

Abstract
Our understanding of biological processes as well as human diseases has improved greatly thanks to studies on model organisms such as yeast. The power of scientific approaches with yeast lies in its relatively simple genome, its facile classical and molecular genetics, as well as the evolutionary conservation of many basic biological mechanisms. However, even in this simple model organism, systems biology studies, especially proteomic studies had been an intimidating task. During the past decade, powerful high-throughput technologies in proteomic research have been developed for yeast including protein microarray technology. The protein microarray technology allows the interrogation of protein–protein, protein–DNA, protein–small molecule interaction networks as well as post-translational modification networks in a large-scale, high-throughput manner. With this technology, many groundbreaking findings have been established in studies with the budding yeast Saccharomyces cerevisiae, most of which could have been unachievable with traditional approaches. Discovery of these networks has profound impact on explicating biological processes with a proteomic point of view, which may lead to a better understanding of normal biological phenomena as well as various human diseases.
Keywords: Yeast proteomics, Protein microarray, Protein interaction detection, Protein modification detection, Systems biology
Graphical Abstract
1. Introduction
Until relatively recently, investigation of the full proteome has been a daunting task. One of the main reasons was the incomplete definition of the proteome due to the lack of comprehensive genomic information, as well as technical limitations for large scale profiling of proteins. During the past few decades, improved and novel technologies have emerged as powerful tools for proteomic studies including shotgun proteomics by mass spectrometry technology [1] and protein microarray technology [2]. Mass spectrometry technology, combined with various sample preparation methods, has been used extensively in biological research such as proteome profiling [3], protein–protein interaction mapping [4], [5] and identification of post-translational modification [6], [7], [8]. The greatest advantage of this technology lies in its capability of high throughput protein identification and quantification. Nevertheless, mass spectrometry still has its limitations, such as the undersampling issue (insufficient coverage of the proteome) [9], [10] as well as bias against low abundant proteins [11]. For the purpose of this review mass spectrometry technology will not be covered here in detail. Protein microarray technology, on the other hand, does not have the above limitations for mass spectrometry owing to its particular platform. Protein microarray technology [12], [13] was developed upon the completion of the genome sequence of multiple organisms, including the budding yeast Saccharomyces cerevisiae [14] and humans (Homo sapiens) [15]. Determination of genomic sequences leads to the annotation of known and predicted open reading frames (ORFs) in these genomes, which makes it possible to express the full or partial proteome with large-scale cloning and gene expression methods [2].
Protein microarray typically contains hundreds to thousands of proteins that are arrayed in an addressable format. Protein microarrays are generally one of two types: analytical/diagnostic microarrays and functional microarrays [16], [17]. One form of analytical/diagnostic microarrays is the antibody microarray, in which specific antibodies against defined target proteins are arrayed on the surface of a support material (such as glass slides), and are used for the detection and quantification of their specific antigens [18], [19]. This type of microarray has great potential for clinical diagnosis and clinical research, and its advantages and disadvantages are reviewed in [17], [20], [21]. The other type of protein microarray is the functional microarray. These arrays usually contain a large number of individually expressed and purified functional, full-length proteins or peptides printed in a high-density format on support surfaces, and can represent the complete or partial proteome of a given organism [22]. This type of protein microarray has been used in studies of protein–protein, protein–DNA, and protein–small molecule interactions as well as protein modifications (Fig. 1 ) [2], [17], [23], [24], [25]. Many of these studies were done in the budding yeast S. cerevisiae, and will be discussed in detail below. Functional protein microarrays containing human proteins have also been widely applied in various clinical studies of cancer, autoimmune diseases and viral infections [26], [27], [28], [29], [30], [31], [32], which will not be covered here.
Fig. 1.

Application of functional protein microarray and novel insights gained in protein interaction and modification. Protein–protein interactions may be detected with either fluorophore-labeled proteins or specific recognition antibodies to the probe protein. Protein–nucleic acid interactions can be visualized with fluorophore-labeled DNA/RNA. Protein–small molecule interactions may be identified with biotinylated small molecules and fluorophore-labeled streptavidin. Posttranslational modifications of proteins are not drawn to scale. P, phosphorylation; U, ubiquitination; Ac, acetylation.
In this article we will review functional protein microarray technologies in yeast. We cover the various methods for manufacturing of protein microarray, and discuss their applications in studying protein–protein, protein–DNA, protein–small molecule interactions, and protein post-translational modifications (phosphorylation S-nitrosylation and ubiquitination) (Fig. 1). We will also discuss the advantages and limitations of this technology.
2. Manufacture of high-density protein microarray
Since our group generated the first protein microarray covering 5800 ORFs of the budding yeast S. cerevisiae [2], various methods have been developed in protein microarray production. A diagram of the manufacture of protein microarray is shown in Fig. 2 .
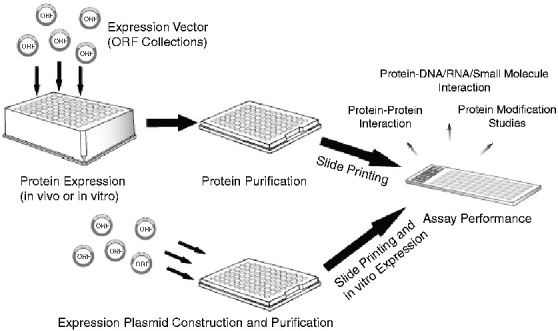
Fig. 2.

Manufacture and application of functional protein microarrays. Functional protein microarrays can either be manufactured by printing a library of in vitro or in vivo expressed, affinity-purified proteins on to coated glass slides with a microarray printer (top), or printing the protein expression plasmids on to the slides followed by on-slide in vitro expression (bottom). The printed microarrays are then ready for various downstream applications, such as protein–protein/DNA/small molecule interaction and protein post-translational modification studies.
There are different support surfaces for making protein microarray, including nanowells [33], solid surfaces (such as glass slides) [2], [34], and absorbent surfaces (such as polyacrylamide gel pads) [35]. The pros and cons of these surfaces are reviewed by Zhu et al. in [17] and [36]. The polyacrylamide gel pad technology has not been applied extensively due to cumbersome slide preparation and inconvenient slide handling (e.g. hard to change buffers); similarly, the nanowell surface is less convenient to make and use [17], [36]. Currently, the most popular high-density protein microarrays are manufactured on chemically modified or coated glass microscope slides (e.g. nitrocellulose or amino-silane-coated slides) using a standard contact printer [22], [25]. This format is compatible with many commercial scanners. It is noteworthy that, even within this format, different surface chemistries have different attributes in terms of their protein binding ability, protein function/folding and background, and one has to carefully choose the optimal surface that best fits the specific need of the experiment [21]. For example, stronger protein attachment, such as gold-coated glass surface [37] or reactive surfaces with bifunctional cross-linkers [2] can retain the proteins firmly on the surface with covalent cross-linking, but may decrease their activity due to steric hindrance or disruption of proper folding; however, these surfaces will allow detection methods such as Surface Plasmon Resonance or mass spectrometry to study the dynamics of biochemical reactions on these proteins [21]. In addition, we found that amino-silane-coated glass slides provide the highest signal-to-noise ratio in kinase studies [25].
