

Abstract
Legionella pneumophila possesses a large arsenal of type IV translocated substrates. Over 100 such proteins have been identified, but the functions of most are unknown. Previous studies have demonstrated that L. pneumophila activates NF-κB, a master transcriptional regulator of the mammalian innate immune response. Activation of NF-κB is dependent on the Legionella Icm/Dot type IV protein translocation system, consistent with the possibility that translocated bacterial proteins contribute to this response. To test this hypothesis, an expression library of 159 known and putative translocated substrates was created to evaluate whether ectopic production of a single L. pneumophila protein could activate NF-κB in mammalian cells. Expression of two of these proteins, LnaB (Legionella NF-κB Activator B) and LegK1, resulted in ~150-fold induction of NF-κB activity in HEK293T cells, levels similar to the strong induction that occurs with ectopic expression of the known activator Nod1. LnaB is a substrate of the Icm/Dot system, and in the absence of this protein, a partial reduction of NF-κB activation in host cells occurs after challenge by post-exponential phase bacteria. These data indicate that LnaB is an Icm/Dot substrate that contributes to NF-κB activation during L. pneumophila infection in host cells.
Introduction
Legionella pneumophila is a Gram negative facultative intracellular bacterial pathogen. Upon inhalation from contaminated water sources L. pneumophila can replicate in human alveolar macrophages and epithelial cells leading to a severe pneumonia known as Legionnaires’ disease (Chiaraviglio et al., 2008, Fraser, 2005). In the host cell, L. pneumophila replicates within a membrane bound vacuole that avoids fusion with late endosomes and lysosomes (Horwitz, 1983b, Horwitz et al., 1984). Instead, the Legionella-containing vacuole (LCV) recruits ER-derived secretory vesicles to its surface, altering the morphology of the vacuole to a compartment resembling rough ER (Derre et al., 2004, Horwitz, 1983a, Kagan et al., 2002, Kagan et al., 2004, Tilney et al., 2001). Intracellular growth and evasion of lysosomal fusion depends on Icm/Dot, a chromosomally encoded type IV secretion system (Segal et al., 1998, Vogel et al., 1998). Icm/Dot is made up twenty-six gene products predicted to assemble into a multi-protein membrane spanning complex (Vincent et al., 2006).
Like other bacterial specialized secretion systems, Legionella’s Icm/Dot system allows the translocation of a large arsenal of proteins known as “Icm/Dot Translocated substrates” (IDTS) (Zusman et al., 2007). Studies over the last six years has led to the identification of approximately 100 proteins that may be delivered by the type IV secretion system into host cells (Kubori et al., 2008, Ninio et al., 2007). Of these IDTS, approximately 15 proteins have described activities that include altering vesicle trafficking (Ingmundson et al., 2007, Liu et al., 2007, Machner et al., 2006, Machner et al., 2007, Murata et al., 2006, Pan et al., 2008, Shohdy et al., 2005), promoting macrophage survival (Banga et al., 2007, Laguna et al., 2006), and inhibiting eukaryotic translation (Belyi et al., 2006, Belyi et al., 2008, de Felipe et al., 2005), and mediating host cell release (Chen et al., 2004). However, the majority of the identified IDTS are of unknown function and furthermore, deletion of a single IDTS gene rarely alters intracellular growth (Luo et al., 2004, Machner et al., 2006, Nagai et al., 2002, Ninio et al., 2005, Shohdy et al., 2005). The lack of phenotype is often attributed to the possibility that IDTS are functionally redundant to one another, with multiple proteins targeting a single pathway required for intracellular growth. This is also a common theme with secreted substrates of other bacterial pathogens. The deletion of individual substrates of the type III secretion systems from Shigella flexneri, Salmonella sp, and Enterohaemorrhagic Escherichia coli rarely has strong effects on the pathogenesis of these bacteria. Therefore, additional tools are needed to identify the activities of these translocated proteins and determine how they manipulate cell signaling to promote intracellular growth.
One strategy researchers have used is genetic screens to identify translocated proteins that interfere with essential pathways in Sacchromyces cerevisiae (Campodonico et al., 2005, Heidtman et al., 2008, Lesser et al., 2001, Shohdy et al., 2005, Sisko et al., 2006). Although S. cerevisiae has served as a useful model system to study bacterial translocated proteins, other expression systems are necessary to evaluate protein activities directed towards pathways that are not conserved in lower eukaryotes. Previous work showed that L. pneumophila activates the mammalian transcription factor, NF-κB (Abu-Zant et al., 2007, Bartfeld et al., 2009, Losick et al., 2006, Shin et al., 2008a). Activation could be shown to occur via two pathways: a Toll-like receptor (TLR) dependent path, as well as one that responds to the Icm/Dot translocation system (Abu-Zant et al., 2007, Bartfeld et al., 2009, Losick et al., 2006, Shin et al., 2008a). All previous studies agree that the TLR-dependent response occurs shortly after contact of bacteria with host cells before being down-modulated, whereas the Icm/Dot-dependent activation was persistent and occurred throughout the replication cycle (Abu-Zant et al., 2007, Bartfeld et al., 2009, Losick et al., 2006, Shin et al., 2008a).
NF-κB is one of the master regulators of the mammalian innate immune response leading to activation of pro-inflammatory cytokines, chemokines, and cell survival genes (Karin et al., 2002). Most research has focused on activation of NF-κB through the detection of pathogen associated molecular patterns (PAMPs) that are common to all bacteria, such as lipopolysacchride, peptidoglycan, flagellin, and lipoproteins. The membrane bound Toll-like receptors (TLR) and the cytoplasmic sensors of the Nod-like receptor family (NLR) have been implicated in detecting these PAMPs (Shaw et al., 2008, Uematsu et al., 2008). However, there is emerging evidence that translocated proteins may also play a role in inducing NF-κB activation. For instance, Helicobacter pylori delivers the protein CagA via its type IV secretion system into mammalian cells (Backert et al., 2000, Stein et al., 2000). CagA was shown to directly contribute to activation of the innate immune response by activating NF-κB (Brandt et al., 2005). Even ectopic expression of CagA in mammalian cells is sufficient to activate the NF-κB pathway (Brandt et al., 2005). More recently it was shown that LegK1, a L. pnenumophila IDTS, activated NF-κB when ectopically expressed in cultured cells, consistent with the idea that Legionella IDTS may be involved in activating this transcription factor (Ge et al., 2009).
L. pneumophila could have multiple proteins contributing to the NF-κB response, so we determined whether the ectopic expression of known or putative L. pneumophila IDTS could be sufficient to activate this pathway. Here we describe the identification of two proteins that can strongly induce an NF-κB response in HEK293T cells.
Results
NF-κB activation is independent of Rip2
Previous studies from our laboratory showed that in bone marrow (BM) macrophages NF-κB activation can occur in the absence of either the TLR signaling adaptor MyD88 or the cytoplasmic peptidoglycan sensor Nod1 (Losick et al., 2006). That signaling can still take place in the absence of TLR signaling is confirmed by the use of macrophages that are lacking the two adaptors MyD88 and Trif, which should eliminate all known TLR signaling (Supplementary Figure S1). Nod1 has been shown to sense the peptidoglycan degradation product iE-DAP released into the host cell cytoplasm (Chamaillard et al., 2003, Girardin et al., 2003a). There is a second peptidoglycan sensor Nod2 (Girardin et al., 2003b, Inohara et al., 2003), and both Nod proteins feed downstream to the protein kinase, Rip2, during peptidoglycan signaling to NF-κB (Kobayashi et al., 2002, Park et al., 2007). To determine whether these cytoplasmic sensors could be functioning redundantly to one another we used BM macrophages from Rip2 knockout mice that retain TLR signaling (Kobayashi et al., 2002).
BM macrophages from C57Bl/6J (B6) or the Rip2 knock out (Rip2KO) were infected with L. pneumophila ΔflaA strains, which are permissive for intracellular growth within this mouse strain background (Machner et al., 2006, Molofsky et al., 2006, Ren et al., 2006). NF-κB activation was measured at 7 hours post infection (hpi) by staining for the subcellular localization of NF-κB in L. pneumophila infected macrophages within the monolayer (Losick et al., 2006). Using this assay we observed that L. pneumophila was still able to cause efficient nuclear translocation of NF-κB independently of Rip2, since ~80% of the macrophages harboring wild-type (Lp02 ΔflaA) bacteria had strong nuclear staining of NF-κB (Fig. 1, data not shown). As was observed previously, the Icm/Dot mutant (dotA ΔflaA) had a reduction in the percent of cells with NF-κB in the nucleus, indicating that NF-κB translocation is dependent on L. pneumophila Icm/Dot system in both B6 and Rip2KO macrophages (Fig. 1; T-test p-value ≤ 0.01). This is consistent with recent findings in Rip2KO macrophages in which it was observed that IκBα, the negative regulator of NF-κB, was degraded in Icm/Dot dependent manner (Shin et al., 2008a). Therefore, we predicted that Icm/Dot translocated substrates (IDTS) were likely contributing to NF-κB activation, since peptidoglycan signaling through Rip2 was not required for this response in BM macrophages.
Figure 1. L. pneumophila induced NF-κB activation is not dependent on Rip2.
