

Abstract
The African swine fever virus (ASFV) DP71L protein is present in all isolates as either a short form of 70 to 72 amino acids or a long form of about 184 amino acids, and both of these share sequence similarity to the C-terminal domain of the herpes simplex virus ICP34.5 protein and cellular protein GADD34. In the present study we expressed DP71L in different mammalian cells and demonstrated that DP71L causes dephosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α) in resting cells and during chemical-induced endoplasmic reticulum stress and acts to enhance expression of cotransfected reporter genes. We showed that DP71L binds to all the three isoforms (α, β, and γ) of the protein phosphatase 1 catalytic subunit (PP1c) and acts by recruiting PP1c to eIF2α. We also showed that DP71L inhibits the induction of ATF4 and its downstream target, CHOP. We investigated the eIF2α phosphorylation status and induction of CHOP in porcine macrophages infected by two ASFV field isolates, Malawi Lil20/1 and Benin 97/1, and two DP71L deletion mutants, MalawiΔNL and E70ΔNL. Our results showed that deletion of the DP71L gene did not cause an increase in the level of eIF2α phosphorylation or induction of CHOP, indicating that DP71L is not the only factor required by the virus to control the phosphorylation level of eIF2α during infection. We therefore hypothesize that ASFV has other mechanisms to prevent the eIF2α phosphorylation and the subsequent protein synthesis inhibition.
In eukaryotic cells, control of the availability of active nonphosphorylated eukaryotic translation initiation factor 2 alpha (eIF2α) by reversible phosphorylation is the key and rate-limiting step regulating global protein synthesis (40). There are four mammalian protein kinases that phosphorylate eIF2α on Ser51: heme-regulated kinase (12), which is probably significant only in erythroid cells; protein kinase R (PKR), which is activated by double-stranded RNA of more than 40 bp in length and is important in the antiviral response (2); PKR-like endoplasmic reticulum (ER) kinase (PERK), which is activated by ER stress (14); and a homologue of the only eIF2 kinase in the yeast Saccharomyces cerevisiae, general control of amino acid biosynthesis kinase (GCN2), which is activated by depletion of certain amino acids (3). eIF2 forms ternary complexes with GTP and initiator Met-tRNA, which are required for preinitiation complex formation and mRNA scanning to recognize the translational start codon. Phosphorylated eIF2α (P-eIF2α) is fully capable of forming an initiation-competent eIF2 ternary complex, but following its release, P-eIF2α-GDP binds to the guanine nucleotide-exchange factor eIF2B with a much greater affinity and sequesters its activity (22). The level of eIF2 ternary complex consequently falls and most mRNA translation is reduced. Paradoxically, translation from certain mRNAs, such as activating transcription factor 4 (ATF4) ATF5, and growth arrest and DNA damage-inducible protein 34 (GADD34), containing upstream open reading frames (uORFs) of appropriate type and position, can actually be increased in the presence of P-eIF2α (24, 29, 37, 43).
Accumulation of unfolded proteins within the ER causes ER stress. Eukaryotic cells employ the unfolded protein response (UPR) to counterbalance this stress. Three ER transmembrane proteins have been identified to sense ER stress and trigger the UPR pathway through distinct but closely cooperative signaling mechanisms (35). These are ATF6, inositol-requiring transmembrane kinase/endonuclease 1 (IRE1), and PERK. Activated PERK phosphorylates and inactivates eIF2α, thereby arresting most mRNA translation to alleviate ER stress. P-eIF2α increases the ATF4 expression level by bypassing the inhibitory uORFs in its mRNA that inhibit translation under resting conditions (37). ATF4 increases the transcription of UPR target genes, including GADD34, which recruits the protein phosphatase 1 catalytic subunit (PP1c) α isoform (PP1α) to dephosphorylate eIF2α and therefore restores global translation (4). CHOP is another ATF4 target gene, and its translation is also upregulated when eIF2α phosphorylation increases (20). CHOP is the key factor causing apoptosis in cells with irrecoverable ER stress (19).
To counter viral infection, host cells have evolved pathways to sense, confine, and clear invading viruses, including the shutoff of translation to inhibit viral protein synthesis. Viruses have evolved sophisticated strategies to mask host sensing of infection, prevent translational shutoff, and prolong host life. For example, vaccinia virus E3L and K3L inhibit PKR-mediated phosphorylation of eIF2α (23), herpes simplex virus (HSV) protein Us11 inhibits PKR activation, and HSV ICP34.5 protein recruits PP1α to dephosphorylate eIF2α (18).