Proteins for functional microarrays are either produced individually prior to printing [2] or produced in situ by coupling in vitro transcription and translation of DNA that is printed directly on the surface of the array (e.g. the Nucleic Acid Programmable Protein Array, NAPPA) [38]. Our group has built two collections of full-length yeast genes cloned in expression plasmids that produce either N-terminal GST (glutathione-S-transferase) -tagged or C-terminal TAP (tandem affinity purification) -tagged fusion proteins [39], [40]. The recombinant yeast proteins are expressed in individual yeast clones in a 96-well format, and purified using corresponding affinity tags (GST or TAP). The purified proteins are then arrayed with a 48-pin ESI contact printer on nitrocellulose or amino-silane-coated glass slides in an addressable, high-density format. The detailed protein microarray manufacture protocols can be found in references [22], [25]. In addition to the yeast functional microarrays, we have also produced an Arabidopsis array [41], [42] and a coronavirus array [26]. Invitrogen has produced a human protein array which currently contains more than 9000 proteins (expressed individually using Baculovirus/sf9 expression systems). With a similar method, the group led by Heng Zhu at the Johns Hopkins University also fabricated an Escherichia coli proteome chip containing 4256 proteins encoded by the K12 strain, which is the first reported prokaryotic proteome microarray [43]. Working with Chuan He's group at the University of Chicago, the authors identified proteins with DNA damage-recognizing activity by probing this array with DNA probes containing a single base-mismatch or an abasic site, which included the known cold-shock DNA-binding protein CspE, and proteins with previous unknown functions, such as YbcN and YbaZ [43]. In this study, the authors further validated the function of YbcN and YbaZ with biochemical analyses, and identified the potential binding partner of YbaZ with the same arrays, demonstrating great potentials of functional protein microarrays. The approach of producing proteins and directly spotting them has the following advantages: first of all, in vivo expression of the proteins may help ensure their proper folding and post-translational modifications; second, the individual expression clones allow us to perform single protein quality control, if needed, over the overexpressed collections compared to the coupled transcription-translation method developed by LaBaer et al. [38]. In contrast, the NAPPA method by LaBaer et al. excels in the simplified steps with coupled in vitro, on-slide transcription and translation of the proteins, which saves considerable time and effort [38].
Various detection and data acquisition methods for protein microarray are used to meet different purposes of proteomic studies. For protein–protein, protein–DNA and protein–small molecule interaction studies, these arrays are usually probed with fluorescently labeled molecules and the signals are then acquired with a confocal laser scanner [2], [44], [45]. These interactions can also be detected with oligonucleotide-conjugated probes followed by Rolling Circle Amplification [46]. For kinase assays, isotope-labeled ATP and autoradiography film exposure are often used for data acquisition [25], [33], [47].
3. Detection of protein–protein, protein–dna and protein–small molecule interactions using protein microarray
Because of its unique features, protein microarrays provide a powerful and convenient platform to study protein–protein, protein–nucleic acid and protein–small molecule interaction networks. Unlike DNA and RNA, whose functional information depends mainly on the sequence of individual molecules (with the exception of ribozymes), the function and proper regulation of proteins often requires their 3-dimensional conformation and post-translational modifications. Moreover, the function of proteins also depends heavily on their binding partners in the functional complexes. Therefore, to completely understand the function of a given protein, it is critical to know the information on its interaction to various molecular species in the living cells, whether they are other proteins, nucleic acids, and small molecules.
Much of the potential protein–protein interaction information in yeast was obtained by yeast two-hybrid approaches [48], [49], which is very labor-intensive. Currently the most comprehensive protein–protein binary interaction data in S. cerevisiae were obtained by Yu et al. [50], by performing high-throughput yeast two-hybrid screening with 3917 bait proteins and 5246 prey proteins, which yielded 1809 interactions among 1278 proteins. Ninety-four randomly chosen interactions were validated with a precision rate of 94–100% in this study. In addition, a high quality binary protein interaction map was generated in combination with other available data sets including the “Uetz-screen” by Uetz et al. [49], the “Ito-score” by Ito et al. [51], and high-throughput affinity purification/mass spectrometry data sets by Gavin et al. [4] and Krogan et al. [5].
Protein microarray has many advantages over the yeast two-hybrid method in studying protein interactions. First, once made, its in vitro nature does not require yeast culture, transformation and mating, which greatly saves time and effort. Second, it uses fluorescently labeled probes instead of reporter genes, therefore the interaction signal can be readily quantified with a laser scanner in a matter of minutes, and the relative fluorescence intensity can also reflect the binding strength of the two interacting proteins. Moreover, protein microarray also allows many types of molecules to be used as probes (e.g. DNA and small molecules) besides proteins or peptides as compared with the yeast two-hybrid method which only allows detection of protein–protein interactions.
Since its inception, our group has long been interested in pursuing protein–protein and protein–small molecule interaction networks [2]. We obtained the binding profile of calmodulin by probing the array with biotinylated calmodulin and Cy3-labeled streptavidin. Calmodulin is an important calcium binding protein which is highly conserved between species and participates in the regulation of many signaling pathways [52]. With this high throughput method, we identified 33 previously unknown potential binding partners of calmodulin, as well as a novel binding-motif, (I/L)QXXK(K/X)GB for this protein (Fig. 2 in ref. [2]). Meanwhile, to test the possibility of detecting protein–small molecule interactions, we also probed these arrays with phosphoinositide (PI), a well known secondary messenger lipid. We identified 150 novel lipid-binding proteins, which include membrane-associated proteins as well as proteins involved in glucose metabolism [2]. Our study was the first to show the capability of protein microarray technology to pursue protein–protein and protein–small molecule interactions.