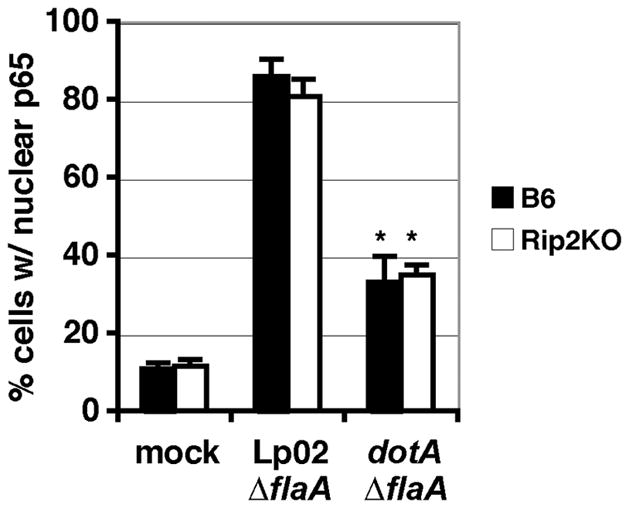
BM macrophages from C57Bl/6J (B6) and Rip2 knockout (Rip2KO) mice were challenged with L. pneumophila strain Lp02 ΔflaA and dot ΔflaA at MOI 1 for 7 hours. Fixed cells were stained for NF-κB, probed with anti- L. pneumophila antibody and the percent of cells harboring bacteria with detectable nuclear NF-κB staining were scored. Data represents the mean ± SE from three independent experiments, with 300 cells scored. * T-test p-value ≤ 0.01.
Construction of a L. pneumophila Icm/Dot translocated substrate library to assay for NF-κB activity
There are approximately 85 proteins identified to have translocation signals recognized by the Icm/Dot system. Studies from our laboratory indicate that an additional 100 or more proteins could be delivered into the host cell via the Icm/Dot system (L. Huang, and R. Isberg, unpublished). We hypothesized that some of these proteins could be contributing to NF-κB activation. To do so, a library of 159 known and candidate L. pneumophila IDTS was created to evaluate whether the ectopic expression of IDTS would be sufficient to activate NF-κB. Eighty of the 159 proteins in the library have been demonstrated to be secreted by at least one of the following approaches: direct immmunofluorescence (Conover et al., 2003, Laguna et al., 2006, Machner et al., 2006), Cre/loxP interbacterial translocation assay (Luo et al., 2004), CyaA reporter system (Chen et al., 2004), β-lactamase translocation assay (de Felipe et al., 2008), and/or a protein fusion translocation assay (Huang, unpublished, Luo et al., 2004, VanRheenen et al., 2006) (Table S1). Other Legionella proteins included were either paralogs of known substrates, proteins with eukaryotic-like domains (de Felipe et al., 2005), proteins regulated by the response regulator, PmrA (Zusman et al., 2007), or hypothetical proteins unique to L. pneumophila (Table S1). L. pneumophila genes were cloned using the Gateway® system and expressed in mammalian cells as fusion proteins to the green fluorescent protein (GFP) (Supplementary Figure S2). The expression of most of these GFP bacterial fusions could be detected by Western blot with anti-GFP antibodies. Of the 159 GFP fusions, full-length proteins were detected for 120 (~75% of total number; data not shown).
NF-κB activation in HEK293T cells is dependent on L. pneumophila’s Icm/Dot translocation system and is independent of flagella recognition
A transfectable mammalian system was required to test whether the ectopic production of the cloned IDTS could stimulate NF-κB. Human embryonic kidney (HEK293T) cells and the NF-κB luciferase reporter were chosen, since HEK293T cells are easily transfectable and the NF-κB luciferase reporter assay is a sensitive detection method. To evaluate this reporter system we first tested the NF-κB response to L. pneumophila in the HEK293T cell-type, to verify that the pattern of NF-κB activation is similar to our previously characterized human and mouse macrophage systems (Losick et al., 2006). As we observed previously in a human macrophage cell line (U937 cells), NF-κB activation in HEK293T increased over a time course of infection (Fig. 2A). By 4 hours post infection (hpi) there was a 5-fold increase in NF-κB activity, and by 10 hpi there was 25-fold increase in activity compared to uninfected cells (Fig. 2A). In agreement with our previous studies in macrophages, NF-κB activation in HEK293T was dependent on the Icm/Dot translocation system. The dotA strain did not significantly stimulate NF-κB activity (Fig. 2A). This was in contrast to previous studies that showed an early response in the absence of Icm/Dot, using much higher MOI than used in this figure (Abu-Zant et al., 2007, Bartfeld et al., 2009).
Figure 2. NF-κB activation by L. pneumophila in HEK293T cells is dependent on the Icm/Dot translocation system and independent of flagellin.
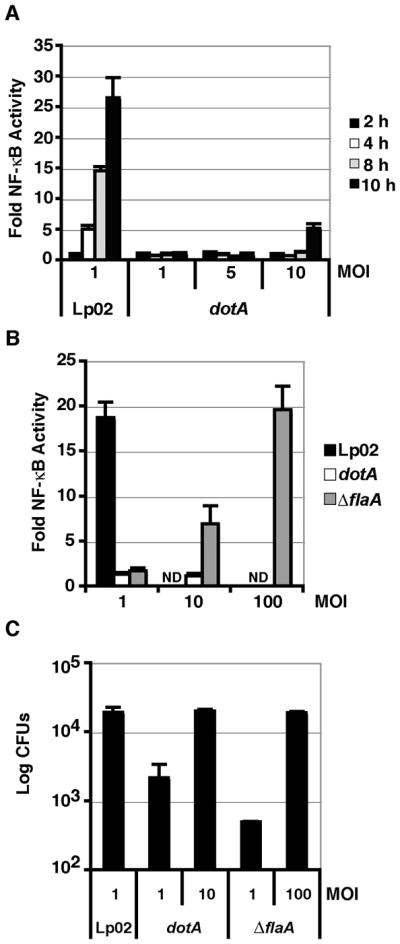
(A) Time course of NF-κB activation at 2 (black bars), 4 (white bars), 6 (gray bars), or 10 (dark gray bars) hours post infection (hpi) with wild-type L. pneumophila (Lp02) or a Icm/Dot mutant (dotA) at the MOI indicated. (B) Dose dependent activation of NF-κB in response to Lp02, dotA, or a flagellin mutant (ΔflaA). MOI is indicated on the x-axis. Luminescence was assayed after 8 hours post infection (hpi). ND: not determined. HEK293T cells were transfected with the NFκB-luciferase reporter plasmid, and then challenged with the L. pneumophila strains indicated. Fold NF-κB activity is expressed as the light units of samples relative to transfected cells incubated in absence of bacteria. (C) High efficiency L. pneumophila uptake into HEK293T cells requires flagellin. Uptake was measured at 2 hpi by lysing cells and enumerating CFUs. Data represent the mean and SE of triplicate samples from a representative experiment. Experiments were repeated in duplicate.
Unlike mouse bone marrow derived macrophages, HEK293T cells are known to express Toll-like receptor 5, which senses bacterial flagellin (Gewirtz et al., 2001). Flagellin is also known to be important for promoting uptake of L. pneumophila (Molofsky et al., 2005), and high MOI infections of an immortalized lung epithelial line have been associated with a flagellin-dependent NFκ-B response (Bartfeld et al., 2009, Molofsky et al., 2005). To determine whether flagellin sensing could be contributing to the NF-κB response in HEK293T cells, we challenged cells with an L. pneumophila flagellin deficient strain, ΔflaA (Ren et al., 2006). At 8 hpi, the ΔflaA strain could induce NF-κB activity, but the induction was dependent on the multiplicity of infection (MOI) (Fig. 2B). Only by infecting cells with ΔflaA strain at MOI=100 was there an equivalent induction of NF-κB activity when compared to the wild-type (Lp02) strain at MOI=1 (Fig. 2B). The lower levels of NF-κB activation in the ΔflaA strain appeared to be due to a defect in cell association. There was a 38-fold defect in the uptake of bacteria at 2 hpi (MOI 1) for the ΔflaA strain, as measured by plating for colony forming units (CFU) (Fig. 2C). When the dose of the ΔflaA strain was increased to MOI = 100, the dose at which NF-κB activation by this strain was identical to Lp02, the uptake efficiencies of the Lp02 and ΔflaA were identical (Fig. 2C). In contrast, there was low NF-κB activation by the Icm/Dot-defective mutant dotA, but increasing the MOI could not increase NF-κB activation levels to that observed in the Lp02 strain (Fig. 2B). When cells were challenged with the dotA strain at MOI=10, there was no uptake defect of the mutant (Fig. 2C). Therefore when these defects are taken into account, the Icm/Dot translocation system, but not flagella, significantly contributes to NF-κB activation consistent with our previous finding in human and mouse macrophages. The HEK293T cell type and the NF-κB luciferase reporter together are a valid system to test whether L. pneumophila IDTS can activate NF-κB.