African swine fever virus (ASFV) is the sole member of the Asfaviridae family. It is a large cytoplasmic DNA virus with similarity to poxviruses in genome organization and replication strategy. ASFV infects domestic pigs and causes an acute disease with a high mortality, for which there is no vaccine. It has a genome of between 170 and 190 kb, depending on the strain, and encodes more than 150 genes, of which many are not essential for virus replication but are involved in modulating virus-host interaction, immune evasion, and pathogenesis (36). The DP71L gene is encoded as either a 184-amino-acid long form, DP71L(l), or a 70- to 72-amino-acid short form, DP71L(s). The ASFV DP71L(l) form has been shown to be expressed late during the replication cycle (11). Deletion of either the DP71L(l) or the DP71L(s) gene did not reduce ASFV replication in either primary pig macrophages or tissue culture cell lines (32, 44). Deleting the short form of this gene from the virulent E70 isolate resulted in a dramatic reduction of virulence following infection of pigs (44). However, deletion of the DP71L gene from other isolates, Malawi Lil20/1 and Pr4, did not significantly reduce virulence (1). The C-terminal 56-amino-acid domain of DP71L is highly conserved among different isolates and shares about 40% amino acid identity with the C terminus of the herpes simplex virus ICP34.5 protein and the cellular protein GADD34. Notably, the PP1 docking site and the regulatory motif Vx(7,8)Rx3Wx5DRxRFxRRx11L are conserved between these proteins, suggesting that DP71L may have a similar function and also acts to direct PP1c to dephosphorylate substrates, including eIF2α. By using the yeast two-hybrid system and by direct binding studies, we have previously shown that DP71L interacts with PP1c (32). ASFV infection of Vero cells was shown to decrease the phosphorylation level of eIF2α at late times postinfection (5, 32), but so far there is no direct evidence to show that DP71L is responsible for this dephosphorylation of eIF2α. We reported previously that extracts from ASFV-infected cells have increased PP1 activity compared to that of mock-infected cells, as assayed by dephosphorylation of the substrate phosphorylase a, and that this is dependent on the presence of the DP71L gene (32). However, the relevance of this increase in PP1 activity to ASFV virulence and pathogenesis is unclear.
In the present study, we demonstrate that DP71L, the host protein GADD34, and the HSV ICP34.5 protein bind to all the three isoforms (α, β, and γ) of PP1c. We also show that expression of DP71L in cell lines reduces the level of phosphorylated eIF2α to undetectable levels, supporting the model that DP71L recruits PP1 to dephosphorylate eIF2α. As predicted from this observation, we show that expression of DP71L enhances the expression of cotransfected reporter genes. We also show that DP71L inhibits the induction of ATF4 and its downstream target, CHOP. We investigated the eIF2α phosphorylation status and CHOP induction in porcine macrophages infected by two field isolates, ASFV Malawi LiL20/1 and ASFV Benin, and two DP71L deletion mutants, MalawiΔNL and E70ΔNL. Our results show that deletion of DP71L did not cause an increase in eIF2α phosphorylation or CHOP induction, indicating that factors in addition to DP71L are involved in control of the phosphorylation level of eIF2α during infection.
MATERIALS AND METHODS
Plasmids.
Vectors used for transient expression were based on the pEF.plink2 plasmid and contain the EF1α promoter (25). The cDNA fragments of the DP71L(s) and DP71L(l) forms of DP71L with a hemagglutinin (HA) tag fused at the 5′ end were amplified by PCR from the constructs pcDNA3.HA.DP71L(s) and pcDNA.HA.DP71L(l) (11), respectively, and were cloned into pEF.plink2 between the NcoI and XbaI sites to generate pEF.HA.DP71L(s) and pEF.HA.DP71L(l), respectively. To make recombinant lentiviruses, the DP71L(s) sequences with and without the 3′ end V5 tag were amplified by PCR and cloned into a bicistronic expression vector derived from the self-inactivating lentivirus vector pHR-SIN-CSGW (10), between the NdeI and SpeI sites, to create pdl.DP71L(s).V5 and pdl.DP71L(s), respectively.