Since its first debut a decade ago, protein microarray technology has been used to discover protein interacting partners/networks in a wide range of applications. For detecting protein–protein interactions, Hesselberth et al. identified interacting proteins of the WW domain in S. cerevisiae [53]. The WW domain is a highly conserved domain in many organisms including yeast and human. Proteins containing these domains are involved in many cellular processes including cell cycle regulation, targeted protein degradation and transcription activation [54], [55], [56]. There are 10 WW domain-containing proteins (with 13 WW domains) in S. cerevisiae, including Prp40 which is involved in mRNA splicing, the ubiquitin ligases Rsp5 and SSm4, the histone methyltransferase Set2, the peptidyl-prolyl isomerase Ess1, and proteins with still unknown functions such as YPR152C, Wwm1, Aus1, Vid30 and Alg9 [53]. Hesselberth et al. determined the binding partners of 12 of the 13 WW domains by probing them on the yeast protein arrays, and identified 587 high-confidence interactions with 207 proteins [53]. These WW-domain interaction proteins are enriched in cofactor metabolism, peroxisome localization, and ER localization with Gene Ontology annotation classifications, suggesting their potential functions. They compared their protein microarray findings with yeast two-hybrid and affinity-purification/mass spectrometry data available at that time and identified 19 unreported interacting partners. Interestingly they also found that some WW-domain containing proteins, such as Rsp5, Alg9, Wwm1 and Ssm4 also have conserved WW-domain binding motifs, which suggested possible multimerization between these proteins. Using a similar approach, Galindo et al. also identified 4 binding partners for the Aeromonas hydrophila cytotoxic enterotoxin Act in yeast, one of which was the vesicle tethering protein Vsp52, indicating that Act might mediate its function through interfering with vesicle transport of the host cells [57]. Protein microarray technology can also be used for specific protein quantification. Park et al. fabricated an antibody protein microarray (named by the authors as “reverse-phase protein microarray”), and quantified the protein N-myc Downstream Regulated Gene 2 (NDRG2) in hepatocellular carcinoma patients and normal controls and observed significant lower expression of this protein in cancerous tissues [58].
Protein microarrays have also been used to identify protein-nucleic acid interactions. Our group probed the yeast protein microarray with Cy3-labeled single-stranded (ssDNA) or double-stranded (dsDNA) yeast genomic DNA and identified 84 ssDNA-binding, 58 dsDNA binding, and 131 ssDNA and dsDNA-binding proteins (schematic design shown in Fig. 1 as well as Fig. 1 in ref. [59]). We chose 8 proteins previously unreported for their DNA-binding activity for further validation, and 3 proteins were confirmed by Chromatin-Immunoprecipitation followed by probing genomic DNA microarrays (ChIP-Chip; [60]). Of the 3 validated proteins, Arg5,6 was found to bind both mitochondrial (15 S ribosomal DNA, COX1, COX3, and COB1) and nuclear genes (PUF4, PHO23 and THI13), and participate in both transcription elongation and RNA-processing. Using a similar approach, Hu et al. identified conserved DNA motif-binding proteins using a protein microarray containing 4191 human proteins [61]. Interestingly, besides known transcription factors, they also identified over 300 previously unknown or unexpected DNA-binding proteins, including ERK2, a multifaceted mitogen-activated protein kinase [62], which also acts as a transcription repressor in the interferon gamma signaling pathway. Another interesting study was reported by Zhu et al. on the identification and validation of binding proteins to the clamped adenine motif (CAM) of Brome mosaic virus (BMV), a model positive-strand RNA plant virus [63]. As BMV can also replicate in S. cerevisiae, the authors probed the yeast proteome array (containing ~ 5000 purified proteins) with both Cy3-labeled CAM-containing RNA and a Cy5-labeled control, and identified 12 hits that showed equal or stronger signals than BMV's own capsid protein (CP). The authors focused on 2 of the 3 strongest binders, Pus4 and App1, and found that they could also inhibit positive-strand RNA accumulation and systemic viral infection in vivo. Their further investigation demonstrated that Pus4 inhibited in vivo viral systemic spread by affecting virion formation through a direct competition with CP. This study shows a good example of the capability of functional protein microarray in the discovery of interactions of in vivo physiological importance, and in this case, components involved in antiviral response.
Besides protein–protein and protein–DNA/RNA discoveries, protein microarrays were also used to identify protein–small molecule interactions. Rapamycin is a small molecule drug that can induce starvation response and inhibit cell growth through its target TOR (Target of Rapamycin), a highly conserved kinase regulating cell proliferation and metabolism, in yeast and humans [64]. Rapamycin analogs are promising chemotherapeutic drugs to treat cancer [65]. Jiang et al. identified small molecules that could enhance (small-molecule enhancers of rapamycin, or SMERs) or inhibit (small-molecule inhibitors of rapamycin, or SMIRs) rapamycin effects in yeast, and discovered intracellular protein targets of two SMIRs, SMIR3 and SMIR4, by probing the yeast protein microarray with biotinylated SMIRs [66]. Among the strongest protein targets identified in this study, two proteins, Tep1p and Nir1p, were shown to be components of the TOR signaling pathway, suggesting that the protein array method is capable of detecting specific protein–small molecule interactions. By probing the protein arrays with biotinylated polyanions (two proteins, two compounds, and DNA) Salamat-Miller et al. also identified a total of 893 polyanion-binding proteins in S. cerevisiae [67]. Interestingly, the polyanions and their binding proteins are found to form a network that is involved in maintaining the structure and activity of the yeast cells.
4. Detection of post-translational modifications using protein microarray
The proteome ultimately carries out most cellular functions, and is regulated by many different types of post-translational modifications. These modifications include phosphorylation, glycosylation, acetylation, ubiquitination, SUMOylation, and S-nitrilation [68], [69], [70]. Therefore, the status of post-translational modification of the proteome is a snapshot of the dynamic activities of the living cell.
The specific platform of protein microarray makes it ideal to identify proteome-wide potential targets for protein post-translational modification enzymes. The high throughput format enables screening of potential substrates in the whole or partial proteome in a single assay, and its in vitro nature allows fine and accurate controls over the assay conditions.
