

Abstract
A convenient synthesis of sidechain-modified phytosterols is achieved via a temporary masking of the stigmasterol 5,6-alkene as an epoxide. Following performance of the desired modification, the alkene is regenerated through a mild deoxygenation. The approach is applied to the syntheses of β-sitosterol and campesterol acetate, and suggests a facile route to the (Z)-isomers of Δ22–23 phytosterols.
1. Introduction
Phytosterols and their derivatives are widely applied in the food and cosmetic industries, and have recently received a great deal of attention as nutraceutical additives [1,2,3]. Phytosterols have also attracted attention as inhibitors of sarcoplasmic reticulum calcium ATPase and potassium ion channels [4,5]. As part of a collaboration investigating the structural influences on uptake and processing of sterol esters[6], we required semipreparative amounts of β-sitosterol. However, β –sitosterol is commercially available in preparative amounts only as mixtures with other phytosterols, including stigmasterol, campesterol, and/or brassicasterol (Figure 1); reported separations are relatively laborious [7,8].
Figure 1.
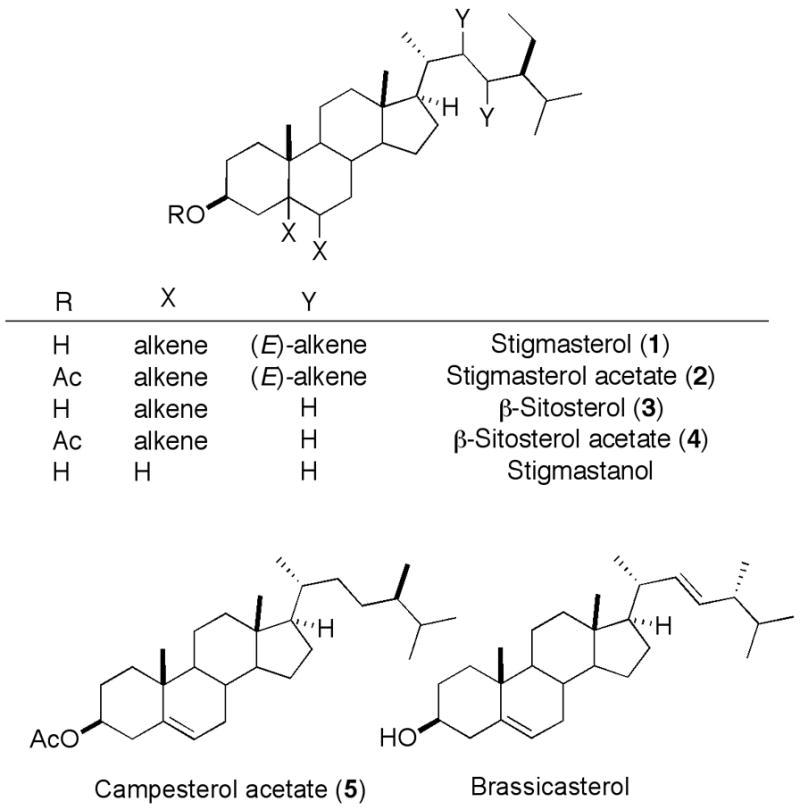
Structural relationship of phytosterols
Two routes have been reported for synthesis of β –sitosterol from stigmasterol, which is available in pure form. The first, selective hydrogenation of the sidechain Δ22–23 alkene [9], was found to produce β –sitosterol contaminated with varying amounts of recovered stigmasterol as well as the fully saturated stigmastanol [10]. The second approach, which has been applied to the synthesis of sitosterol and related sterols (Figure 2), circumvents the need for selective hydrogenation by protecting the Δ5–6 alkene as a cyclopropyl carbinyl ether [11,12]. Following hydrogenation of the Δ22–23 double bond, solvolysis of the cyclopropane reintroduces both the C3-alcohol and the Δ5–6 alkene. Although we found the latter approach very useful as a means of obtaining very pure samples of β –sitosterol, semipreparative applications were challenging in terms of removal of sterol methyl ether byproducts.
Figure 2.