For yeast two-hybrid assays, cDNAs were cloned into pGBKT7 (Clontech) or pHON1 (a derivative of pGBKT7 with the kanamycin resistance gene replaced by an ampicillin resistance gene) for expression of proteins as GAL4 DNA binding domain (DBD) fusions, into pGADT7 (Clontech) for expression of proteins as GAL4 activation domain (AD) fusions, or into pHON7, a vector designed to permit nuclear expression of a nonfusion protein (the bridging or “third” protein) in yeast (8). Full-length cDNAs for these cloning steps were generated by PCR using oligonucleotides containing NcoI and EcoRI sites at the 5′ and 3′ ends, respectively. The sequences cloned were as follows: DP71L(s), from the Pr4 strain sequence of ASFV; DP71L(l), from the Malawi Lil-20/1 strain of ASFV; HSV type 1 (HSV-1) ICP34.5, from a BamHI fragment of genomic HSV-1; GADD34; eIF2α; and the three isoforms of protein phosphatase from full-length clones obtained from the mammalian genome consortium.
Cells and viruses.
Primary porcine macrophages were prepared by lung lavage or as the adherent cell population from bone marrow cells. The former were maintained in RPMI 1640 medium containing 10% fetal bovine serum and the latter in Earl's saline supplemented with 10% porcine serum. The ASFV Malawi Lil20/1 and Benin 97/1 isolates were described previously (6, 15). DP71L gene-knockout mutants MalΔNL and E70ΔNL were kind gifts from L. Zsak and D. Rock (1). Deletion of the DP71L genes from these viruses was confirmed by PCR (data not shown). Macrophages were infected with different ASFV isolates at a multiplicity of infection (MOI) of about 5.
Vero, PK15, HEp2, and 293FT cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum. Transfection was carried out using Lipofectamine (Invitrogen) for plasmids and Lipofectamine 2000 (Invitrogen) for small interfering RNAs (siRNAs).
To generate recombinant lentiviral particles, 293FT cells were transfected with pdl.DP71L(s).V5 or pdl.DP71L(s), pCMVR8.91 (expressing the gal/pol, tat, and rev genes of human immunodeficiency virus type 1), and pMD-G (expressing the vesicular stomatitis virus G gene). Two days after transfection, viruses were harvested from the culture medium and filtered through 0.45-μm-pore-size Tuffryn membrane filters (Invitrogen). To make cell lines expressing DP71L(s), 293FT, PK15, and HEp2 cells were transduced with the harvested recombinant lentiviral particles at an MOI of about 1 in the presence of Polybrene (8 μg/ml; Sigma). At 2 days posttransduction, pools of transformed cells were selected by resistance to puromycin at a concentration of 3.0 μg/ml for 293FT and PK15 cells or 0.5 μg/ml for HEp2 cells.
Reporter gene assays.
Lysates were prepared from cells that had been transfected with plasmids expressing reporter genes and analyzed for luciferase and β-galactosidase (β-Gal) activities, as described previously (8). The luciferase reporter plasmid pTK(-l05)lucter with the firefly luciferase gene under the control of the HSV thymidine kinase (tk) promoter and a constitutively expressed β-galactosidase reporter plasmid, pJATlacZ, have been described previously (21, 26).
siRNA delivery.
Stealth Select RNA interference (RNAi) siRNAs targeting different isoforms of PP1c (HSS143413, HSS143414, HSS143415, HSS108340, HSS108341, HSS108342, HSS108343, HSS108344, and HSS108345; Invitrogen) were transfected into HEp2 or PK15 cells using Lipofectamine 2000 to knock down the expression of the respective PP1c isoform. Stealth RNAi siRNA negative control (Invitrogen) was included in the experiments.
Antibodies.
Antibodies used included mouse monoclonal antibody (MAb) against total eIF2α (MAb L57A5; Cell Signaling Technology), used at a dilution of 1:500 for Western blotting, and rabbit monoclonal antibody against eIF2α phosphorylated at Ser51 (ab32157; Abcam), used at dilutions of 1:50 and 1:200 for confocal microscopy and Western blotting, respectively. The V5 tag antibody (ab53418; Abcam) was diluted 1:100 and 1:1,000 for confocal microscopy and Western blotting, respectively. The following antibodies were from Santa Cruz Biotechnology and used according to the manufacturer's recommendations: PP1α (sc-6104), PP1β (sc-6107), PP1γ (sc-6108), GADD153/CHOP (sc-575 and sc-7351), and ATF4 (sc-22800). Mouse MAbs against ASFV late protein VP72 (4H3) were described previously (9). The following horseradish peroxidase (HRP)-conjugated secondary antibodies were used for Western blotting, according to the manufacturer's instructions: the goat anti-mouse IgG (H+L) HRP conjugate (W4021; Promega), the donkey anti-goat IgG HRP conjugate (V8051; Promega), and the goat anti-rabbit IgG (H+L) HRP conjugate (7074; Cell Signaling Technology). Both the Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 568 goat anti-mouse IgG (A1108 and A1104, respectively; Molecular Probes) were used at a dilution of 1:500 in the confocal microscopy staining.