Of all post-translational modifications, phosphorylation is the most studied. The kinome, or collection of all kinases in the cell, is responsible for the dynamic phosphorylation of proteins and controls many different cellular processes. Levels of kinase activities are likely to be very tightly controlled; indeed Sopko et al. examined the influence of individual overexpression of more than 80% of all yeast genes and observed enrichment of kinases and phosphatases in the list of “toxic gene set”; a set of genes whose overexpression significantly slowed yeast growth [71]. Several groups including ours have carried out large-scale studies to identify the substrates of yeast kinases at the proteomic level. In 2000 our group screened 119 of the 122 yeast kinases with 17 different substrates (including the kinases themselves for monitoring autophosphorylation) on a prototype of protein microarray [33]. A schematic design was shown in Fig. 3 . The substrates were immobilized onto nanowell protein chips and phosphorylation events were identified by adding 33P-γ-ATP and a specific yeast kinase and exposing the chip to a phosphoimager. We discovered that more than 60% of the kinases autophosphorylated themselves, and 94% of the tested kinases had at least one substrate in vitro, with 32 of them specifically phosphorylating one or two substrates. Twenty-seven kinases were found to phosphorylate poly(Tyr-Glu), which quadrupled the number of identified tyrosine kinases (seven) reported at that time. Moreover, these tyrosine kinases preferentially contain 3 conserved lysines and one conserved methionine near the catalytic region, indicating their potential roles in substrate selection. The same method was later used to identify Hrr25p as a kinase for the zinc-finger transcription factor Crz1, which turned out to negatively regulate Crz1 activity and nuclear localization by phosphorylation in vivo [72]. Our group later expanded the study to search for the substrates of 87 different S. cerevisiae kinases in a large set of more than 4400 full-length, functional yeast proteins with a yeast protein microarray containing 4400 yeast proteins [47]. In this study we discovered about 4200 phosphorylation events affecting 1325 proteins and generated the first version of the phosphorylation network in yeast (schematic design shown in Fig. 3). There were many interesting findings in this work. We found that each kinase had a unique substrate profile and most phosphorylation substrates were recognized by 3 or fewer kinases. Even within closely related kinases such as Tpk1, Tpk2 and Tpk3, there seem to be a low overlap between their substrate profiles (only 12.3% of all their identified substrates could be recognized by more than one of the three kinases). Moreover, we also found that kinases might display distinct substrate preferences when they form complexes with different proteins. We determined the consensus motifs for 11 kinases in this study using a method developed by Jonassen et al. [73], and further investigated this in a larger scale in a separate study with a rapid peptide screening approach (schematic design shown in Fig. 3), in which we determined the consensus phosphorylation motifs for 61 kinases [74]. Lastly, by combining our profiling data with available transcription factor binding and protein interaction data sets [49], [51], [75], [76], [77], [78], [79], we discovered 8 regulatory modules in the phosphorylation network. All of the above indicated that kinases have evolved to have their unique, specific roles in carrying out molecular and cellular functions in the eukaryotic cells.
Fig. 3.

Schematic diagram for protein kinase target identification, consensus phosphorylation motif identification and determination.
Besides protein microarrays, large-scale profiling of the protein phosphorylation networks was also approached with other methods. Instead of direct proteomic approaches, Fiedler et al. determined the epistasis of binary interactions between all yeast kinases, phosphatases and key cellular regulators by building an epistatic miniarray profile (E-MAP) through the generation of double mutants [80]. In this study, by analyzing “Triplet Genetic Motifs”, the authors detected both known (such as Cak1 and its substrates Ctk1 and Smk1) and previously undiscovered (e.g. involvement of Cak1 and Fus3 in the Set2/Rpd3C(S) pathway) interactions with this method. Moreover, Korf et al. generated an antibody-based protein microarray, which can be used to monitor time-dependent quantitative changes in the phosphorylation states of individual kinases [81]. This method could be applied potentially to monitor many targets at the same time; however, like other antibody protein microarrays, this method significantly depends on the quality and specificity of the antibody pairs for each protein [21]. Alternatively, the phospho-proteome can also be profiled with quantitative mass spectrometry [82], [83]. Soufi et al. identified 5534 unique phospho-peptides associated with the osmotic response of S. cerevisiae with a quantitative high-resolution mass spectrometry of their phospho-proteome, and observed that greater than 15% of these changes occured within 5 min after the start of the treatment, suggesting that phosphorylation of a group of specific proteins is part of the early response to osmotic changes [82].
Other post-translational modifications have also been investigated with protein microarray technology. By probing the yeast protein microarray with Concanavalin A or Wheat-Germ Agglutinin, both of which are lectins that bind glycans on proteins, Kung et al. identified 534 glycosylated yeast proteins including 30 mitochondrial proteins and a number of nuclear proteins; 406 of the glycosylated protein were unreported previously [84]. Of the novel identifications, they validated 45 by gel mobility shift assay. Interestingly, they also found that inhibition of glycosylation by N-linked glycosylation inhibitor tunicamycin affected the localization of some mitochondrial proteins, such as Ydr065wp and Lpe10p, which suggests that proper protein glycosylation is important for their normal localization and function. In addition to glycosylation, Tao et al. used an analogous approach to examine poly(ADP-ribosyl)ation in yeast with protein microarray [85]. Poly(ADP-ribosyl)ation of proteins is usually carried out by the enzyme poly(ADP-ribose) polymerase-1 (PARP-1), a pluripotent enzyme involved in both DNA damage sensing mechanism and regulation of gene expression [86], [87]. The authors identified 33 PARP-1 substrates in this study, including six proteins involved in ribosome biogenesis, indicating that poly(ADP-ribosyl)ation may be required for the normal biogenesis process of the ribosomes.
Acetylation and deacetylation of the lysine residues in proteins, especially histones are important events in regulating chromatin-related processes and gene expression, although not much was known for the non-chromatin-associated substrates of these histone acetyltransferases and deacetylases [88], [89]. Of the many different histone acetyltransferases, only the catalytic enzyme Esa1p in the essential nucleosome acetyltransferase of H4 (NuA4) complex was found required for the viability of the cells, indicating this complex may have additional roles beyond gene expression regulation [90]. By incubating the NuA4 complex with yeast proteome microarrays in the presence of 14C labeled Acetyl-CoA, Lin et al. discovered a large number of novel non-histone substrates for NuA4 and confirmed 13 of them with standard in vitro activity assays [88]; and more importantly, the authors also validated these substrates with in vivo experiments [91]: the levels of acetylated substrates were found decreased dramatically in ESA1 Ts (esa1-531) mutant cells when they were grown at the nonpermissive temperature, which proved that these proteins are real in vivo targets of Esa1p. The authors then focused on Pck1p (phosphoenolpyruvate carboxykinase), a metabolic enzyme involved in gluconeogenesis, and found that the acetylation of Pck1p is essential for its enzymatic activity and the life span of yeast. This study is one of the first to examine an important acetyltransferase complex for non-histone-related regulatory functions in the cell and suggests that histone acetyltransferases and deacetylases may also have other important cellular roles besides chromatin-related processes. Moreover, in vivo validation of the acetylation targets of the NuA4 complex suggested that using enzyme complexes instead of individual proteins in protein microarray studies may help identify substrates that are more physiologically relevant.
