis, the separated proteins were transferred onto a nitrocellulose membrane (Biorad, Hercules, CA). The resulting blots were blocked within 10% milk for 1 h, and then incubated with the primary antibody overnight at 4°C. The secondary antibody used in the immunoblot was a 1:2000 dilution of HRP-linked anti-IgG. The ECL reagent (Amersham Biosciences, Piscataway, NJ) was used as the substrate for detection. For immunoprecipitation-coupled western blotting, the cleared cell lysates were immunoprecipitated with the anti-FLAG M2 affinity resin (Sigma) and washed with lysis buffer. Eluted samples were separated by 9–12% SDS-PAGEs and immunoblotted with indicated antibodies.
Production of Fc fusion proteins
To clone individual fragments, CD81 LEL (aa 116–202), CLDN1 EL1 (aa 29–81), CLDN1 EL2 (aa 139–164), CLDN6 EL1 (aa 29–81), CLDN7 EL1 (aa 29–81), OCLN EL1 (aa 92–132) and EL2 (aa 196–244), sE2 (aa 384–661 from H77 clone) were amplified by PCR using corresponding primers and cloned in the pFuse-IL-2ss-hFc vector (Invivogen) between EcoRV/BglII sites.
80% confluent 293T cells were transfected with 5 μg DNA plasmids expressing Fc fusion proteins by 10 μl of PEI (1μg/μl) in 60mm plates. 4~5 hours post-transfection, media were replaced with the serum-free 293 freestyle expression medium (Invitrogen). After an additional 48 hours’ incubation, supernatants were collected and cleared by centrifugation at 12,000 rpm for 15 minutes. The supernatants were purified through protein A affinity beads and stored at −80°C in the presence of proteinase inhibitors (Roche) until use.
To perform in vitro pull-down assays, 12 μl of Protein G beads were added to a 1.5ml tube and pre-equilibrated in 800 μl ice-cold 1×PBS. We then added around 200 μg protein lysates to the beads and placed the tube on a rotator in a cold room for 3–4 hrs at a slow speed. After incubation, the beads were spun down at 4000 rpm and washed three times in 800 μl ice-cold 1× PBS. Bound proteins were eluted by boiling the beads in 35 μl of 2× SDS sample buffer and then assayed by western blotting.
Immunofluorescence staining and Confocal Microscopy
Transfected Lenti-X 293T were fixed in 2% paraformaldehyde (15 min, RT) and directly imaged. For PHHs, cells were fixed in ice-cold methanol (5 min, RT), and then permeablized with 0.1% (v/v) Triton X-100 in PBS three times (PBST; 10 min, RT), blocked in 0.3% (w/v) bovine serum albumin (BSA) in PBS (1h, RT), and subsequently incubated in PBS (1h, RT) with appropriately diluted primary antibodies against CLDN1 (1:80), OCLN (1:100), CD81 (1:250), CD26 (1:200), E-cadherin (1:100). Samples were washed in PBS and subsequently incubated with Alexa 488 or Alexa 568 secondary antibodies diluted in PBS (1:500). Samples were mounted on a slide using a homemade Gelvatol mounting medium. Images were captured on a Carl Zeiss Meta LSM 510 confocal microscope (40× Plan-Neo/1.3 NA Oil) and edited by Photoshop (Adobe). Nucleus was stained with Draq5 (Biostatus, United Kingdom). Z-stack images were acquired at 0.3 μm intervals. Orthogonal views (XY, XZ, YZ) were presented in the figure.