L. pneumophila IDTS can activate NF-κB
HEK293T cells were co-transfected with plasmids encoding the NFκB-luciferase reporter and GFP fusion proteins (Fig. 3A), then assayed for luminescence (Experimental Procedures). As a positive control, the luciferase reporter construct was co-transfected with a vector encoding GFP-Nod1, which strongly stimulates NF-κB (Inohara et al., 1999, Bertin et al., 1999). Of the 159 proteins tested, the expression of 13 L. pneumophila proteins each caused more than 3-fold increase in NF-κB activity relative to empty vector. These were placed in three groups based on expression levels observed in HEK293T cells (Fig. 3A). Those causing milder effects included known translocated substrates LidA, SidM, SidE, SidA, SdbA, VpdA, SidH, LegA5 and LegA12, as well as candidate substrates, SdeB (a paralog of SidE) and SdhB (a paralog of SidH) (Figs. 3B and 3C). Of the ones causing mild effects, the most striking IDTS was VpdA, which showed 14-fold induction of the reporter despite being poorly expressed (Fig. 3A and 3B).
Figure 3. Candidate IDTS can activate NF-κB.
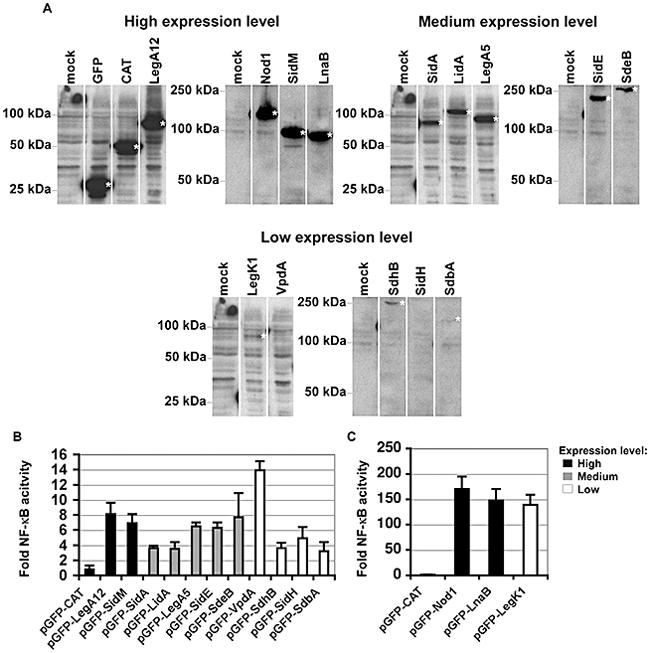
(A). Varied expression levels of L. pneumophila IDTS expressed in 293T cells. Protein expression levels of the indicated GFP fusions that had enhanced NF-κB activation are displayed on immunoblots, probing with anti-GFP. To allow clearer comparisons of the relative increases in NFκB activity, IDTS are grouped according to their expression levels in HEK293T cells. (B) Low level activation of NF-κB by L. pneumophila IDTS. Legionella NF-κB activators fused to GFP are marked on the x-axis. Shown are those IDTS that show statistically significant activation of NF-κB (T-test p-value ≤ 0.05). Bars representing those IDTS expressed at high levels are marked in black. Medium and low expression levels are shown in grey and white, respectively. (C) High level NF-κB activation by two Legionella IDTS. HEK293T cells were co-transfected with 200 ng of pNFκB-luciferase reporter and either 50 ng of the control plasmid (pGFP) or the indicated pGFP fusions and luminescence was assayed at 24 hours. pGFP-CAT (chloramphenicol aminotransferase) served as a negative control and pGFP-Nod1 was used as a positive control for NF-κB activation. Fold NF-κB activity is the relative light units of pGFP fusion compared to the pGFP control. Data represents the mean ± SE from three independent experiments preformed in triplicate.
Of the 159 L. pneumophila genes screened, expression of two candidate L. pneumophila IDTS LegK1 and Lpg2527 caused ~150-fold induction of NF-κB activity (Figs. 3A and 3C). As was true of VpdA, LegK1 was poorly expressed, but it generated 10-fold higher induction relative to VpdA, consistent with previous observations (Ge et al., 2009). The gene for the candidate IDTS Lpg2527, which was more efficiently expressed than LegK1, was named lnaB for Legionella NF-κB activator B, and encodes a predicted protein that has no sequence similarity to any known proteins. LegK1, on the other hand, is predicted to be a serine/threonine kinase (de Felipe et al., 2005), with a conserved catalytic asparatic acid residue at amino acid 223. As previously demonstrated, mutation of this residue to alanine in the GFP-LegK1 construct (D223A) eliminated induction of the reporter encoding LegK1 in HEK293T cells (Fig. 4).
Figure 4. The ser/thr kinase activity of LegK1 is required for NF-κB activation.
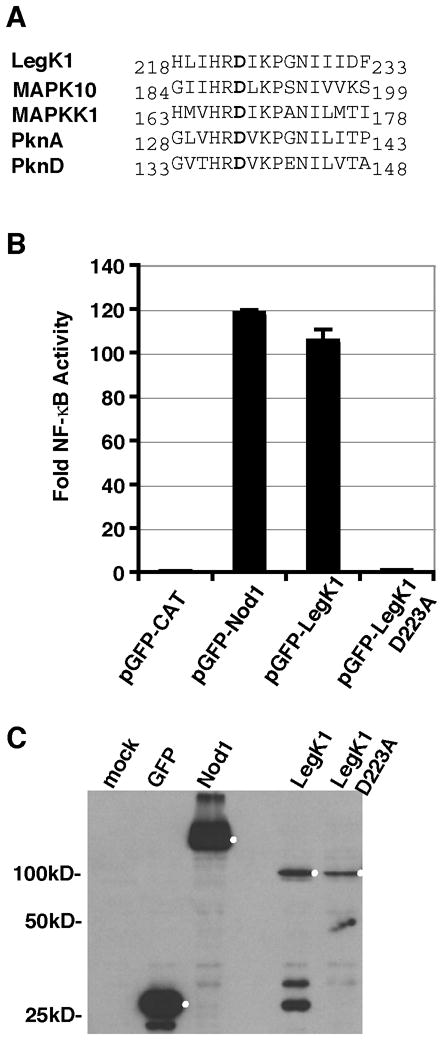
(A) Alignment of the predicted catalytic domain of L. pneumophila LegK1 with other ser/thr kinase domain containing proteins: Human MAPK10 (NP_002744), Chlamydomonas reinhardtii MAPKK (XP_001696437), Mycobacterium tuberculosis PknA (NP_214529) and PknB (NP_214528). The catalytic aspartate (shown in bold) is preceded by an arginine in all the ser/thr kinases. (B) Mutation of the putative LegK1 catalytic aspartate to alanine eliminates NF-κB activation. HEK293T cells were co-transfected with 200 ng per well of pNFκB-luciferase and either 50ng of pGFP fused to CAT (negative control), Nod1 (positive control), LegK1, or catalytically inactive mutant LegK1 D223A. Data represent the mean ± SE from triplicate samples of a representative experiment. The experiment was preformed in triplicate. (C) Western blot of the protein expression levels of GFP, GFP-Nod1, GFP-LegK1, and GFP-LegK1-D223A probed with anti-GFP antibodies. HEK293T cells were transfected for 48 hours with 200 ng of the indicated plasmids; the expected protein based on molecular weight are marked by a white dot.
The coiled-coil domain in LnaB is required for NF-κB activation
LnaB is a 558 amino acid protein with a small putative coiled-coil domain located between 361-401 amino acids. Truncation and deletion constructs of LnaB were generated to determine whether NF-κB activity was dependent on the coiled-coil domain. LnaB was divided into regions that included or excluded the coiled-coil domain, and an in-frame deletion of the coiled domain (ΔCC). All constructs were designed to be in-frame with EGFP at the N-terminus (Fig. 5A). Co-transfection of HEK293T cells with the NF-κB reporter and EGFP-LnaB fusions revealed that the N-terminal region of LnaB including the coiled-coil domain (amino acids 1-401) was sufficient to induce NF-κB activity (Fig. 5C). However, the level of NF-κB activity in the presence of this N-terminal fragment was reduced to one third of the activity observed for full-length protein: the full-length EGFP-LnaB fusion resulted in ~120 fold induction, whereas the EGFP-LnaB1-401 construction showed only a ~40 fold increase in luminescence (Fig. 5C). The coiled-coil domain was required for activation, because expression of the ΔCC construct, or one containing the amino terminus truncated at the amino terminus of this domain (amino acid residues 1-361) showed almost no NF-κB induction (Fig. 5C). Each of these fusions showed very similar amounts of protein expression, arguing that the differences in phenotypes were due to the loss of specific amino acid residues (Fig. 5B). Similarly, shorter fusions also failed to show induction (Fig. 5C), and these showed even more robust expression than the wild type fusion (Fig. 5B).
Figure 5. The coiled-coil domain of LnaB is required for NF-κB activity.