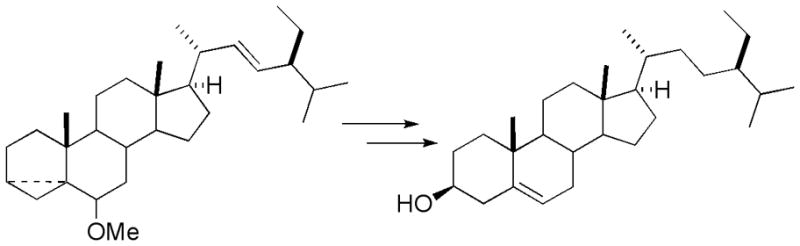
Selective saturation of cyclopropyl carbinyl ether
We now report a new strategy for the synthesis of side-chain modified phytosterols based upon protection of the Δ5–6 alkene as an epoxide. The approach is illustrated with syntheses of β-sitosterol and campesterol acetate.
2. Experimental
2.1. General Experimental Procedures
AlI3 and Cu(MnO4)2 were prepared by literature procedures [13,14]. All other reagents and solvents were used as supplied commercially, except CH2Cl2 (CaH2) and THF (Na, Ph2CO) which were distilled from the indicated reagent under an atmosphere of N2. Melting points are uncorrected. Unless noted, NMR spectra were acquired at 400 MHz (1H) or 100 MHz (13C) in CDCl3; individual peaks are reported as: multiplicity, integration, coupling constant in Hz. IR spectra were recorded as neat films on a ZnSe crystal with selected absorbances reported in cm−1. Mass spectroscopy was conducted at the Nebraska Center for Mass Spectrometry.
2.2. Stigmasterol acetate (2)
Stigmasterol acetate was prepared as a white solid (97%, mp 138–140 °C) by a variant of the procedure of Wang [15]. Other physical and spectral data were identical to literature values.
2.3. 5α , 6α - and 5β , 6β-Epoxides of stigmasterol acetate (6a, 6b)
A mixture of KMnO4 (10 g, 60 mmol) and CuSO4 · 5H2O (5.0 g, 20 mmol) was finely ground in a mortar and pestle [14]. Water (0.5 mL) was added, and the slightly wet mixture was transferred to the reaction flask. To the stirred suspension of this mixture in 25 mL CH2Cl2 was added stigmasterol acetate (2, 2.12 g, 4.51 mmol), followed by t-BuOH (2.5 mL). The reaction was heated to reflux for 1 hour and cooled to room temperature. The reaction mixture was filtered through a silica pad, which was washed with ether. The residue obtained after concentration was recrystallized from CH3OH to give a white solid (1.59 g, 75%) with mp 125–126 °C. NMR data indicated the product was a 1 : 6 mixture of the α - (6a) and β - (6b) epoxides of stigmasterol acetate [14]. Repeating this reaction with 2.23 g of stigmasterol acetate afforded 1.80 g (78%) of a 1:6 mixture of 6a and 6b.
Approach to 5α , 6α - and 5β , 6β-Epoxides of stigmasterol acetate (6a, 6b) via peracid epoxidation: To a 0° C solution of 2 (0.308 g, 0.66 mmol) in CH2Cl2 (20 mL) was added mCPBA (0.170 g, 0.76 mmol). After 4 h at 0 °C, the reaction was diluted with sat. aq K2CO3 (80mL) and the aqueous layer was extracted with CH2Cl2 (50 mL x 3). The combined organic layers were dried over anhydrous Na2SO4 and the filtrate was concentrated in vacuo. The residue was purified by flash chromatography (hexane/EtOAc, 90:10) to afford 0.257 g (83%) of a white solid which was a 2.6:1 mixture of α - (6a) and β-isomers (6b) according to 1H NMR.
2.4. 5α , 6α - and 5β , 6β-Epoxy sitosterol acetate (7a, 7b)
To a solution of the 1 : 6 mixture of 6a and 6b (1.35 g, 3.0 mmol) in EtOAc (150 mL) was added 10% Pd/C (0.32 g), and the stirred reaction mixture was placed under an atmosphere of H2 (balloon) for 12 h. The reaction mixture was filtered through a Celite pad, and the filtrate evaporated to furnish white solid (1.28 g, 96%, mp 113–114 °C) as a 1:6 mixture of epoxides 7a and 7b [8]. Repeating this reaction on 1.76 g of 6a/6b afforded 1.75 g (99%) of a 1:6 mixture of 7a and 7b.