Western blotting.
Proteins in cell lysates were fractionated by reducing SDS-PAGE before transfer to Hybond ECL membranes (RPN303D; GE Healthcare). The membranes were blocked in phosphate-buffered saline (PBS) with 5% skim milk powder for 0.5 h and incubated with the primary and HRP-conjugated secondary antibodies diluted in the same blocking buffer overnight at 4°C and 1 h at room temperature, respectively. The membranes were washed copiously with PBS containing 0.5% Tween 20 after each antibody staining. The bound antibodies were detected by using LumiGLO reagent (7003; Cell Signaling Technology) and exposure to X-ray films.
Confocal microscopy.
Cells were grown on coverslips. After mock treatment, transfection, or infection, cells were fixed with 4% paraformaldehyde and then permeabilized with 0.5% Triton X-100. The fixed and permeabilized cells were blocked with 0.5% bovine serum albumin for 30 min before antibody staining. Assays with primary and secondary antibody controls were carried out to ensure that the staining was specific. The DNA was counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Initial confocal microscope images were obtained using the manufacturer's software (Leica confocal software).
Coimmunoprecipitation (CoIP).
Cells expressing DP71L(s)-V5 fusion protein and control nontransfected cells were lysed in Pierce IP lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol) supplemented with protease inhibitor cocktail (Thermo Scientific Halt). The lysate, clarified by centrifugation, was incubated with anti-V5 agarose affinity gel (A3745; Sigma) overnight at 4°C with rotation, and the resin was washed with the lysis buffer before it was boiled in 2× SDS-PAGE sample buffer to release the proteins from the resin. Proteins in the supernatant were separated by SDS-PAGE and analyzed by Western blotting using antibodies against total eIF2α, PP1α, PP1β, and PP1γ. The antibody against the NF-κB p50 subunit (sc-1190; Santa Cruz Biotechnology) was used as a negative control.
Yeast two-hybrid assays.
Combinations of GAL4 DBD and GAL4 AD fusion plasmids, in the presence or absence of a plasmid expressing nuclear targeted nonfusion proteins (pHON7; see above), were introduced into Saccharomyces cerevisiae strain PJ69-4α using polyethylene glycol-lithium acetate-mediated transformation and selected on synthetic dropout medium lacking leucine and tryptophan (SD-LW) for two-hybrid combinations or synthetic dropout medium lacking leucine, tryptophan, and uracil (SD-LWU) for three-plasmid combinations. Individual colonies were subsequently streaked onto the appropriate medium also lacking histidine (i.e., SD-LWH or SD-LWUH) and containing 5 mM 3-aminotriazole. Growth was monitored for 4 to 10 days at 30°C.
RESULTS
DP71L(s) expression causes a decrease in phosphorylation of eIF2α.
DP71L has the conserved PP1c docking motif (RVxF) and the 38-amino-acid motif [Vx (7,8)Rx3Wx5DRxRFxRRx11L] common to GADD34 and ICP34.5, which are required for PP1c binding and eIF2α phosphatase activity (16, 17). To determine the effect of DP71L expression on eIF2α phosphorylation in cells, we compared the levels of eIF2α phosphorylated on serine 51 (P-eIF2α) in cells expressing DP71L with those in cells not expressing DP71L. PK15 cells were transduced with lentiviral vectors expressing DP71L(s), and cells expressing DP71L(s) were selected by growth in the presence of puromycin. As shown in Fig. 1, under normal culture conditions, the basal level of P-eIF2α could be detected by Western blotting in control nontransduced PK15 cells (lane 1) but not in cells transduced with lentiviral vectors expressing DP71L(s) (lane 2). The same results were obtained using DP71L(s) tagged at the C terminus with the V5 tag (Fig. 1, lane 3), demonstrating that the V5 tag did not affect the function of DP71L(s). Similar results were observed in experiments using HEp2 and 293FT cells (data not shown).