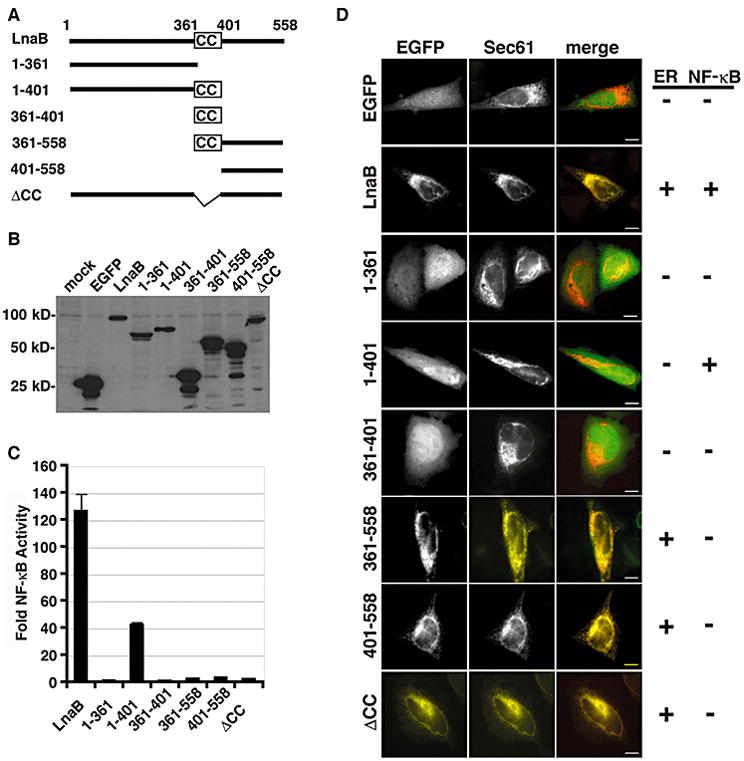
(A) Schematic of EGFP-LnaB fusions. Full-length LnaB is 558 amino acids and contains a putative coiled-coil domain (CC) between amino acids 361-401. (B) Expression of EGFP fusions, 24 hours after transfection of HEK293T cells with the corresponding plasmids as detected by Western blot with anti-GFP. (C) HEK293T cells were co-transfected with 200 ng of pNFκB-luciferase and 50 ng of indicated EGFP fusion and luminescence was assayed after 24 hours. Fold NF-κB activity is the relative light units of pEGFP fusions compared to the pEGFP control. Data represents the mean ± SE from triplicate samples of a representative experiment. The experiment was preformed in triplicate. (D) Subcellular localization of indicated EGFP-LnaB fusions in HeLa cells expressing the ER marker Sec61 (red) and EGFP (green) in the merged image. Constructs that co-localized with Sec61 or activated NF-κB are marked with a +. Scale bar = 2μm.
Unlike GFP-LegK1, which localized diffusely in the cytoplasm of HEK293T cells (data not shown), GFP-LnaB localized to the nuclear envelope and to a perinuclear region reminiscent of an ER compartment in all mammalian cell lines tested including HEK293T and Hela cells (Fig. 5D; data not shown). To determine whether NF-κB activity correlated with ER localization, Hela cells were co-transfected with the EGFP-LnaB fusions and the ER membrane protein Sec61 fused to the fluorescent protein mCherry. By fluorescence microscopy it appeared that the 401-558 amino acid C-terminal region was sufficient for ER localization (Fig. 5D, EGFP panel). In contrast, the residue 1-401 fragment that has NF-κB activating activity was diffuse within the cell and did not co-localize with Sec61 (Fig. 5D, EGFP panel). Therefore, the region of the protein that promotes NF-κB activation can be separated from the region that allows ER localization, indicating that ER localization of LnaB is not required for NF-κB activation.
LnaB is a substrate of L. pneumophila’s Icm/Dot translocation system
LnaB was fused to the catalytic domain of Bordetella pertussis adenylate cyclase protein, CyaA, to determine whether this protein is a substrate of L. pneumophila’s Icm/Dot translocation system. The cytoplasmic activation of CyaA by calmodulin leads to an increase in cAMP in the host cell (Sory et al., 1994). The cAMP level can then be quantified by ELISA, thereby acting as an indicator of the translocation of the CyaA fused protein. The human U937 cell-line was incubated with L. pneumophila expressing CyaA (negative control), CyaA-RalF (positive control), or CyaA-LnaB, and assayed for cAMP levels after 1 hour. The Icm/Dot deficient strain, dotA, was used as negative control. Both Lp02 CyaA-RalF and Lp02 CyaA-LnaB resulted in a ~30-fold increase in cAMP as compared to uninfected or Lp02 CyaA infected U937 cells (Fig. 6A). The cAMP induction by CyaA-RalF and CyaA-LnaB was dependent on the Icm/Dot system, since there was no significant increase in cAMP with the dotA strains (Fig. 6A). CyaA fusions were expressed at high levels in all bacterial strains before infection (Fig. 6B). These data demonstrate that LnaB has an Icm/Dot translocation signal. A recent study also presented evidence that LnaB (designated lpg2527) is a Icm/Dot substrate (Kubori et al., 2008), supporting our independent observations.
Figure 6. LnaB is a substrate of the Icm/Dot translocation system.

(A) U937 cells were challenged with the indicated L. pneumophila strains expressing CyaA (negative control), CyaA-RalF (positive control), CyaA-LnaB. After a 1 hour incubation, cAMP was extracted and relative cAMP levels were measured. Data represent the mean ± SE of triplicate samples from a representative experiment performed in triplicate. (B) Protein expression of CyaA, CyaA-RalF, CyaA-LnaB in the corresponding L. pneumophila strains prior to challenge of U937 cells. Isocitrate dehydrogenase (ICDH) was used as a loading control.
LnaB and legK1 are dispensable for intracellular growth, but lnaB contributes to NF-κB activity
As LnaB and LegK1 can activate NF-κB when ectopically expressed, we next determined if knockout mutations in ΔlnaB or ΔlegK1 affect L. pneumophila intracellular growth and/or NF-κB activation. The in-frame deletions ΔlnaB or ΔlegK1 were generated in the wild-type Lp02 strain and assayed for the ability to grow intracellularly. Mouse macrophages were challenged with Lp02, ΔlnaB, or ΔlegK1 mutants to assay for their efficiencies of intracellular growth. L. pneumophila growth was monitored over a three-day growth curve by lysing macrophages and enumerating CFUs. As is true of many strains harboring single IDTS mutation (Luo et al., 2004, Machner et al., 2006, Nagai et al., 2002, Ninio et al., 2005, Shohdy et al., 2005, VanRheenen et al., 2006), the absence of ΔlnaB or ΔlegK1 did not appear to influence L. pneumophila intracellular growth. The growth of both deletion strains was equivalent to wild-type Lp02 (Fig. 7A, B).
Figure 7. LnaB is dispensable for intracellular growth and protection from cell death.

(A and B). Intracellular growth of L. pneumophila within A/J mouse BM macrophages. Displayed is the wild-type (Lp02) strain compared to ΔlegK1 (A) or ΔlnaB (B). At 2, 24, 48, and 72 hpi macrophages were lysed and colony forming units (CFUs) were enumerated (Experimental Procedures). Data represents the mean ± SE from three independent infections of a representative experiment preformed in duplicate. (C,D) Bacterial replication at 1 hpi and 9 hpi. The percentage of A/J mouse BM macrophages harboring the denoted number of bacteria is shown. The wild type (Lp02) was compared to ΔlnaB, ΔlegK1 and ΔlnaBΔlegK1 after infection of MOI=1. (E) Host cell survival. At the denoted time points post infection with wild type (Lp02), ΔlnaB, ΔlegK1 and ΔlnaBΔlegK1, condensed nuclei (cell death) were observed by Hoechst DNA staining and immunofluorescence microscopy. Average and SE of three cover slips of the respective experiments are displayed. (F). Intracellular targeting of the L. pneumophila replication vacuole in mutants defective for NF-κB activators. Bone marrow-derived macrophages from A/J mice were challenged with the noted L. pneumophila strains at MOI = 1.0 for one hour (Experimental Procedures). Cells were fixed, probed as described for L. pneumophila and late endocytic marker LAMP-1 (Experimental Procedures) and visualized by indirect immunofluorescence microscopy. Data are the mean ± SD of nine coverslips from three experiments for samples except ΔlegK1 which was the mean ± SD of 6 coverslips from two experiments.
To investigate whether strains lacking lnaB and legK1 are defective for initiating intracellular replication, bacterial replication at the single cell level was followed after low MOI challenge at time points in which NF-κB activation was maximal (Fig. 7C,D). At 9 h post-infection, the wild type strain (Lp02), mutants lacking either lnaB or legK1, as well as a double mutant lacking both genes, were equally efficient at initiating replication in bone marrow derived macrophages. For each strain, almost the entire population of infected macrophages showed multiple intracellular bacteria, with intracellular loads nearly equivalent for each of the strains (Fig. 7C,D). Consistent with this observation, the lack of these two translocated substrates had little effect on the survival of macrophages harboring bacteria. For each of the single mutants, as well as the double ΔlnaBΔlegK1 mutant, greater than 60% of the infected macrophages showed normal nuclei after 9 hours incubation with bacteria, with similar kinetics of cell death after challenge with each strain (Fig. 7E). Similarly, there was little effect of the mutations on avoiding targeting to a late endosomal compartment (Fig. 7F), based on colocalization of the late endosomal marker LAMP-1. For each of the mutants, there were similar levels of co-localization of the replication vacuole with LAMP-1, although strains harboring theΔlnaBΔlegK1 mutations showed a small increase relative to wild type (Fig. 7F).