Ubiquitination is another critical post-translational modification involved in various cellular processes. It facilitates proteosome-mediated protein degradation/recycling [92] as well as activation of cell signaling molecules [93]. Gupta et al. identified the substrates of Rsp5, a ubiquitin-protein ligase (E3) in yeast by both in vitro ubiquitination and protein interaction assays [94]. To identify substrates that could be ubiquitinated by Rsp5, the authors incubated the yeast protein microarrays with Rsp5 and FITC-labeled ubiquitin; alternatively, the arrays were probed with Alexa Fluor 647-labeled Rsp5 to identify interacting proteins. One hundred and fifty proteins were identified in the ubiquitination assay and 155 in the interaction assay, of which 64 proteins were identified in both sets of experiments. Of these identified substrates, the authors discovered more details of the known PY motif for Rsp5 binding. They found that Prolines, Serines and Alanines were enriched in the (L/P)PxY motif (at position x) which may also contribute to substrate specificity.
Protein microarray technology was also used for the discovery of other protein post-translational modifications such as S-nitrosylation [95]. This type of post-translational modification plays an important role in nitric oxide (NO) and endogenous S-nitrosothiol (SNO) signaling, abnormal regulation of which is associated with diseases such as neurological dysfunction and cancer [95], [96], [97]. Using a biotin switch technique that converts SNO to S-biotinylated Cys [98], which is then readily detectable with anti-biotin antibody, Foster et al. identified several hundred SNO-proteins after S-nitrosocysteine treatment of the yeast protein microarrays [95]. These proteins were enriched with proteins with active-site Cys thiols, however, neither secondary structures nor Cys thiol nucleophilicity could explain substrate specificity. The authors then found out that S-nitrosylation efficiency might depend on the stereochemistry and structure of the NO-donor as well as allosteric effectors.
5. Advantages and limitations of protein microarray technology
As reviewed above, protein microarray technology serves as a powerful and convenient tool in a large-scale, high-throughput manner for proteomic studies such as protein interactions and modifications. The advantages of the protein microarray technology are (also refer to review by Zhu et al. [17]): its convenient and straightforward probing using a wide variety of standard detection platforms, the ability to control and fine-tune experimental conditions, and its unbiased protein target selection (regardless of their endogenous cellular abundance) due to its particular fashion of manufacture. The latter enables the inclusion of normally underrepresented proteins such as low abundant proteins, membrane and secreted proteins, and conditionally expressed proteins.
Protein microarray technology also have limitations. First, the scope of protein microarray relies heavily on the availability of genomic information of the specific organisms. Therefore, this technology is not capable of covering unknown ORFs/splicing variants, and is unavailable for organisms whose genome information is unknown. A related limitation is that for most genes usually only one splicing variant is used to represent the specific gene, therefore the splicing diversity of the represented proteome is limited [12]. Second, as proteins are purified from the cells, they may contain mixed post-translational modifications or even co-purified interacting proteins, which may interfere with the protein binding, kinase assays and post-translational modification detection. In vitro expression methods may avoid contamination from other tightly interacting proteins; however, proteins produced this way may not be properly folded or modified. Third, as protein microarray-based studies are in vitro studies, off-target interactions and modifications that do not normally happen in cell may also occur. For this reason protein microarray findings should be validated with more direct methods such as immunoprecipitation, immunoblotting and in vitro and in vivo activity assays. Furthermore, as the cost for manufacturing protein microarrays is still high, it can be expensive to study large proteomes such as the human proteome. Therefore in studies on these proteomes they are often partially represented. Lastly, the specific platform of protein microarray usually allows only one or two probes/enzymes per each assay on a single proteome array, which, with the cost of manufacturing the protein microarrays, limits researchers to investigate only one or a few probes/enzymes at a time. This limitation will be significantly alleviated, however, with the drop of manufacturing cost as well as improvements in multiplexing technologies.
6. Closing remarks
Since its debut a decade ago, protein microarray technology has evolved into a powerful tool in proteomic studies. Application of this technology in the model organism S. cerevisiae has led to numerous significant scientific findings at the proteomic level, including protein–protein, protein–DNA and protein–small molecule interactions as well as post-translational modifications. Since many of these mechanisms are conserved through evolution, some of these findings are expected to be relevant to those occurring in other organisms, such as plant and human [99]. In fact, multiple studies mentioned above have already confirmed the counterparts of their findings in humans [57], [85], [88].
The proteome is only a part of the complex network in a living organism. It coordinates with the other systems including the genome, the transcriptome, and the metabolome, to carry out complex cellular functions and intercellular communication [100]. With the emergence of new high throughput technologies such as next generation high-throughput sequencing [101] and improved mass spectrometry technologies [1], it becomes possible to simultaneously generate integrated networks at the systems level from the same sample sets in a time dependent manner, if desired. Time-resolved global networks generated from quantitative data sets of the proteome, the genome, the methylome, the transcriptome and the metabolome, with environmental factors such as the microbiome (Fig. 4 ), will enable a comprehensive view of miscellaneous biological processes, as well as their roles in natural courses such as development and aging, and in pathogenesis of human diseases such as cancer.
Fig. 4.

Comprehensive understanding of biological processes through integrated information of biological systems and environmental factors.
Acknowledgements
We thank Drs. Hogune Im and Linfeng Wu for their thoughtful comments and help in proofreading this manuscript. Research in the Snyder lab is supported by grants from the National Institutes of Health.