2.5. β-Sitosterol acetate (4)
The 1 : 6 mixture of epoxides 7a and 7b (470 mg, 1.0 mmol) was dissolved in 2:1 CH3CN/CH2Cl2 (30 mL). Aluminum triiodide was added (610 mg, 1.5 mmol) and the resulting mixture was stirred at room temperature for 10 minutes. The reaction was quenched with aq. 10% Na2S2O3 (100 mL) and the resulting mixture was extracted with CH2Cl2 (3 x 100 mL). The combined organic layers were dried over Na2SO4, and the residue from the concentrated filtrate was purified by flash chromatography (hexane/EtOAc, 95:5) to give 360 mg (80%) of 4 as a white solid: mp 111–112 °C, [α ]D = −34.5 (CHCl3, c = 1.0). Other physical data were identical to values reported in the literature [11]. Repeating this reaction on 1.70 g of 7a/7b afforded 1.40 g (85%) of 4.
2.6. β-Sitosterol (3)
To a solution of β-sitosterol acetate (4, 240 mg, 0.47 mmol) in 1:1 CH3OH:CH2Cl2 (30 mL) was added K2CO3 (140 mg, 1.01 mmol). The reaction mixture was stirred at room temperature for 12 hours and then concentrated under vacuum. The residue was extracted with 30 mL CH2Cl2. The organic layer was washed with 30 mL water and dried over Na2SO4. The filtered organic layer was concentrated and the residue was purified through flash chromatography (hexane/EtOAc, 80:20) to give 220 mg (93%) of β -sitosterol 3 as a white solid. Mp 134–135 °C, [α ]D = −37.0 (CHCl3, c = 1.0). Elemental analysis calculated for C29H50O: C 83.60, H 11.96; found: 83.99, 12.15. Other spectral properties were identical to values reported in the literature [11]. Repeating this reaction on 1.35 g of 8 afforded 1.20 g (98%) of beta sitosterol (3).
2.7. (S)-2,3-Dimethylbutan-1-ol (8)
(S)-2,3-Dimethylbutan-1-ol 8 was prepared as a colorless liquid (overall yield 60%, [α ]D = 4.4 (CHCl3, c = 1.0) by the procedure of Tietze, affording a product with spectral data identical to literature values [16].
2.8. (S)-2-(2,3-Dimethylbutylthio)benzothiazole (9)
To a mixture of dimethylbutanol 8 (102 mg, 1.00 mmol), 2-mercaptobenzothiazole (183 mg, 1.10 mmol) and PPh3 (288 mg, 1.10 mmol) in freshly distilled THF (4 mL) was added diisopropy azodicarboxylate (DIAD, 0.21 mL, 1.10 mmol) dropwise at 0°C under argon. The reaction was stirred for 3 h at 0 °C and then quenched with water. The aqueous layer was extracted with EtOAc (10mL x 3) and the combined organic layers were dried over anhydrous Na2SO4. The filtered organic layer was concentrated in vacuo and the residue purified by flash chromatography (hexane/EtOAc, 99:1) to afford thioether 9 (228 mg, 91%) as a light yellow oil. [α ]D = 42.3 (CHCl3, c = 1.6); IR 2957, 1455, 1426, 1057, 991, 752 cm−1; 1H NMR: Δ 7.89 (d, J= 8.1 , 1H), 7.75 (d, J= 8.1 , 1H), 7.42 (t, J= 7.2 , 1H), 7.29 (t, J= 7.2 , 1H), 3.50 (dd, J= 12.7 , 4.8 , 1H), 3.18 (dd, J= 12.7, 8.2 , 1H), 1.88–1.77 (m, 2H), 1.04 (d, J= 6.7 , 3H), 0.99 (d, J= 6.6 , 3H), 0.94 (d, J= 6.6 , 3H); 13C NMR: Δ 167.75, 153.38, 135.15, 125.99, 124.08, 121.44, 120.91, 38.87, 38.78, 31.62, 20.37, 17.96, 15.29; HRFAB-MS (m/z) [M-H]+ calcd for [C13H18NS2]+: 252.0881, found: 252.0875.