To assay for NF-κB activation, HEK293T cells transfected with the NF-κB reporter were challenged with L. pneumophila strains for 7 hours and luciferase activity was determined and normalized to CFU to ensure that the readout was not affected by bacterial load (Experimental Procedures). The deletion of ΔlegK1 did not affect NF-κB activity when assayed in this system (Fig. 8A). In contrast, the absence of ΔlnaB modestly reduced NF-κB activation, and this phenotype could be complemented by re-integration of lnaB+ on the chromosome (Fig. 8B; T-test p-value < 0.05). The double mutant lacking both lnaB and legK1 showed no further reduction in NF-κB activation (data not shown). Western blots of extracts probed with anti-LnaB serum demonstrated that the deletion mutant expressed no detectable LnaB, whereas the strain having a chromosomally encoded lnaB+ showed levels of expression equivalent to the wild-type Lp02 (Fig. 8C). The strains were grown to post-exponential phase prior to infection when LnaB is most highly expressed (Fig. 8D). Therefore the delivery of LnaB during L. pneumophila infection contributes to NF-κB activation when L. pneumophila is grown to post-exponential phase.
Figure 8. LnaB is required for full activation of NF-κB.

(A and B) HEK293T cells were transfected with the NF-κB-luciferase reporter plasmid and then challenged with the wild-type L. pneumophila (Lp02), the ΔlegK1 (A) or the ΔlnaB (B) strains at MOI = 1 for 7 hours. Fold NF-κB activity* is the relative light units of cells incubated with bacteria compared to cells incubated without bacteria. Samples were normalized to bacterial uptake at 2 hpi (Experimental Procedures). Data represents the mean ± SE of triplicate samples from a representative experiment performed in duplicate (A) or the mean ± SE from 4 independent experiments performed in triplicate (B) in which the maximum NF-κB activity was normalized to 100%. *T-test p-value < 0.05. (C) LnaB production in wild-type (Lp02::SR47s), ΔlnaB::SR47s, and ΔlnaB-LnaB+ complement strains. (D) LnaB expression is induced in post-exponential phase of growth. Lp02 was grown overnight to indicated A600 and immunoblotted with anti-LnaB or anti-ICDH as loading control.
Discussion
In this study we identified Legionella pneumophila Icm/Dot translocated substrates (IDTS) that have an impact on host cell signaling when expressed within mammalian cells. A large swath of known and potential IDTS was chosen, based on either bioinformatics or the presence of an experimentally identified translocation signal, and plasmids encoding these genes were introduced into a screening system that allowed rapid identification of a phenotype. The NF-κB-luciferase screening procedure described here was developed because previous studies demonstrated that NF-κB activation was dependent on the presence of the Icm/Dot translocation system (Abu-Zant et al., 2007, Losick et al., 2006, Shin et al., 2008a). Here we demonstrate that several known IDTS modestly activated an NF-κB-luciferase reporter, whereas two candidate IDTS expressed in cultured cells, LnaB and LegK1, resulted in robust activation. Full activation of this promoter after challenge of cells with post-exponential phase L. pneumophila required the presence of LnaB.
In an approach that was similar to the one used here, LegK1 was shown to induce expression of an NF-κB reporter in cultured cells (Ge et al., 2009). Interestingly LegK1 was shown to phosphorylate IκB in mammalians cells, although it is unclear whether this activity occurs during a natural infection, particularly in the natural host amoebae, which have no known NF-κB signaling systems. When legK1 was deleted, bacteria lacking this protein showed normal levels of NF-κB activation, suggesting that LegK1 may be a minor player in inducing NF-κB in vivo. Therefore, the exact role of this protein during Legionella infection is not known.
The activation of NF-κB in response to low doses of L. pneumophila has been hypothesized to positively contribute to intracellular growth by upregulating genes encoding antagonists of apoptosis (Abu-Zant et al., 2007, Losick et al., 2006, Bartfeld et al., 2009, Shin et al. 2008a). In addition, a conflicting property of this level of control is that NF-κB activates an anti-microbial response. The best characterized routes of NF-κB activation in response to bacterial infection occurs through the stimulation of surface localized Toll-like receptors (TLR) and/or the cytoplasmic peptidoglycan sensors Nod1 and Nod2 via common molecules liberated by both pathogenic and nonpathogenic microbes (Shaw et al., 2008, Uematsu et al., 2008). L. pneumophila activation of NF-κB can occur independently of TLR signaling, as macrophages lacking the TLR adaptor MyD88 (Losick et al., 2006), or double knockouts of both required TLR adaptors MyD88 and Trif (Supplementary Figure S1) still support NF-κB nuclear localization in response to L. pneumophila.
The role of Nod proteins in Icm/Dot-dependent signaling to NF-κB is not clearcut. Rip2 knock-out macrophages, missing the downstream kinase necessary for Nod1 and Nod2 signaling to NF-κB (Inohara et al., 2000, Kim et al., 2008, Ogura et al., 2001) supported Icm/Dot-dependent NF-κB nuclear translocation (Fig. 1), paralleling our previous results with Nod1 knock out macrophages (Losick et al., 2006). Therefore, there exists a peptidoglycan-independent, Icm/Dot-dependent signal to NF-κB in bone marrow macrophages. However, recent results from Shin et al., (2008a) suggest that the Rip2-independent, Icm/Dot-dependent response appears to require the TLR system in bone marrow macrophages. We tested NF-κB activation in HEK293T cells knocked down for Nod1 and found that NF-κB activation was reduced in response to L. pneumophila infection (Supplementary Figure S3). HEK293T cells do not express a number of TLRs present in bone marrow macrophages suggesting there may indeed be cross talk between TLR and Nod signaling to activate NF-κB in response to L. pneumophila. So perhaps the Rip2-independent activation in bone marrow macrophages results from cross talk between an IDTS and TLR-signaling, or due to an IDTS preventing downregulation of TLR signaling. This emphasizes the hypothesis that multiple IDTS could affect NF-κB activation via mechanistically distinct, but redundant, strategies.
The novel strong NF-κB activator we identified was LnaB (Lpg2527). We and Kubori et al., 2008 have demonstrated that Lpg2527 is translocated into host cells via the L. pneumophila Icm/Dot translocation system. In addition, eight of the eleven modest NF-κB activators are previously characterized IDTS (LidA, SidM, SidA, SidE, SidH, VpdA, LegA12, and LegA5) (Cambronne et al., 2007, Luo et al., 2004, Machner et al., 2006, Murata et al., 2006, VanRheenen et al., 2006). Therefore multiple L. pneumophila translocated proteins could contribute to NF-κB activation when introduced into the host cell during infection, indicating that there may be more than one route that leads to manipulation of the NF-κB signaling pathway. In line with this argument is the behavior of an L. pneumophila ΔlnaB mutant. Using the HEK293T NF-κB reporter assay there was a partial defect in the activation of this transcription factor. There must be other microbial molecules that contribute to activation, because LnaB is only expressed in post-exponential phase (Fig. 8D).
There is precedence for NF-κB activation by translocated bacterial proteins. The Helicobacter pylori type IV secretion system delivers the CagA protein which, when ectopically expressed in mammalian cells, strongly induces NF-κB activation (Brandt et al., 2005, Hatakeyama, 2008). CagA has been shown to influence a diverse set of host cell signaling pathways including, cell motility and cell proliferation. Any of these CagA stimulated pathways could feed into NF-κB signaling. Therefore it is possible that like CagA, LnaB acts indirectly to stimulate the NF-κB pathway in mammalian cells. Alternatively, the protein may be “sensed” by a host cell receptor to stimulate this pathway. It is important to point out that the peak of Icm/Dot-dependent induction of NF-κB occurs between 6–8 hr after challenge with L. pneumophila, and this timing is independent of the host cell type challenged by the bacteria (Losick et al., 2006, Bartfeld et al., 2009). This result is supported by other work showing waves of NF-κB induction in response to L. pneumophila (Bartfeld et al., 2009). This delay could also be explained by accumulation of translocated substrates or peptidoglycan fragments (Shin et al., 2008a) within the cell, or due to paracrine signaling from outside the cell, as inflammatory mediators accumulate in the culture medium dependent on Icm/Dot activity.
In plants there are Nod-like receptor (NLR) proteins known as R-proteins that either interact directly with injected proteins from bacterial pathogens or recognize the activities of these proteins to trigger a defense response (Grant et al., 2006). Plants may have more than a hundred R-proteins that can recognize the activities of these diverse bacterial translocated proteins. In mammalian cells, with a more limited number of NLRs, sensing appears to be limited to common microbe-associated molecules presented into the host cell cytoplasm (Jones et al., 2006, Proell et al., 2008). However, alterations to the actin cytoskeleton (Kustermans et al., 2005, Legrand-Poels et al., 2007) perturbations of the plasma membrane and disruption of ER homeostasis have been shown to activate NF-κB (Schroder, 2008). Many bacterial secreted proteins do just that, inhibiting the actin cytoskeleton for the purpose of blocking phagocytosis (Trosky et al., 2008), disrupting the plasma membrane or altering membrane trafficking events (Ramsden et al., 2007, Shin et al., 2008b). Thus, in response to microbial attack, it is conceivable that the disruption or manipulation of these cellular pathways by bacterial translocated proteins serve as triggers for activation of mammalian innate immune responses as well. Furthermore, the response of host cells to common microbial molecules, such as peptidoglycan fragments, could be amplified as a result of misregulation of cellular processes by translocated substrates. In support of the possibility that IDTS could either cause or potentiate an existing NF-κB response, we observed that LidA and SidM, two IDTS that modulate the host cell vesicle trafficking GTPase Rab1 (Machner et al., 2006, Murata et al., 2006), can also induce NF-κB activity when ectopically expressed.