References
- 1.Wu L., Han D.K. Overcoming the dynamic range problem in mass spectrometry-based shotgun proteomics. Expert Rev Proteomics. 2006;3:611–619. doi: 10.1586/14789450.3.6.611. [DOI] [PubMed] [Google Scholar]
- 2.Zhu H., Bilgin M., Bangham R., Hall D., Casamayor A., Bertone P. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 3.Swaney D.L., Wenger C.D., Coon J.J. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J Proteome Res. 2010;9:1323–1329. doi: 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gavin A.C., Aloy P., Grandi P., Krause R., Boesche M., Marzioch M. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 5.Krogan N.J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 6.Chen S.H., Albuquerque C.P., Liang J., Suhandynata R.T., Zhou H. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J Biol Chem. 2010 doi: 10.1074/jbc.M110.106989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kristjansdottir K., Wolfgeher D., Lucius N., Angulo D.S., Kron S.J. Phosphoprotein profiling by PA-GeLC-MS/MS. J Proteome Res. 2008;7:2812–2824. doi: 10.1021/pr700816k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu P., Duong D.M., Seyfried N.T., Cheng D., Xie Y., Robert J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nesatyy V.J., Groh K., Nestler H., Suter M.J. On the acquisition of + 1 charge states during high-throughput proteomics: implications on reproducibility, number and confidence of protein identifications. J Proteomics. 2009;72:761–770. doi: 10.1016/j.jprot.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Liu H., Sadygov R.G., Yates J.R., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 11.Boratyn G.M., Merchant M.L., Klein J.B. Utlization of human expert techniques for detection of low-abundant peaks in high-resolution mass spectra. Conf Proc IEEE Eng Med Biol Soc. 2006;1:5798–5801. doi: 10.1109/IEMBS.2006.260190. [DOI] [PubMed] [Google Scholar]
- 12.Phizicky E., Bastiaens P.I.H., Zhu H., Snyder M., Fields S. Protein analysis on a proteomic scale. Nature. 2003;422:208–215. doi: 10.1038/nature01512. [DOI] [PubMed] [Google Scholar]
- 13.Wolf-Yadlin A., Sevecka M., MacBeath G. Dissecting protein function and signaling using protein microarrays. Curr Opin Chem Biol. 2009;13:398–405. doi: 10.1016/j.cbpa.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goffeau A., Barrell B.G., Bussey H., Davis R.W., Dujon B., Feldmann H. Life with 6000 genes. Science. 1996;274(546):63–67. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 15.Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 16.Guan H., Kiss-Toth E. Advanced technologies for studies on protein interactomes. Adv Biochem Eng Biotechnol. 2008;110:1–24. doi: 10.1007/10_2007_092. [DOI] [PubMed] [Google Scholar]
- 17.Zhu H., Bilgin M., Snyder M. Proteomics Annu Rev Biochem. 2003;72:783–812. doi: 10.1146/annurev.biochem.72.121801.161511. [DOI] [PubMed] [Google Scholar]
- 18.Sreekumar A., Nyati M.K., Varambally S., Barrette T.R., Ghosh D., Lawrence T.S. Profiling of cancer cells using protein microarrays: discovery of novel radiation-regulated proteins. Cancer Res. 2001;61:7585–7593. [PubMed] [Google Scholar]
- 19.Haab B.B., Dunham M.J., Brown P.O. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001:2. doi: 10.1186/gb-2001-2-2-research0004. RESEARCH0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borrebaeck C.A., Wingren C. Design of high-density antibody microarrays for disease proteomics: key technological issues. J Proteomics. 2009;72:928–935. doi: 10.1016/j.jprot.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 21.Zhu H., Snyder M. Protein chip technology. Curr Opin Chem Biol. 2003;7:55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 22.Fasolo J., Snyder M. Protein microarrays. Methods Mol Biol. 2009;548:209–222. doi: 10.1007/978-1-59745-540-4_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong W., He K., Covington M., Dinesh-Kumar S.P., Snyder M., Harmer S.L. The development of protein microarrays and their applications in DNA-protein and protein–protein interaction analyses of Arabidopsis transcription factors. Mol Plant. 2008;1:27–41. doi: 10.1093/mp/ssm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacBeath G. Protein microarrays and proteomics. Nat Genet. 2002;32(Suppl):526–532. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
- 25.Mok J., Im H., Snyder M. Global identification of protein kinase substrates by protein microarray analysis. Nat Protoc. 2009;4:1820–1827. doi: 10.1038/nprot.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu H., Hu S., Jona G., Zhu X., Kreiswirth N., Willey B.M. Severe acute respiratory syndrome diagnostics using a coronavirus protein microarray. Proc Natl Acad Sci USA. 2006;103:4011–4016. doi: 10.1073/pnas.0510921103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hudson M.E., Pozdnyakova I., Haines K., Mor G., Snyder M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc Natl Acad Sci USA. 2007;104:17494–17499. doi: 10.1073/pnas.0708572104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Q., Liu G., Hu S., Zhang Y., Tao Y., Han Y. Novel autoimmune hepatitis-specific autoantigens identified using protein microarray technology. J Proteome Res. 2010;9:30–39. doi: 10.1021/pr900131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright C., Sibani S., Trudgian D., Fischer R., Kessler B., Labaer J. Detection of multiple autoantibodies in patients with ankylosing spondylitis using nucleic acid programmable protein arrays. Mol Cell Proteomics. 2010 doi: 10.1074/mcp.M9.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michaud G.A., Salcius M., Zhou F., Bangham R., Bonin J., Guo H. Analyzing antibody specificity with whole proteome microarrays. Nat Biotechnol. 2003;21:1509–1512. doi: 10.1038/nbt910. [DOI] [PubMed] [Google Scholar]
- 31.Michaud G.A., Salcius M., Zhou F., Papov V.V., Merkel J., Murtha M. Applications of protein arrays for small molecule drug discovery and characterization. Biotechnol Genet Eng Rev. 2006;22:197–211. doi: 10.1080/02648725.2006.10648071. [DOI] [PubMed] [Google Scholar]
- 32.Sharon D., Chen R., Snyder M. Systems biology approaches to disease marker discovery. Dis Markers. 2010;28:209–224. doi: 10.3233/DMA-2010-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu H., Klemic J.F., Chang S., Bertone P., Casamayor A., Klemic K.G. Analysis of yeast protein kinases using protein chips. Nat Genet. 2000;26:283–289. doi: 10.1038/81576. [DOI] [PubMed] [Google Scholar]
- 34.MacBeath G., Schreiber S.L. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 35.Arenkov P., Kukhtin A., Gemmell A., Voloshchuk S., Chupeeva V., Mirzabekov A. Protein microchips: use for immunoassay and enzymatic reactions. Anal Biochem. 2000;278:123–131. doi: 10.1006/abio.1999.4363. [DOI] [PubMed] [Google Scholar]