2.9 (S)-2-(2,3-Dimethylbutylsulfonyl)benzothiazole (10)
A 0°C solution of 9 (183 mg, 0.73 mmol) in EtOH (10 mL) was oxidized with ammonium heptamolybdate tetrahydrate (1.8 g, 1.46 mmol) and 30% H2O2 (2.5 mL, 21.9 mmol) for 2 hours. The mixture was extracted with EtOAc (10 mL x 3) and the combined organic extracts were washed with brine (10 mL x 3). The dried organic layers was filtered and the residue obtained upon concentration was purified by flash chromatography (hexane/ EtOAc, 90:10) to afford sulfone 10 (177mg, 86%) as a pale yellow oil. [α ]D = 15.5 (CHCl3, c = 3.4); IR 2961, 1470, 1324, 1140, 1085, 758 cm−1; 1H NMR: Δ 8.23 (d, J= 7.9, 1H), 8.03 (d, J= 7.9, 1H), 7.68–7.59 (m, 2H), 3.59(dd, J= 14.4, 3.5, 1H), 3.31(dd, J= 14.1, 8.9, 1H), 2.29–2.19(m, 1H), 1.82–1.73(m, 1H), 1.10(d, J= 6.9, 3H), 0.89(d, J= 6.8, 3H), 0.85(d, J= 6.9, 3H); 13C NMR: Δ 166.66, 152.70, 136.74, 128.00, 127.67, 125.43, 122.38, 58.83, 38.68, 32.47, 19.23, 17.89, 15.93; HRFAB-MS (m/z) [M-H]+ calcd for [C13H18NO2S2]+: 284.0779, found: 284.0778.
2.10. (3β ,5α ,6α )- and (3β ,5β ,6β )- Pregnane-20α-carboxaldehyde-5,6-epoxy-3-yl acetate (11a, b)
A −78 °C solution of 6a, 6b (~ 1:6 mixture, 100 mg, 0.21 mmol) in 10 mL of 50/50 CH2Cl2/MeOH was treated with a gaseous stream of ozone (2% O3/O2) for 5 minutes. The solution was purged with pure oxygen and then solvent was removed under vacuum. The residue was redissolved in 10 mL of 10/90 H2O/AcOH and treated with zinc powder (55 mg, 0.84 mmol). The reaction mixture was stirred for 2 hours at room temperature and then extracted with 50 mL CH2Cl2. The organic layer was washed with water (25 mL x 3), then dried over anhydrous Na2SO4. The filtered organic layer was concentrated and the residue purified by flash chromatography (hexane/ EtOAc, 90:10) to afford a 1:6 mixture of epoxides 11a and 11b as a white solid (81 mg, 99%), mp 87–8 °C. IR: 2950, 1727, 1367, 1262, 1238, 1042, 783 cm−1; 1H NMR: Δ 9.57 (d, J= 3.3 , 0.76H, β ), 9.55 (d, J= 3.3 , 0.16H, α ), 4.99–4.91 (m, 0.14H, α ), 4.81–4.73(m, 0.87H, β ), 3.09(d, J= 2.2, 0.88H, β ), 2.90 (d, J= 4.2, 0.13H, α ), 2.38–2.31(m, 1H), 2.13–1.82(m, 9H), 1.54–0.89(m, 20H), 0.7(s, 3H); 13C NMR: Δ 204.95, 170.52, 71.25, 63.41, 62.48, 55.39, 51.05, 50.93, 49.42, 42.89, 39.43, 37.95, 36.67, 35.06, 32.41, 29.74, 27.17, 26.97, 24.54, 21.84, 21.30, 17.03, 13.40, 12.11; HRFAB-MS (m/z) [M-Li]+ calcd for [C24H36LiO4]+: 395.2774, found: 395.2778.
2.11. (3β ,5α ,6α ,22Z)- and (3β ,5β ,6β ,22Z)- Ergost-5,6-epoxy-22-en-3-yl acetate (12a, 12b)
To a 78 °C solution of sulfone 10 (62 mg, 0.22 mmol) in THF (5 mL) was dropwise added LiHMDS (0.22 mL, nominally 1M in THF, 0.22 mmol). The reaction was stirred for 1 h, whereupon the mixture of aldehydes 11a and 11b (~1:6, 85 mg, 0.22 mmol) was added in 5 mL of THF. Stirring was continued for 1 hour, and reaction was gradually warmed to room temperature. The reaction was quenched by 15 mL water and extracted with EtOAc (10 mL x 3). The combined organic layers were dried over anhydrous Na2SO4 and filtered. The residue obtained upon concentration was purified by flash chromatography (hexane/ EtOAc, 95:5) to afford a 1:16 mixture of epoxides 12a and 12b as a white solid (90 mg, 90%), mp 145–147 °C. IR: 2950, 2867, 1743, 1368, 1037, 764 cm−1; 1H NMR: Δ5.02(dd, J= 10.9, 9.9 , 2H), 4.83–4.73(m, 1H), 3.09(d, J= 2.0 , 0.95H, β ), 2.91(d, J= 4.4, 0.06, a), 2.43–2.33(m, 1H), 2.21–1.80(m, 9H), 1.68–0.83(m, 31H), 0.69(s, 3H); 13C NMR: Δ170.56, 135.13, 131.22, 71.34, 63.58, 62.52, 56.27, 56.02, 51.02, 42.18, 39.72, 38.32, 38.01, 36.68, 35.04, 34.48, 33.35, 32.43, 29.74, 28.32, 27.20, 24.15, 21.92, 21.34, 20.55, 20.36, 19.94, 18.63, 17.06, 12.06; HRFAB-MS (m/z) [M-H]+ calcd for [C30H49O3]+: 457.3682, found: 457.3668.