LnaB associates with the ER when ectopically expressed, however the protein contains no predicted transmembrane domains by a Kyle Doolittle hydropathy plot (data not shown). We found that the C-terminal region between amino acids 401-558 was sufficient for ER localization, but not NF-κB activation. Therefore ER localization is not sufficient to induce NF-κB activity. In addition, more than thirty other GFP-fusions in our library co-localized to an ER compartment but did not cause activation of the NF-κB sensitive promoter when ectopically expressed (Losick V.P., et al., unpublished), further indicating that ER localization is not sufficient to induce this response. In contrast, the coiled-coil domain of LnaB is required for NF-κB activation and the N-terminal region containing the coiled-coil domain (from amino acids 1-401) is sufficient to induce this activity despite being localized to the cytoplasm. We propose that LnaB is likely to have a binding partner that targets the coiled-coil domain of this protein. Interaction with this region either is important for an activity that results in the NF-κB response, or the protein directly interacts with a component of a pathway upstream of this transcriptional regulator.
In conclusion, we have identified several L. pneumophila Icm/Dot translocated proteins that can stimulate NF-κB activation when ectopically produced in a mammalian cell-line. In particular the IDTS LnaB strongly activates NF-κB and a deletion of ΔlnaB is sufficient to cause reduction of transcription of this pathway when post-exponential bacteria challenge mammalian cells. The mechanism by which LnaB induces NF-κB activity still remains to be elucidated, but our study lends further support to the role of translocated proteins in contributing to activation of the innate immune response in mammalian cells.
Experimental Procedures
Bacterial strains, media, and plasmids
L. pneumophila Philadelphia-1 strains Lp02 (thyA) and Lp03 (dotA3 point mutant; referred to as dotA) are derivatives of Lp01 (hsdR rpsL) (Berger and Isberg, 1993). Lp02 ΔflaA was generously provided by Drs. Tao Ren and William Dietrich (Ren et al., 2006). L. pneumophila strains were maintained on buffered charcoal yeast extract (BCYE) solid medium and ACES-buffered yeast extract (AYE) broth culture media (Feeley et al., 1979, Gabay et al., 1985, Swanson et al., 1995). For infections, Legionella was patched from a single colony onto BCYE containing 100 μg/ml of thymidine and/or antibiotics: 5 μg/ml of chlroamphenicol, 20 μg/ml of kanamycin, if required. After 2 days at 37°C, patches were used to inoculate AYE broth culture. Cultures were grown overnight in AYE broth with appropriate additives and grown to post-exponential phase (A600 ~ 4.0 – 4.5) prior to infection. The in-frame deletion of ΔlnaB (VPL2) and ΔlegK1 (VPL1) in Lp02 were constructed as previously described (Luo et al., 2004, Roy et al., 1997). The VPL11 (Lp02::SR47s) and VPL12 (ΔlnaB::SR47s) were constructed by selecting for KanR colonies that integrated the SR47s suicide vector into the lnaB chromosomal locus. The complementing strain VPL13 (ΔlnaB::SR47s-LnaB+) was constructed by selecting for KanR colonies that integrated the suicide vector containing the full-length lnaB under its endogenous promoter onto the chromosome.
pJB-CyaA and CyaA translocation assay plasmids and related procedures were generously provided by Dr. Joseph Vogel (Bardill et al., 2005). All L. pneumophila plasmids were constructed using PCR fragments amplified from Lp02 genomic DNA isolated using DNeasy kit, Qiagen. The cyaA plasmids pcyaA-ralF, pcyaA-lnaB, and pcyaA-legK1 were cloned in-frame with cyaA at BamHI and SalI sites. The mammalian expression plasmid pEGFP-C1 (Clontech) has a constitutive CMV promoter. LnaB was cloned in-frame with EGFP using EcoRI and BamHI sites. Competent DH5α and DH5αλpir were prepared by the Tufts GRASP center. See Table S2, S3, and S4 for more information and details on primers, plasmids, and strains used in this study.
Cell culture
HEK293T and HeLa cells were maintained in DMEM plus 10% fetal bovine serum (FBS). U937 cells (ATCC) were grown in RPMI 1640 (Invitrogen) supplemented with 10% FBS and 1 mM glutamine (Invitrogen). 5 × 107 U937 cells were differentiated using 10 ng/ml 12-tetradecanoyl phorbol 13-acetate (TPA) for 48 hr, after which cells were washed, replated with fresh media in the absence of TPA, and used for infections with L. pneumophila ~16 hours later. Bone marrow (BM) macrophages were flushed from the femurs of 6- week to 3-month old mice and differentiated in bone marrow macrophage media, BMM (RPMI, 1mM glutamine, 10% FBS, 30% L-cell supernatant) (Swanson et al., 1995). BM macrophages were differentiated for 7–8 days, collected, and frozen for use in multiple experiments in medium containing 20% serum, 10% DMS0. BM macrophages were replated in BMM media containing 200 μg/ml of thymidine.
L. pneumophila growth curves in BM macrophages
BM macrophages from A/J mice were plated at a density of 4 × 105 cells per well of a 24 well plate and allowed to settle overnight. L. pneumophila strains were grown to post-exponential phase, as determined by the presence of highly motile bacteria, and incubated with macrophages at MOI = 0.05. After infection, plates were spun at 1,000 rpm for 5 minutes at room temperature to promote contact of bacteria with the macrophages. Incubations proceeded at 37°C, 5% C02 for 2 hours, after which the monolayers were washed three times with pre-warmed BMM medium plus 200 μg/ml of thymidine. After 2, 24, 48, and 72 hours, three independent wells were lysed with 0.2% saponin, and then wells were washed with 1ml of sterile water. CFUs were enumerated on BCYE plates and colonies were counted 4 days post-plating.
NF-κB nuclear translocation assay
NF-κB nuclear translocation in bone marrow-derived macrophages was performed as previously described (Losick et al., 2006). Cells were considered positive if there was intense staining for NF-κB in the nucleus at 7 hours post infection (hpi).
NF-κB luciferase reporter assay
HEK293T cells were replated at a density of 3 × 104 cells per well of 96-well white Corning clear-bottomed plates and left overnight to adhere. Cells were co-transfected with 50 ng of the indicated plasmid and 200 ng of ultra pure NFκB-luciferase (Stratagene) reporter plasmid per well using 0.5 μl of Lipofectamine 2000 according to manufacturer’s instructions (Invitrogen) for ~24 hours. For L. pneumophila infections after 16 hours of tranfection with the reporter plasmid, HEK293T cells were incubated with Legionella pneumophila grown to post-exponential phase at MOI indicated. Plates were spun at 1,000 rpm for 5 minutes at room temperature. After 2 hours post infection, cells were gently washed twice with warm DMEM plus 10% FBS and 200 μg/ml thymidine. Incubations were allowed to proceed for times indicated, after which the medium was aspirated and replaced by 50 μl of sterile PBS plus 50 μl of Steadylite HTS (Perkin-Elmer) luminescent reagent. The plates were read immediately in a 96-well plate reader. For each well, the relative light units (RLU) reporter was the mean data from nine points within the well, scanned for 1 second each. The fold NF-κB activity is the mean RLU of the indicated infection versus uninfected cells. Standard error is based on triplicate wells from a representative experiment.
If indicated, the NF-κB activity was normalized to mean bacterial uptake from three independent wells to ensure that activity was not affected by subtle differences in bacterial load. HEK293T cells were replated and infected as described above, except at 2 hpi three independent wells were lysed with 0.2% saponin and washed with equal volume of sterile water. CFUs were enumerated on BCYE plates and colonies were counted after 4 days at 37°C.
Quick-change mutagenesis of LegK1 D223A
The PAGE purified primer, 5′GCATTTGATTCATCGAGCTATAAAACCAGG-3′ (IDT), was used to mutate the putative catalytic asparatic acid residue (D223) of the LegK1 Ser/Thr kinase domain to an alanine according to the Quick Change protocol described by Stratagene. Underlined is the nucleotide change used for this mutation. Plasmids were screened by digest and sequenced to confirm the mutation.
CyaA translocation assay
TPA treated U937 cells were plated at density of 1 – 2 ×106 cells per well of 24-well plate. L. pneumophila pCyaA strains were grown overnight in AYE plus 100 μg/ml thymidine and 5 μg/ml chloramphenicol to A600 ~2.5. CyaA protein expression was induced by adding 100 μM IPTG and strains were allowed to grow for additional 2 to 3 hours until the bacteria were in post-exponential phase and motile. The equivalent of 1 OD unit of cells was pelleted, resuspended and boiled in 150 ul 1 × SDS sample buffer. 1/20th of the sample was used for Western blot analysis of CyaA fusions with anti-CyaA. The CyaA expressing L. pneumophila strains were used to infect U937 cells at MOI = 1. After addition of the bacteria, 24 well plates were spun at 1,000 rpm for 5 minutes and incubated for 1 hour at 37°C, 5% CO2. Cells were washed 3 times with PBS and cells were lysed by adding 200 μl of lysis buffer (50mM HCl, 0.1% Triton X-100) and incubated on ice for 10 minutes. Lysates were collected and boiled immediately for 5 minutes, then neutralized by addition of 12 μl of 0.5 M NaOH. The cAMP present in the extracts was precipitated by adding 400 μl of cold 95% EtOH (65% final) and incubated on ice for 5 minutes. Insoluble material was removed by spinning samples in a microfuge for 5 minutes at 4°C at 13,000 rpm. Supernatants containing the cAMP were dried under vacuum and resuspended in 200 μl dH20. The cAMP concentration was measured using Amersham Biotrak cAMP ELISA Kit according to manufactures instructions.