Cell surface biotinylation assay
293T cells (6×106) were first transfected with indicated OCLN constructs. 48 hours post-transfection, cells were washed three times with ice cold PBS and resuspended in PBS at a density of 25×106 cells/ml. Freshly prepared Sulfo-NHS-SS-biotin (Pierce) was added to the cells (final concentration 0.5μg/ml) and allowed to incubate at 4°C for 30minutes. Cells were then washed three times with ice cold PBS. 25mM Tris (pH8.0) was added in the initial wash to quench any non-reacted biotin reagent. Following cell lysis in RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, 2 mM Na3VO4 and Pierce protease inhibitor cocktail), lysates were cleared by centrifugation at 13,000 × g for 15 min at 4°C. The cleared lysates were used for immunoprecipitation using a 1:1 mixture of Streptavidin beads (Pierce). Beads were washed three times with RIPA buffer, and bound proteins were eluted by boiling the samples in SDS-PAGE sample buffer and then resolved on 9% SDS-PAGE. Biotinylated proteins were detected by anti-Flag or anti-Occludin antibodies.
Cell viability assay
PHHs (105 per well) were treated with Dynasore (80 μM) for the indicated period of time. After removal of Dynasore, cells were incubated for an additional 48 hrs in 24-well plates. The numbers of viable cells in culture were determined using the CellTiter-Glo Cell Viability Luminescent Assay kit according to the manufacturer’s instruction (Promega). This luciferase-based assay system quantitatively measures the cellular ATP level, an indicator of metabolically active cells.
Statistical analysis
Bar graphs were plotted to show mean ± standard deviation (SD). Statistical analyses were performed using SigmaPlot 10. A p value of <0.05 in the Student’s test was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Drs. T. Wakita, H. Greenberg, C. Rice, F. Chisari, F. Cosset, G. Luo, R. Bartenschlager, G. Gao, J. Dubuisson, S. Foung, C. Coyne, G. Fanning-Heidecker, and J. McKeating for providing cell lines and reagents. This work was supported by grants from University of Pittsburgh Faculty Research Development Funds and NIH (R21AI083389) (to T.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balda MS, Matter K. Tight junctions at a glance. J Cell Sci. 2008;121(Pt 22):3677–82. doi: 10.1242/jcs.023887. [DOI] [PubMed] [Google Scholar]
- Bamforth SD, Kniesel U, Wolburg H, Engelhardt B, Risau W. A dominant mutant of occludin disrupts tight junction structure and function. J Cell Sci. 1999;112 (Pt 12):1879–88. doi: 10.1242/jcs.112.12.1879. [DOI] [PubMed] [Google Scholar]
- Bartosch B, Cosset FL. Cell entry of hepatitis C virus. Virology. 2006;348(1):1–12. doi: 10.1016/j.virol.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med. 2003;197(5):633–42. [Google Scholar]
- Benedicto I, Molina-Jimenez F, Barreiro O, Maldonado-Rodriguez A, Prieto J, Moreno-Otero R, Aldabe R, Lopez-Cabrera M, Majano PL. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology. 2008;48(4):1044–53. doi: 10.1002/hep.22465. [DOI] [PubMed] [Google Scholar]
- Benedicto I, Molina-Jimenez F, Bartosch B, Cosset FL, Lavillette D, Prieto J, Moreno-Otero R, Valenzuela-Fernandez A, Aldabe R, Lopez-Cabrera M, Majano PL. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J Virol. 2009;83(16):8012–20. doi: 10.1128/JVI.00038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80(14):6964–72. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazzoli M, Bianchi A, Filippini S, Weiner A, Zhu Q, Pizza M, Crotta S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J Virol. 2008;82(17):8316–29. doi: 10.1128/JVI.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. Faseb J. 2005;19(8):923–33. doi: 10.1096/fj.04-3260com. [DOI] [PubMed] [Google Scholar]
- Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788(4):864–71. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua J, Rikhy R, Lippincott-Schwartz J. Dynamin 2 orchestrates the global actomyosin cytoskeleton for epithelial maintenance and apical constriction. Proc Natl Acad Sci U S A. 2009;106 (49):20770–20775. doi: 10.1073/pnas.0909812106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquerel L, Duvet S, Meunier JC, Pillez A, Cacan R, Wychowski C, Dubuisson J. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J Virol. 1999;73(4):2641–9. doi: 10.1128/jvi.73.4.2641-2649.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocquerel L, Kuo CC, Dubuisson J, Levy S. CD81-dependent binding of hepatitis C virus E1E2 heterodimers. J Virol. 2003;77(19):10677–83. doi: 10.1128/JVI.77.19.10677-10683.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codran A, Royer C, Jaeck D, Bastien-Valle M, Baumert TF, Kieny MP, Pereira CA, Martin JP. Entry of hepatitis C virus pseudotypes into primary human hepatocytes by clathrin-dependent endocytosis. J Gen Virol. 2006;87(Pt 9):2583–93. doi: 10.1099/vir.0.81710-0. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Kim KS, Bergelson JM. Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. Embo J. 2007;26(17):4016–28. doi: 10.1038/sj.emboj.7601831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, Shen L, Turner JR, Bergelson JM. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe. 2007;2 (3):181–92. doi: 10.1016/j.chom.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Massol RH, Saffarian S, Kirchhausen TL, Whelan SP. Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog. 2009;5(4):e1000394. doi: 10.1371/journal.ppat.1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446(7137):801–5. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- Feldman GJ, Mullin JM, Ryan MP. Occludin: structure, function and regulation. Adv Drug Deliv Rev. 2005;57(6):883–917. doi: 10.1016/j.addr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Flint M, Dubuisson J, Maidens C, Harrop R, Guile GR, Borrow P, McKeating JA. Functional characterization of intracellular and secreted forms of a truncated hepatitis C virus E2 glycoprotein. J Virol. 2000;74(2):702–9. doi: 10.1128/jvi.74.2.702-709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, Higginbottom A, Levy S, McKeating JA. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J Virol. 1999;73(8):6235–44. doi: 10.1128/jvi.73.8.6235-6244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tsukita S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett. 2000;476(3):258–61. doi: 10.1016/s0014-5793(00)01744-0. [DOI] [PubMed] [Google Scholar]
- Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123(6 Pt 2):1777–88. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris HJ, Davis C, Mullins JG, Hu K, Goodall M, Farquhar MJ, Mee CJ, McCaffrey K, Young S, Drummer H, Balfe P, McKeating JA. Claudin association with CD81 defines hepatitis C virus entry. J Biol Chem. 2010 Apr 7; doi: 10.1074/jbc.M110.104836. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris HJ, Farquhar MJ, Mee CJ, Davis C, Reynolds GM, Jennings A, Hu K, Yuan F, Deng H, Hubscher SG, Han JH, Balfe P, McKeating JA. CD81 and claudin 1 coreceptor association: role in hepatitis C virus entry. J Virol. 2008;82(10):5007–20. doi: 10.1128/JVI.02286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A. 2003;100(12):7271–6. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AI, Nusrat A, Parkos CA. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell. 2004;15(1):176–88. doi: 10.1091/mbc.E03-05-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohaar I, Ploss A, Korol E, Mu K, Schoggins JW, O’Brien TR, Rice CM, Prokunina-Olsson L. Splicing diversity of human OCLN gene and its biological significance for hepatitis C virus (HCV) entry. J Virol. 2010 May 12; doi: 10.1128/JVI.00196-10. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komoroski BJ, Zhang S, Cai H, Hutzler JM, Frye R, Tracy TS, Strom SC, Lehmann T, Ang CY, Cui YY, Venkataramanan R. Induction and inhibition of cytochromes P450 by the St. John’s wort constituent hyperforin in human hepatocyte cultures. Drug Metab Dispos. 2004;32(5):512–8. doi: 10.1124/dmd.32.5.512. [DOI] [PubMed] [Google Scholar]
- Kovalenko OV, Yang XH, Hemler ME. A Novel Cysteine Cross-linking Method Reveals a Direct Association between Claudin-1 and Tetraspanin CD9. Mol Cell Proteomics. 2007;6 (11):1855–67. doi: 10.1074/mcp.M700183-MCP200. [DOI] [PubMed] [Google Scholar]