2.12. 5α , 6α - and 5β , 6β-Epoxides of campesterol acetate (13a, 13b)
The mixture of epoxides 12a and 12b (30mg, 0.065 mmol) was dissolved in 5mL EtOAc. 10% Pd/C (7 mg) was added, and the reaction mixture was stirred at room temperature under an atmosphere of H2 (balloon) for 12 h. The reaction mixture was filtered through a Celite pad, and the filtrate evaporated to a white solid (28 mg, 94%, mp 110–111 °C) as 1:9 mixture of epoxides 13a and 13b. IR: 2953, 2867, 1729, 1367, 1263, 1043, 784 cm−1; 1H NMR: Δ5.01–4.93(m, 0.15H, ■), 4.83–4.73(m, 0.96H, β ), 3.09(d, J=2.1 , 0.9H, β ), 2.90(d, J= 4.4 , 0.1H, ■), 2.12–1.8(m, 8H), 1.58–0.77(m, 37H), 0.65(s, 3H); 13C NMR: Δ 170.54, 71.34, 63.58, 62.51, 56.19, 56.14, 50.97, 42.28, 39.78, 38.81, 38.01, 36.66, 35.82, 35.03, 33.65, 32.47, 32.41, 30.26, 29.73, 28.14, 27.21, 24.18, 21.92, 21.31, 20.19, 18.66, 18.24, 17.03, 15.37, 11.76; HRFAB-MS (m/z) [M-H]+ calcd for [C30H51O3]+: 459.3838, found: 459.3820.
2.13. Campesterol acetate (5)
The mixture of epoxides 13a and 13b (28 mg, 0.061 mmol) was dissolved in 2:1 CH3CN/CH2Cl2 (3 mL). Aluminum triiodide (37 mg, 0.091 mmol) was added and the resulting mixture was stirred at room temperature for 40 minutes. The reaction was quenched with aq. 10% Na2S2O3 (10 mL) and the resulting mixture was extracted with CH2Cl2 (3 x 10 mL). The combined organic layers were dried over anhydrous Na2SO4, and the residue from the concentrated filtrate was purified by flash chromatography (hexane/EtOAc, 95:5) to give 24mg (91%) campesterol acetate (5) as a white solid. Mp 130–131 °C, [α ]D = −32 (CHCl3, c = 0.7); IR 2954, 1730, 1367, 1247, 1037, 735 cm−1; 1H NMR: Δ 5.39(d, J= 4.8, 1H), 4.66–4.58(m, 1H), 2.33(d, J= 7.9, 2H), 2.05(s, 3H), 1.90–1.84(m, 2H), 1.59–0.78(m, 38H), 0.68(s, 3H); 13C NMR: Δ170.56, 139.66, 122.66, 73.99, 56.69, 56.08, 50.02, 42.31, 39.73, 38.84, 38.12, 36.99, 36.59, 35.90, 33.70, 32.43, 31.90, 31.86, 30.27, 28.24, 27.78, 24.29, 21.46, 21.03, 20.22, 19.32, 18.70, 18.26, 15.38, 11.87 ; HRFAB–MS (m/z) [M-Na]+ calcd for [C30H50O2Na]+: 465.3709, found: 465.3703. Elemental analysis calculated for C30H50O2: C 81.20, H 11.38; found: 81.39, 11.39. The 1H NMR data matched that of a literature report [17].
3. Results and Discussion
Our synthesis of β-sitosterol (3) is illustrated in Scheme 1. Selective epoxidation of the Δ5–6 alkene of stigmasterol acetate (2) with copper permanangate formed a 6:1 mixture of the 5β ,6β - and 5α ,6α epoxides 6b and 6a [14,18]. Hydrogenation over Pd/C cleanly furnished a 1:6 mixture of sitosterol epoxides 7a and 7b. Deoxygenation of the saturated epoxides with AlI3 [13] proceeded rapidly to furnish a good yield of β-sitosteryl acetate 4. Saponification afforded pure β-sitosterol (3), with mp 134–135 °C and [α] D = − 37.0 [8,19].