Western blotting of GFP fusions and other proteins
To analyze expression in cultured cells of the GFP fusions expressed from pDEST53, HEK293T cells were plated at a density of 5 × 104 cells per well in 96-well plates and 200 ng plasmid was transfected using 0.5 μl Lipofectamine 2000 for 24 – 48 hours. Samples were analyzed by Western blotting using rabbit anti-GFP (Invitrogen) (1:5000). Detection of full-length GFP-fusions were marked as positive (+) (Table S1). The other primary antibodies used in this study included rabbit anti-Myc (Santa Cruz) (1:3000); mouse anti-Tubulin (Sigma) (1:10,000); rabbit anti-CyaA (Santa Cruz) (1:10,000); rabbit anti-LnaB affinity purified (1:2000); rabbit anti-ICDH (1:10,000) was kind gift Dr. Abraham Sonenshein at Tufts University.
To generate anti-LnaB antibodies, lnaB (lpg2527) was introduced into pDEST17 to generate an N-terminal His fusion under the control of arabinose according to manufacturer’s instructions (Invitrogen). The His-LnaB fusion protein expressed in E. coli strain BL21-A1 was insoluble, so the recombinant protein was isolated from the pellet. One liter of BL21-A1 expressing His-LnaB was pelleted and resuspended in 50 ml of 6 M guandine hydrochloride in 100 mM NaH2PO4, 10 mM Tris-HCl, pH 7.5. His-LnaB was bound to Ni-NTA beads (Qiagen) by incubating at 4°C overnight. His-LnaB was eluted with 6 M guandine hydrochloride buffer containing 500 mM imidazole at pH 7.5. The eluant was dialyzed overnight into 6 M urea buffer containing 20 mM Tris-HCl pH 7.4, 20% glycerol, 500 mM NaCl, then again in a two-fold stepwise dialysis to remove the urea. The fusion protein, which was now soluble in the absence of denaturant, was used to immunize rabbits by the Quick draw protocol from Pocono Rabbit Farm (Canadensis PA).
Subcellular localization
1 ×105 Hela cells were co-transfected with 0.5 μg of EGFP fusion plasmids and 0.2 μg of pmCherry-Sec61 with 1 μl of lipofectamine 2000. After 24 hours, the transfected cells were fixed for 15 minutes at room temperature in 4% paraformaldhyde in PBS. Representative images were taken using a 63X lens on a Zeiss IM200 fluorescent microscope. For all images the contrast and brightness of individual channels were adjusted linearly in Adobe Photoshop.
LAMP-1 targeting assay
L. pneumophila strains were tested for avoidance of a LAMP-1-containing compartment as targeting described, challenging bone marrow-derived macrophages from A/J mice (Conover et al., 2003). The number of LAMP-1-positive phagosomes per bacterial strain was quantified by counting 100 bacteria-associated macrophages on each of three coverslips. The mean and standard error were calculated a typical experiment.
Supplementary Material
Acknowledgments
We would like to thank Joyce Yang, Molly Bergman, Matt Heidtman, Elizabeth Creasey, Tamara O’Conner for careful review of the manuscript. We thank Joe Vogel, Elizabeth Creasey, Sina Mohammadi, and Matt Heidtman for providing plasmids and strains used in this work. This study was supported by the Howard Hughes Medical Institute. VL was supported by T32AI007422 and EH was supported by the German Academic Exchange Service (DAAD). The authors have declared that no competing financial interests exist.
References
- Abu-Zant A, Jones S, Asare R, Suttles J, Price C, Graham J, Kwaik YA. Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell Microbiol. 2007;9:246–264. doi: 10.1111/j.1462-5822.2006.00785.x. [DOI] [PubMed] [Google Scholar]
- Backert S, Ziska E, Brinkmann V, Zimny-Arndt U, Fauconnier A, Jungblut PR, et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2000;2:155–164. doi: 10.1046/j.1462-5822.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- Banga S, Gao P, Shen X, Fiscus V, Zong WX, Chen L, Luo ZQ. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc Natl Acad Sci U S A. 2007;104:5121–5126. doi: 10.1073/pnas.0611030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardill JP, Miller JL, Vogel JP. IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol Microbiol. 2005;56:90–103. doi: 10.1111/j.1365-2958.2005.04539.x. [DOI] [PubMed] [Google Scholar]
- Bartfeld S, Engels C, Bauer B, Aurass P, Flieger A, Bruggemann H, Meyer TF. Temporal resolution of two-tracked NF-κB activation by Legionella pneumophila. Cell Microbiol. 2009;11:1638–1651. doi: 10.1111/j.1462-5822.2009.01354.x. [DOI] [PubMed] [Google Scholar]
- Belyi Y, Niggeweg R, Opitz B, Vogelsgesang M, Hippenstiel S, Wilm M, Aktories K. Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc Natl Acad Sci U S A. 2006;103:16953–16958. doi: 10.1073/pnas.0601562103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyi Y, Tabakova I, Stahl M, Aktories K. Lgt: a family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J Bacteriol. 2008;190:3026–3035. doi: 10.1128/JB.01798-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin J, Nir WJ, Fischer CM, Tayber OV, Errada PR, Grant JR, et al. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-κB. J Biol Chem. 1999;274:12955–12958. doi: 10.1074/jbc.274.19.12955. [DOI] [PubMed] [Google Scholar]
- Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne ED, Roy CR. The Legionella pneumophila IcmSW Complex Interacts with Multiple Dot/Icm Effectors to Facilitate Type IV Translocation. PLoS Pathog. 2007;3:e188. doi: 10.1371/journal.ppat.0030188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campodonico EM, Chesnel L, Roy CR. A yeast genetic system for the identification and characterization of substrate proteins transferred into host cells by the Legionella pneumophila Dot/Icm system. Mol Microbiol. 2005;56:918–933. doi: 10.1111/j.1365-2958.2005.04595.x. [DOI] [PubMed] [Google Scholar]
- Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4:702–707. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- Chen J, de Felipe KS, Clarke M, Lu H, Anderson OR, Segal G, Shuman HA. Legionella effectors that promote nonlytic release from protozoa. Science. 2004;303:1358–1361. doi: 10.1126/science.1094226. [DOI] [PubMed] [Google Scholar]
- Chiaraviglio L, Brown DA, Kirby JE. Infection of cultured human endothelial cells by Legionella pneumophila. PLoS ONE. 2008;3:e2012. doi: 10.1371/journal.pone.0002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover GM, Derre I, Vogel JP, Isberg RR. The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol Microbiol. 2003;48:305–321. doi: 10.1046/j.1365-2958.2003.03400.x. [DOI] [PubMed] [Google Scholar]
- de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, Shuman HA. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 2008;4:e1000117. doi: 10.1371/journal.ppat.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe KS, Pampou S, Jovanovic OS, Pericone CD, Ye SF, Kalachikov S, Shuman HA. Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J Bacteriol. 2005;187:7716–7726. doi: 10.1128/JB.187.22.7716-7726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derre I, Isberg RR. Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect Immun. 2004;72:3048–3053. doi: 10.1128/IAI.72.5.3048-3053.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeley JC, Gibson RJ, Gorman GW, Langford NC, Rasheed JK, Mackel DC, Baine WB. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J Clin Microbiol. 1979;10:437–441. doi: 10.1128/jcm.10.4.437-441.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser DW. The challenges were legion. Lancet Infect Dis. 2005;5:237–241. doi: 10.1016/S1473-3099(05)70054-2. [DOI] [PubMed] [Google Scholar]
- Gabay JE, Blake M, Niles WD, Horwitz MA. Purification of Legionella pneumophila major outer membrane protein and demonstration that it is a porin. J Bacteriol. 1985;162:85–91. doi: 10.1128/jb.162.1.85-91.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Xu H, Li T, Zhou Y, Zhang Z, Li S, et al. A Legionella type IV effector activates the NF-κB pathway by phosphorylating the IκB family of inhibitors. Proc Natl Acad Sci U S A. 2009;106:13725–13730. doi: 10.1073/pnas.0907200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from Gram-negative bacterial peptidoglycan. Science. 2003a;300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003b;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- Grant SR, Fisher EJ, Chang JH, Mole BM, Dangl JL. Subterfuge and manipulation: type III effector proteins of phytopathogenic bacteria. Annu Rev Microbiol. 2006;60:425–449. doi: 10.1146/annurev.micro.60.080805.142251. [DOI] [PubMed] [Google Scholar]
- Hatakeyama M. SagA of CagA in Helicobacter pylori pathogenesis. Curr Opin Microbiol. 2008;11:30–37. doi: 10.1016/j.mib.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Heidtman M, Chen EJ, Moy MY, Isberg RR. Large-scale identification of Legionella pneumophila Dot/Icm substrates that modulate host cell vesicle trafficking pathways. Cell Microbiol. 2009;11:230–248. doi: 10.1111/j.1462-5822.2008.01249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz MA. Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J Exp Med. 1983a;158:1319–1331. doi: 10.1084/jem.158.4.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz MA. The Legionnaires’ disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J Exp Med. 1983b;158:2108–2126. doi: 10.1084/jem.158.6.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz MA, Maxfield FR. Legionella pneumophila inhibits acidification of its phagosome in human monocytes. J Cell Biol. 1984;99:1936–1943. doi: 10.1083/jcb.99.6.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LC, O’Conner T, Isberg RR. (unpublished) [Google Scholar]