- 36.Zhu H., Snyder M. Protein arrays and microarrays. Curr Opin Chem Biol. 2001;5:40–45. doi: 10.1016/s1367-5931(00)00170-8. [DOI] [PubMed] [Google Scholar]
- 37.Houseman B.T., Huh J.H., Kron S.J., Mrksich M. Peptide chips for the quantitative evaluation of protein kinase activity. Nat Biotechnol. 2002;20:270–274. doi: 10.1038/nbt0302-270. [DOI] [PubMed] [Google Scholar]
- 38.Ramachandran N., Raphael J.V., Hainsworth E., Demirkan G., Fuentes M.G., Rolfs A. Next-generation high-density self-assembling functional protein arrays. Nat Methods. 2008;5:535–538. doi: 10.1038/nmeth.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gelperin D.M., White M.A., Wilkinson M.L., Kon Y., Kung L.A., Wise K.J. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ptacek J., Snyder M. Charging it up: global analysis of protein phosphorylation. Trends Genet. 2006;22:545–554. doi: 10.1016/j.tig.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 41.Popescu S.C., Popescu G.V., Bachan S., Zhang Z., Gerstein M., Snyder M. MAPK target networks in Arabidopsis thaliana revealed using functional protein microarrays. Genes Dev. 2009;23:80–92. doi: 10.1101/gad.1740009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Popescu S.C., Snyder M., Dinesh-Kumar S. Arabidopsis protein microarrays for the high-throughput identification of protein–protein interactions. Plant Signal Behav. 2007;2:416–420. doi: 10.4161/psb.2.5.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen C.S., Korobkova E., Chen H., Zhu J., Jian X., Tao S.C. A proteome chip approach reveals new DNA damage recognition activities in Escherichia coli. Nat Methods. 2008;5:69–74. doi: 10.1038/NMETH1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith M.G., Jona G., Ptacek J., Devgan G., Zhu H., Zhu X. Global analysis of protein function using protein microarrays. Mech Ageing Dev. 2005;126:171–175. doi: 10.1016/j.mad.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 45.Huang J., Zhu H., Haggarty S.J., Spring D.R., Hwang H., Jin F. Finding new components of the target of rapamycin (TOR) signaling network through chemical genetics and proteome chips. Proc Natl Acad Sci USA. 2004;101:16594–16599. doi: 10.1073/pnas.0407117101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schweitzer B., Wiltshire S., Lambert J., O'Malley S., Kukanskis K., Zhu Z. Inaugural article: immunoassays with rolling circle DNA amplification: a versatile platform for ultrasensitive antigen detection. Proc Natl Acad Sci USA. 2000;97:10113–10119. doi: 10.1073/pnas.170237197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ptacek J., Devgan G., Michaud G., Zhu H., Zhu X., Fasolo J. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 48.Ito T., Tashiro K., Muta S., Ozawa R., Chiba T., Nishizawa M. Toward a protein–protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc Natl Acad Sci USA. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uetz P., Giot L., Cagney G., Mansfield T.A., Judson R.S., Knight J.R. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 50.Yu H., Braun P., Yildirim M.A., Lemmens I., Venkatesan K., Sahalie J. High-quality binary protein interaction map of the yeast interactome network. Science. 2008;322:104–110. doi: 10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ito T., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hook S.S., Means A.R. Ca(2+)/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- 53.Hesselberth J.R., Miller J.P., Golob A., Stajich J.E., Michaud G.A., Fields S. Comparative analysis of Saccharomyces cerevisiae WW domains and their interacting proteins. Genome Biol. 2006;7:R30. doi: 10.1186/gb-2006-7-4-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sudol M., Hunter T. NeW wrinkles for an old domain. Cell. 2000;103:1001–1004. doi: 10.1016/s0092-8674(00)00203-8. [DOI] [PubMed] [Google Scholar]
- 55.Sudol M., Sliwa K., Russo T. Functions of WW domains in the nucleus. FEBS Lett. 2001;490:190–195. doi: 10.1016/s0014-5793(01)02122-6. [DOI] [PubMed] [Google Scholar]
- 56.Kay B.K., Williamson M.P., Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- 57.Galindo C.L., Gutierrez C., Chopra A.K. Potential involvement of galectin-3 and SNAP23 in Aeromonas hydrophila cytotoxic enterotoxin-induced host cell apoptosis. Microb Pathog. 2006;40:56–68. doi: 10.1016/j.micpath.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 58.Park M.Y., Choi S.C., Lee H.S., Kim D., Baek K.E., Kim J.T. A quantitative analysis of N-myc downstream regulated gene 2 (NDRG 2) in human tissues and cell lysates by reverse-phase protein microarray. Clin Chim Acta. 2008;387:84–89. doi: 10.1016/j.cca.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 59.Hall D.A., Zhu H., Zhu X., Royce T., Gerstein M., Snyder M. Regulation of gene expression by a metabolic enzyme. Science. 2004;306:482–484. doi: 10.1126/science.1096773. [DOI] [PubMed] [Google Scholar]
- 60.Horak C.E., Snyder M. ChIP-chip: a genomic approach for identifying transcription factor binding sites. Methods Enzymol. 2002;350:469–483. doi: 10.1016/s0076-6879(02)50979-4. [DOI] [PubMed] [Google Scholar]
- 61.Hu S., Xie Z., Onishi A., Yu X., Jiang L., Lin J. Profiling the human protein–DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell. 2009;139:610–622. doi: 10.1016/j.cell.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seger R., Krebs E.G. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 63.Zhu J., Gopinath K., Murali A., Yi G., Hayward S.D., Zhu H. RNA-binding proteins that inhibit RNA virus infection. Proc Natl Acad Sci USA. 2007;104:3129–3134. doi: 10.1073/pnas.0611617104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Butcher R.A., Bhullar B.S., Perlstein E.O., Marsischky G., LaBaer J., Schreiber S.L. Microarray-based method for monitoring yeast overexpression strains reveals small-molecule targets in TOR pathway. Nat Chem Biol. 2006;2:103–109. doi: 10.1038/nchembio762. [DOI] [PubMed] [Google Scholar]
- 65.Inoki K., Corradetti M.N., Guan K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 66.Huang J., Zhu H., Haggarty S.J., Spring D.R., Hwang H., Jin F. Finding new components of the target of rapamycin (TOR) signaling network through chemical genetics and proteome chips. Proc Natl Acad Sci USA. 2004;101:16594–16599. doi: 10.1073/pnas.0407117101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salamat-Miller N., Fang J., Seidel C.W., Smalter A.M., Assenov Y., Albrecht M. A network-based analysis of polyanion-binding proteins utilizing yeast protein arrays. Mol Cell Proteomics. 2006;5:2263–2278. doi: 10.1074/mcp.M600240-MCP200. [DOI] [PubMed] [Google Scholar]