Scheme 1.
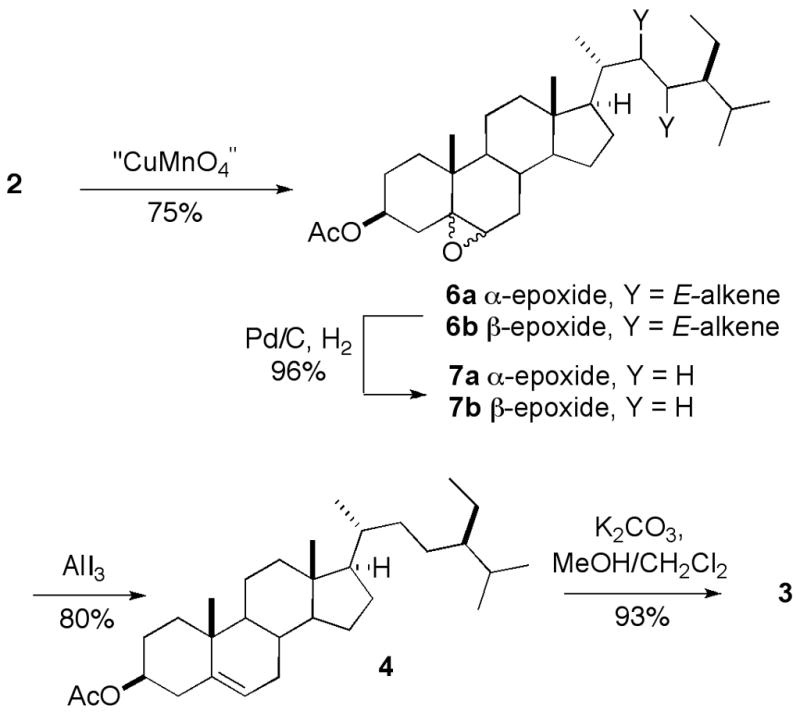
Synthesis of sitosterol
Epoxidation with the commercially available peracid mCPBA also gave good selectivity for the Δ5–6 alkene, but now produced a 2.6:1 mixture of stigmasterol oxides favoring the α-isomer (6a). Hydrogenation proceeded uneventfully to furnish the corresponding mixture of sitosterol oxides 7a and 7b. However, attempted deoxygenation under the same conditions as employed earlier (AlI3, 10 min, CH3CN/CH2Cl2) now furnished only 33% of β-sitosterol acetate (4), accompanied by a significant amount (estimated > 60% by mass) of a more polar product which yellowed immediately upon exposure to room light. The formation of the byproduct could be avoided almost completely by allowing the deoxygenation to proceed for 40 min. Alternatively, the byproduct could be converted to 4 by treatment with additional AlI3. The results suggest that the deoxygenation of the α - and β - epoxides proceeds at very different rates, with the 5α , 6α diastereomer (6a) reacting via the intermediacy of a semistable iodohydrin.
As illustrated in Scheme 2, our strategy also provides a facile means of preparing other sidechain-modified phytosterols. For example, ozonolysis of the mixture of 6a/6b furnished an approximately 1:6 mixture of aldehydes 11a and 11b. Julia-Kocienski olefination, using an enantiomerically pure sulfone (10) derived from (S)-2,3-dimethylbutanol (8) [16] furnished exclusively alkene [20], corresponding to the monoepoxide of the Z-isomer of crinosterol [21]. Hydrogenation, followed by deoxygenation of the epoxide as before, furnished campesterol acetate (5).
Scheme 2.

Synthesis of campesterol acetate
The selective formation of Z-alkenes is unusual in Julia couplings [22] and we investigated the olefination of 11a,b with a known sulfone derived from isobutyl alcohol (Figure 3) [23]. This reaction also selectively furnished the Z-alkene; the lower selectivity (2:1) compared with that observed for the synthesis of 12a,b may reflect the reduced degree of steric encumbrance in this model system. The ability to readily prepare Z-isomers of phytosterols opens the door to a number of steroid analogs not available from synthetic routes based upon Claisen rearrangements of C22 allylic alcohols [12].
Figure 3.