- Ingmundson A, Delprato A, Lambright DG, Roy CR. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature. 2007;450:365–369. doi: 10.1038/nature06336. [DOI] [PubMed] [Google Scholar]
- Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-κB. J Biol Chem. 1999;274:14560–14567. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, et al. An induced proximity model for NF-κ B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- Jones JD, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Roy CR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol. 2002;4:945–954. doi: 10.1038/ncb883. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Stein MP, Pypaert M, Roy CR. Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J Exp Med. 2004;199:1201–1211. doi: 10.1084/jem.20031706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, Nunez G. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity. 2008;28:246–257. doi: 10.1016/j.immuni.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- Kubori T, Hyakutake A, Nagai H. Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol Microbiol. 2008;67:1307–1319. doi: 10.1111/j.1365-2958.2008.06124.x. [DOI] [PubMed] [Google Scholar]
- Kustermans G, El Benna J, Piette J, Legrand-Poels S. Perturbation of actin dynamics induces NF-κB activation in myelomonocytic cells through an NADPH oxidase-dependent pathway. Biochem J. 2005;387:531–540. doi: 10.1042/BJ20041318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc Natl Acad Sci U S A. 2006;103:18745–18750. doi: 10.1073/pnas.0609012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand-Poels S, Kustermans G, Bex F, Kremmer E, Kufer TA, Piette J. Modulation of Nod2-dependent NF-κB signaling by the actin cytoskeleton. J Cell Sci. 2007;120:1299–1310. doi: 10.1242/jcs.03424. [DOI] [PubMed] [Google Scholar]
- Lesser CF, Miller SI. Expression of microbial virulence proteins in Saccharomyces cerevisiae models mammalian infection. Embo J. 2001;20:1840–1849. doi: 10.1093/emboj/20.8.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Luo ZQ. The Legionella pneumophila effector SidJ is required for efficient recruitment of endoplasmic reticulum proteins to the bacterial phagosome. Infect Immun. 2007;75:592–603. doi: 10.1128/IAI.01278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick VP, Isberg RR. NF-κB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J Exp Med. 2006;203:2177–2189. doi: 10.1084/jem.20060766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZQ, Isberg RR. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc Natl Acad Sci U S A. 2004;101:841–846. doi: 10.1073/pnas.0304916101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machner MP, Isberg RR. Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev Cell. 2006;11:47–56. doi: 10.1016/j.devcel.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Machner MP, Isberg RR. A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science. 2007;318:974–977. doi: 10.1126/science.1149121. [DOI] [PubMed] [Google Scholar]
- Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, Swanson MS. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med. 2006;203:1093–1104. doi: 10.1084/jem.20051659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AB, Shetron-Rama LM, Swanson MS. Components of the Legionella pneumophila flagellar regulon contribute to multiple virulence traits, including lysosome avoidance and macrophage death. Infect Immun. 2005;73:5720–5734. doi: 10.1128/IAI.73.9.5720-5734.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Delprato A, Ingmundson A, Toomre DK, Lambright DG, Roy CR. The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat Cell Biol. 2006;8:971–977. doi: 10.1038/ncb1463. [DOI] [PubMed] [Google Scholar]
- Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science. 2002;295:679–682. doi: 10.1126/science.1067025. [DOI] [PubMed] [Google Scholar]
- Ninio S, Roy CR. Effector proteins translocated by Legionella pneumophila: strength in numbers. Trends Microbiol. 2007;15:372–380. doi: 10.1016/j.tim.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Ninio S, Zuckman-Cholon DM, Cambronne ED, Roy CR. The Legionella IcmS-IcmW protein complex is important for Dot/Icm-mediated protein translocation. Mol Microbiol. 2005;55:912–926. doi: 10.1111/j.1365-2958.2004.04435.x. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- Pan X, Luhrmann A, Satoh A, Laskowski-Arce MA, Roy CR. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science. 2008;320:1651–1654. doi: 10.1126/science.1158160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178:2380–2386. doi: 10.4049/jimmunol.178.4.2380. [DOI] [PubMed] [Google Scholar]
- Proell M, Riedl SJ, Fritz JH, Rojas AM, Schwarzenbacher R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS ONE. 2008;3:e2119. doi: 10.1371/journal.pone.0002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden AE, Holden DW, Mota LJ. Membrane dynamics and spatial distribution of Salmonella-containing vacuoles. Trends Microbiol. 2007;15:516–524. doi: 10.1016/j.tim.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2006;2:e18. doi: 10.1371/journal.ppat.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy CR, Isberg RR. Topology of Legionella pneumophila DotA: an inner membrane protein required for replication in macrophages. Infect Immun. 1997;65:571–578. doi: 10.1128/iai.65.2.571-578.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci. 2008;65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal G, Purcell M, Shuman HA. Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc Natl Acad Sci U S A. 1998;95:1669–1674. doi: 10.1073/pnas.95.4.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw MH, Reimer T, Kim YG, Nunez G. NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr Opin Immunol. 2008;20:377–382. doi: 10.1016/j.coi.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Case CL, Archer KA, Nogueira CV, Kobayashi KS, Flavell RA, et al. Type IV Secretion-Dependent Activation of Host MAP Kinases Induces an Increased Proinflammatory Cytokine Response to Legionella pneumophila. PLoS Pathog. 2008a;4:e1000220. doi: 10.1371/journal.ppat.1000220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Roy CR. Host cell processes that influence the intracellular survival of Legionella pneumophila. Cell Microbiol. 2008b;10:1209–1220. doi: 10.1111/j.1462-5822.2008.01145.x. [DOI] [PubMed] [Google Scholar]
- Shohdy N, Efe JA, Emr SD, Shuman HA. Pathogen effector proteinscreening in yeast identifies Legionella factors that interfere with membrane trafficking. Proc Natl Acad Sci U S A. 2005;102:4866–4871. doi: 10.1073/pnas.0501315102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisko JL, Spaeth K, Kumar Y, Valdivia RH. Multifunctional analysis of Chlamydia-specific genes in a yeast expression system. Mol Microbiol. 2006;60:51–66. doi: 10.1111/j.1365-2958.2006.05074.x. [DOI] [PubMed] [Google Scholar]
- Sory MP, Cornelis GR. Translocation of a hybrid YopE-adenylate cyclase from Yersinia enterocolitica into HeLa cells. Mol Microbiol. 1994;14:583–594. doi: 10.1111/j.1365-2958.1994.tb02191.x. [DOI] [PubMed] [Google Scholar]
- Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci U S A. 2000;97:1263–1268. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63:3609–3620. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Harb OS, Connelly PS, Robinson CG, Roy CR. How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J Cell Sci. 2001;114:4637–4650. doi: 10.1242/jcs.114.24.4637. [DOI] [PubMed] [Google Scholar]
- Trosky JE, Liverman AD, Orth K. Yersinia outer proteins: Yops. Cell Microbiol. 2008;10:557–565. doi: 10.1111/j.1462-5822.2007.01109.x. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Akira S. Toll-Like receptors (TLRs) and their ligands. Handb Exp Pharmacol. 2008:1–20. doi: 10.1007/978-3-540-72167-3_1. [DOI] [PubMed] [Google Scholar]
- VanRheenen SM, Luo ZQ, O’Connor T, Isberg RR. Members of a Legionella pneumophila family of proteins with ExoU (phospholipase A) active sites are translocated to target cells. Infect Immun. 2006;74:3597–3606. doi: 10.1128/IAI.02060-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent CD, Friedman JR, Jeong KC, Buford EC, Miller JL, Vogel JP. Identification of the core transmembrane complex of the Legionella Dot/Icm type IV secretion system. Mol Microbiol. 2006;62:1278–1291. doi: 10.1111/j.1365-2958.2006.05446.x. [DOI] [PubMed] [Google Scholar]
- Vogel JP, Andrews HL, Wong SK, Isberg RR. Conjugative transfer by the virulence system of Legionella pneumophila. Science. 1998;279:873–876. doi: 10.1126/science.279.5352.873. [DOI] [PubMed] [Google Scholar]
- Zusman T, Aloni G, Halperin E, Kotzer H, Degtyar E, Feldman M, Segal G. The response regulator PmrA is a major regulator of the icm/dot type IV secretion system in Legionella pneumophila and Coxiella burnetii. Mol Microbiol. 2007;63:1508–1523. doi: 10.1111/j.1365-2958.2007.05604.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.