- Lie PP, Xia W, Wang CQ, Mruk DD, Yan HH, Wong CH, Lee WM, Cheng CY. Dynamin II interacts with the cadherin- and occludin-based protein complexes at the blood-testis barrier in adult rat testes. J Endocrinol. 2006;191(3):571–86. doi: 10.1677/joe.1.06996. [DOI] [PubMed] [Google Scholar]
- Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T. Tight Junction Proteins Claudin-1 and Occludin Control Hepatitis C Virus Entry and are Downregulated during Infection to Prevent Superinfection. J Virol. 2009;83(4):2011–2014. doi: 10.1128/JVI.01888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter K, Balda MS. Biogenesis of tight junctions: the C-terminal domain of occludin mediates basolateral targeting. J Cell Sci. 1998;111 (Pt 4):511–9. doi: 10.1242/jcs.111.4.511. [DOI] [PubMed] [Google Scholar]
- Mee CJ, Grove J, Harris HJ, Hu K, Balfe P, McKeating JA. Effect of cell polarization on hepatitis C virus entry. J Virol. 2008;82(1):461–70. doi: 10.1128/JVI.01894-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L, Bertaux C, Dragic T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol. 2006;80 (23):11571–8. doi: 10.1128/JVI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. Binding of hepatitis C virus to CD81. Science. 1998;282(5390):938–41. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- Ploss A, Evans Matthew J, Gaysinskaya Valeriya A, Panis Maryline, You Hana, de Jong Ype P, Rice Charles M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–6. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5(2):133–47. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- Rothwangl KB, Manicassamy B, Uprichard SL, Rong L. Dissecting the role of putative CD81 binding regions of E2 in mediating HCV entry: putative CD81 binding region 1 is not involved in CD81 binding. Virol J. 2008;5:46. doi: 10.1186/1743-422X-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11(12):4131–42. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. Embo J. 2002;21(19):5017–25. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulzke JD, Gitter AH, Mankertz J, Spiegel S, Seidler U, Amasheh S, Saitou M, Tsukita S, Fromm M. Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta. 2005;1669(1):34–42. doi: 10.1016/j.bbamem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Strom SC, Pisarov LA, Dorko K, Thompson MT, Schuetz JD, Schuetz EG. Use of human hepatocytes to study P450 gene induction. Methods Enzymol. 1996;272:388–401. doi: 10.1016/s0076-6879(96)72044-x. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Yamazaki Y, Katsuno T, Tamura A. Tight junction-based epithelial microenvironment and cell proliferation. Oncogene. 2008;27(55):6930–8. doi: 10.1038/onc.2008.344. [DOI] [PubMed] [Google Scholar]
- Van Itallie CM, Anderson JM. Occludin confers adhesiveness when expressed in fibroblasts. J Cell Sci. 1997;110 (Pt 9):1113–21. doi: 10.1242/jcs.110.9.1113. [DOI] [PubMed] [Google Scholar]
- Wang Z, Mandell KJ, Parkos CA, Mrsny RJ, Nusrat A. The second loop of occludin is required for suppression of Raf1-induced tumor growth. Oncogene. 2005;24(27):4412–20. doi: 10.1038/sj.onc.1208634. [DOI] [PubMed] [Google Scholar]
- Wong V, Gumbiner BM. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997;136(2):399–409. doi: 10.1083/jcb.136.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hood Brian L, Chadwick Sara L, Liu Shufeng, Watkins Simon C, Luo Guangxiang, Conrads Thomas P, Wang Tianyi. Fatty acid synthase is upregulated during HCV infection and regulates HCV entry and production. Hepatology. 2008;48(5):1396–403. doi: 10.1002/hep.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Qiu C, Biswas N, Jin J, Watkins SC, Montelaro RC, Coyne CB, Wang T. Correlation of the tight junction-like distribution of Claudin-1 to the cellular tropism of hepatitis C virus. J Biol Chem. 2008;283(13):8643–53. doi: 10.1074/jbc.M709824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102(26):9294–9. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.