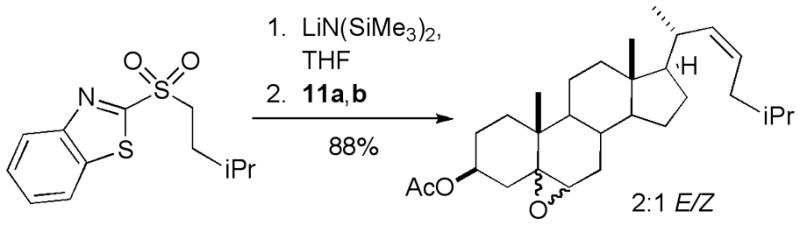
Generality of Z-selective olefination
Conclusion
The formation of β-sitosterol has been achieved in 52% overall yield from commercially available stigmasterol using relatively simple chemistry and via easily purified intermediates. The core strategy, protection of the Δ5–6 alkene as an epoxide, holds potential for synthesis of other phytosterols as well as unnatural analogs.
Supplementary Material
Acknowledgments
We thank Professor Tim Carr (University of Nebraska–Lincoln, Department of Nutrition and Health Sciences) for useful discussions. This research was supported by a USDA-NRI competitive grant (2007-35200-18298). Portions of this work were conducted in facilities remodeled with support from NIH (RR016544-01). NMR spectra were acquired, in part, on spectrometers purchased with NSF support (MRI 0079750, CHE 0091975).
Appendix A. Supplementary data
Supplementary data for this article, consisting of 1H and 13C NMR spectra for compounds 3-13, can be found in the online version at doi 10.1016/j.steroids.2010.xxxx.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moghadasian MH. Pharmacolgical properties of plant sterols: In vivo and in vitro observations. Life Sci. 2000;67:605–15. doi: 10.1016/s0024-3205(00)00665-2. [DOI] [PubMed] [Google Scholar]
- 2.Piironen V, Lindsay DG, Miettinen TA. Plant sterols: Biosynthesis, biological function and their importance to human nutrition. J Sci Food Agric. 2000;80:939–66. [Google Scholar]
- 3.Ling WH, Jone PJH. Dietary phytosterols: A review of metabolism, benefits and side effects. Life Sci. 1995;57:195–206. doi: 10.1016/0024-3205(95)00263-6. [DOI] [PubMed] [Google Scholar]
- 4.Bao L, Li Y, Deng SX, Landry D, Tabas I. Sitosterol-containing Lipoproteins Trigger Free Sterol-induced Caspase-independent Death in ACAT-competent Macrophages. J Biol Chem. 2006;281:33635–49. doi: 10.1074/jbc.M606339200. [DOI] [PubMed] [Google Scholar]
- 5.Promprom W, Kupittayanant P, Indrapichate K, Wray S, Kupittayanant S. The Effects of Pomegranate Seed Extract and β-Sitosterol on Rat Uterine Contractions. Reprod Sci. 2010;17:288–96. doi: 10.1177/1933719109352687. [DOI] [PubMed] [Google Scholar]
- 6.a) Brown AW, Hang J, Dussault PH, Carr T. Plant sterol and stanol substrate specificity of pancreatic cholesterol ester lipase. J Nutr Biochem. 2009 doi: 10.1016/j.jnutbio.2009.04.008. Available online 16 July 2009. [DOI] [PubMed] [Google Scholar]; b) Rasmussen HE, Guderian DM, Jr, Wray CA, Dussault PH, Schlegel VL, Carr T. Reduction in cholesterol absorption is enhanced by stearate-enriched plant sterol esters in hamsters. J Nutr. 2006;136:2722–7. doi: 10.1093/jn/136.11.2722. [DOI] [PubMed] [Google Scholar]
- 7.Holman RT, Lundberg WO, Malkin T. Progress in the Chemistry of Fats and Other Lipids. Vol. 1. Pergamon Press; London: 1952. Chapter 2. [Google Scholar]
- 8.Zhang X, Geoffroy P, Miesch M, Julien-David D, Raul F, Aoud′e-Werner D, Marchioni E. Gram-scale chromatographic purification of β - sitosterol : Synthesis and characterization of β - sitosteroloxides. Steroids. 2005;70:886–895. doi: 10.1016/j.steroids.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Kircher HW, Rosenstein FU. Purification of sitosterol. Lipids. 1973;8:97–110. [Google Scholar]
- 10.For an overview of stigmasterol hydrogenation, see: Geoffroy P, Julien-David D, Marchioni E, Raul F, Aoud’e-Werner D, Miesch M. Synthesis of highly pure oxyphytosterols and (oxy)phytosterol esters: Part I. Regioselective hydrogenation of stigmasterol: An easy access to oxyphytosterols. Steroids. 2008;73:702–7. doi: 10.1016/j.steroids.2008.02.004.