- 68.Zhao Y., Jensen O.N. Modification-specific proteomics: strategies for characterization of post-translational modifications using enrichment techniques. Proteomics. 2009;9:4632–4641. doi: 10.1002/pmic.200900398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Geoffroy M.C., Hay R.T. An additional role for SUMO in ubiquitin-mediated proteolysis. Nat Rev Mol Cell Biol. 2009;10:564–568. doi: 10.1038/nrm2707. [DOI] [PubMed] [Google Scholar]
- 70.Foster M.W., Liu L., Zeng M., Hess D.T., Stamler J.S. A genetic analysis of nitrosative stress. Biochemistry. 2009;48:792–799. doi: 10.1021/bi801813n. [DOI] [PubMed] [Google Scholar]
- 71.Sopko R., Huang D., Preston N., Chua G., Papp B., Kafadar K. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell. 2006;21:319–330. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 72.Kafadar K.A., Zhu H., Snyder M., Cyert M.S. Negative regulation of calcineurin signaling by Hrr25p, a yeast homolog of casein kinase I. Genes Dev. 2003;17:2698–2708. doi: 10.1101/gad.1140603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jonassen I., Collins J.F., Higgins D.G. Finding flexible patterns in unaligned protein sequences. Protein Sci. 1995;4:1587–1595. doi: 10.1002/pro.5560040817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mok J., Kim P.M., Lam H.Y.K., Piccirillo S., Zhou X., Jeschke G.R. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci Signal. 2010:3. doi: 10.1126/scisignal.2000482. ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gavin A.C., Bosche M., Krause R., Grandi P., Marzioch M., Bauer A. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 76.Xenarios I., Rice D.W., Salwinski L., Baron M.K., Marcotte E.M., Eisenberg D. DIP: the database of interacting proteins. Nucleic Acids Res. 2000;28:289–291. doi: 10.1093/nar/28.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bader G.D., Hogue C.W. BIND–a data specification for storing and describing biomolecular interactions, molecular complexes and pathways. Bioinformatics. 2000;16:465–477. doi: 10.1093/bioinformatics/16.5.465. [DOI] [PubMed] [Google Scholar]
- 78.Horak C.E., Luscombe N.M., Qian J., Bertone P., Piccirrillo S., Gerstein M. Complex transcriptional circuitry at the G1/S transition in Saccharomyces cerevisiae. Genes Dev. 2002;16:3017–3033. doi: 10.1101/gad.1039602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 80.Fiedler D., Braberg H., Mehta M., Chechik G., Cagney G., Mukherjee P. Functional organization of the S. cerevisiae phosphorylation network. Cell. 2009;136:952–963. doi: 10.1016/j.cell.2008.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korf U., Derdak S., Tresch A., Henjes F., Schumacher S., Schmidt C. Quantitative protein microarrays for time-resolved measurements of protein phosphorylation. Proteomics. 2008;8:4603–4612. doi: 10.1002/pmic.200800112. [DOI] [PubMed] [Google Scholar]
- 82.Soufi B., Kelstrup C.D., Stoehr G., Frohlich F., Walther T.C., Olsen J.V. Global analysis of the yeast osmotic stress response by quantitative proteomics. Mol Biosyst. 2009;5:1337–1346. doi: 10.1039/b902256b. [DOI] [PubMed] [Google Scholar]
- 83.Van Hoof D., Munoz J., Braam S.R., Pinkse M.W., Linding R., Heck A.J. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell. 2009;5:214–226. doi: 10.1016/j.stem.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 84.Kung L.A., Tao S.C., Qian J., Smith M.G., Snyder M., Zhu H. Global analysis of the glycoproteome in Saccharomyces cerevisiae reveals new roles for protein glycosylation in eukaryotes. Mol Syst Biol. 2009;5:308. doi: 10.1038/msb.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tao Z., Gao P., Liu H.W. Studies of the expression of human poly(ADP-ribose) polymerase-1 in Saccharomyces cerevisiae and identification of PARP-1 substrates by yeast proteome microarray screening. Biochemistry. 2009;48:11745–11754. doi: 10.1021/bi901387k. [DOI] [PubMed] [Google Scholar]
- 86.Meyer-Ficca M.L., Meyer R.G., Jacobson E.L., Jacobson M.K. Poly(ADP-ribose) polymerases: managing genome stability. Int J Biochem Cell Biol. 2005;37:920–926. doi: 10.1016/j.biocel.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 87.Kim M.Y., Mauro S., Gevry N., Lis J.T., Kraus W.L. NAD + -dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004;119:803–814. doi: 10.1016/j.cell.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 88.Lin Y.Y., Lu J.Y., Zhang J., Walter W., Dang W., Wan J. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell. 2009;136:1073–1084. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Millar C.B., Grunstein M. Genome-wide patterns of histone modifications in yeast. Nat Rev Mol Cell Biol. 2006;7:657–666. doi: 10.1038/nrm1986. [DOI] [PubMed] [Google Scholar]
- 90.Smith E.R., Eisen A., Gu W., Sattah M., Pannuti A., Zhou J. ESA1 is a histone acetyltransferase that is essential for growth in yeast. Proc Natl Acad Sci USA. 1998;95:3561–3565. doi: 10.1073/pnas.95.7.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin Y.Y., Qi Y., Lu J.Y., Pan X., Yuan D.S., Zhao Y. A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev. 2008;22:2062–2074. doi: 10.1101/gad.1679508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Glickman M.H., Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 93.Adhikari A., Xu M., Chen Z.J. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26:3214–3226. doi: 10.1038/sj.onc.1210413. [DOI] [PubMed] [Google Scholar]
- 94.Gupta R., Kus B., Fladd C., Wasmuth J., Tonikian R., Sidhu S. Ubiquitination screen using protein microarrays for comprehensive identification of Rsp5 substrates in yeast. Mol Syst Biol. 2007;3:116. doi: 10.1038/msb4100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Foster M.W., Forrester M.T., Stamler J.S. A protein microarray-based analysis of S-nitrosylation. Proc Natl Acad Sci USA. 2009;106:18948–18953. doi: 10.1073/pnas.0900729106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Foster M.W., Hess D.T., Stamler J.S. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hess D.T., Matsumoto A., Kim S.O., Marshall H.E., Stamler J.S. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 98.Forrester M.T., Foster M.W., Benhar M., Stamler J.S. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic Biol Med. 2009;46:119–126. doi: 10.1016/j.freeradbiomed.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith M.G., Snyder M. Yeast as a model for human disease. Curr Protoc Hum Genet. 2006 doi: 10.1002/0471142905.hg1506s48. Chapter 15:Unit 15 6. [DOI] [PubMed] [Google Scholar]
- 100.Snyder M., Gallagher J.E. Systems biology from a yeast omics perspective. FEBS Lett. 2009;583:3895–3899. doi: 10.1016/j.febslet.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]