- 11.Steele JA, Mosettig E. J Org Chem. 1963;28:571–2. [Google Scholar]; McCarthy FO, Chopra J, Ford A. Synthesis, isolation and characterization of β-sitosterol and β-sitosterol oxide derivatives. Org Biomol Chem. 2005;3:3059–65. doi: 10.1039/b505069c. [DOI] [PubMed] [Google Scholar]
- 12.For an application to sidechain-modified steroids, see: Khripach VA, Zhabinskii VN, Konstantinova OV, Khripach NB, Antonchick AV, Antonchick AP, Schneider B. Preparation of (25R)- and (25S)-26-functionalized steroids as tools for biosynthetic studies of cholic acids. Steroids. 2005;70:551–62. doi: 10.1016/j.steroids.2005.02.014.
- 13.Sarmah P, Barua NC. Aluminum triiodide: a convenient reagent for deoxygenation of oxiranes. Tetrahedron Lett. 1988;29:5815–6. [Google Scholar]
- 14.Syamala MS, Das JJ. A novel and highly β-selective epoxidation of Δ5-unsaturated steroids with permanganate ion. J Org Chem. 1992;57:1928–30. [Google Scholar]; Baqi Y, Giroux S, Corey EJ. A study of the epoxidation of cycloolefins by the t-BuOH copper-permanganate system. Org Lett. 2009;11:959–61. doi: 10.1021/ol802923n. [DOI] [PubMed] [Google Scholar]
- 15.Wang SM, Zhang YB, Liu HM, Yu GB, Wang KR. Mild and selective deprotection method of acetylated steroids and diterpenes by dibutyltin oxide. Steroids. 2007;72:26–30. doi: 10.1016/j.steroids.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Tietze LF, Raith C, Brazel CC, Hoelsken S, Magull J. Enantioselective Synthesis of 2-Substituted Alcohols Using (+)-(1S,2S)-Pseudoephedrine as Chiral Auxiliary. Synthesis. 2008:229–36. [Google Scholar]
- 17.Akihisa T, Tanaka N, Yokota T, Tanno N, Tamura T. 5α-Cholest-8(14)-en-3β-ol and three 24-alkyl-Δ8(14)-sterols from the bulbils of Dioscorea batatas. Phytochemistry. 1991;30:2369–2372. [Google Scholar]
- 18.Attempts to perform the Cu(MnO4)2 oxidation on unprotected stigmasterol resulted in formation of numerous byproducts.
- 19.Khripach VA, Zhabinskii VN, Konstantinova OV, Khripach NB, Antonchick AP, Schneider B. [3, 3 ] - Claisen rearrangements in 2 4α - methyl steroid synthesis : Application to campesterol, crinosterol, and Δ2 5 - crinosterol side chain construction. Steroids. 2002;67:597–603. doi: 10.1016/s0039-128x(02)00007-7. [DOI] [PubMed] [Google Scholar]
- 20.Assigned from the 10.6 Hz coupling constant for H22–H23.
- 21.See, for example: Murakami K, Watanabe B, Nishida R, Mori N, Kuwahara Y. Identification of crinosterol from astigamatid mites. Insect Biochem Mol Biol. 2007;37:506–511. doi: 10.1016/j.ibmb.2007.02.007.
- 22.Blakemore PR. The Modified Julia Olefination: Alkene Synthesis via the Condensation of Metallated Heteroarylalkylsulfones with Carbonyl Compounds. Perkins. 2002;1:2563–85. [Google Scholar]
- 23.Sutoris V, Foltinova P, Gaplovsky A. Benzothiazole compounds. XVII. 2-Alkyl- and 2-aralkylsulfonylbenzothiazoles and their antimicrobial activity. Chemicke Zvesti. 1980;34:404–12. [Google Scholar]; Bourdon B, Corbet M, Fontaine P, Goekjian PG, Gueyrard D. Synthesis of enol ethers from lactones using modified Julia olefination reagents: application to the preparation of tri- and tetrasubstituted exoglycals. Tetrahedron Lett. 2008;49:747–749. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.