FIG. 1.
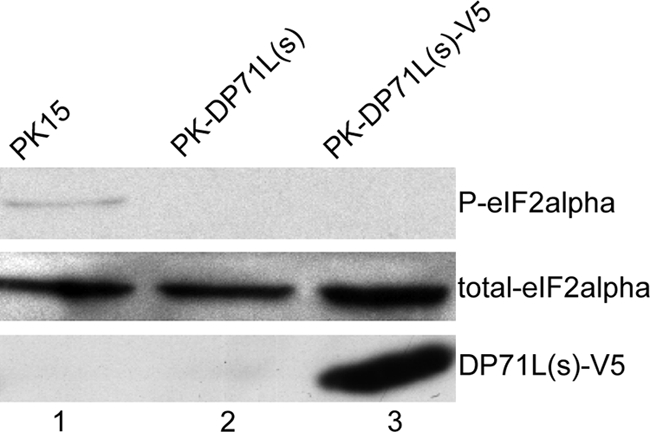
ASFV DP71L(s) mediates dephosphorylation of eIF2α. Western blotting was used to analyze the levels of P-eIF2α, total eIF2α, and V5-tagged DP71L(s) [DP71L(s)-V5] proteins (indicated at the right) under resting conditions in PK15 cells (lane 1), cells stably expressing untagged ASFV DP71L(s) (lane 2), and cells stably expressing V5-tagged ASFV DP71L(s) (lane 3). The data shown here are representative of those from more than three independent experiments. Similar results were also obtained with HEp2 and 293FT cells (data not shown).
DP71L is an enhancer of gene expression.
Reversible phosphorylation of eIF2α regulates global protein synthesis rates (40), and therefore, it was predicted that expression of DP71L would cause an increase in translation (41). To test the effect of DP71L on gene expression, we transfected a reporter plasmid expressing the β-Gal gene under the control of the rat actin promoter into nontransduced PK15 or PK15 cells transduced with the lentiviral vector expressing DP71L(s) and measured the β-Gal activity in the cell lysates at 24 h posttransfection. As shown in Fig. 2 A, the β-Gal activity in the lysates from cells expressing DP71L(s) was over twice that detected in lysates from PK15 cells. To further establish the role of DP71L(s) in enhancing gene expression, we cotransfected a fixed amount of plasmids expressing reporter genes and an equal amount of plasmid pEF.DP71L(s) expressing DP71L(s) or the vector plasmid pEF.plink2 into Vero cells and measured the yield of enzyme derived from expression of the reporter gene. Reporter plasmids expressing β-Gal under the control of the constitutive promoter described in Materials and Methods and in the Fig. 2B legend and the plasmid expressing the luciferase gene under the control of the constitutive thymidine kinase promoter (Fig. 2C) were used. Expression of DP71L(s) increased the yields of both reporters. Similar results were observed in transient transfection experiments in 293FT cells (data not shown). Hence, as expected, DP71L(s) enhances expression of other target genes.
FIG. 2.

ASFV DP71L enhances the expression of reporter genes. (A) PK15 control cells and PK15 cells transduced with the lentiviral vector expressing DP71L(s)-V5 (indicated on the x axis) were plated at the same density and transfected overnight with an equal amount of the β-Gal expression construct pJATlacZ per cell numbers before the β-Gal activity (shown on the y axis) was measured, as described in the Materials and Methods. (B and C) Vero cells were cotransfected with an equal amount of the pEF.DP71L(s) construct or the vector plasmid pEF.plink2 (indicated on the x axis) and a fixed amount of the reporter gene construct pJATlacZ (B) or pTK(-l05)lucter (C). Reporter assays were carried out after 24 h. All experiments were repeated at least 3 times. The results are shown in arbitrary units and expressed as means ± standard deviations. (B and C) The reporter activities are normalized to the values from cells cotransfected with vector pEF.plink2, which are set equal to 1. Student's t test was used to analyze the data, and the P values are shown in the charts.
DP71L(s) associates with all three isoforms of PP1c and with eIF2α.
To determine whether DP71L(s) may be present in a complex with PP1c and eIF2α, we first carried out CoIP experiments in mammalian cells transduced with the lentiviral vector expressing the DP71L(s)-V5 fusion protein and the control cells. Cell lysates were incubated with anti-V5 antibody coupled to agarose beads. After the beads were washed to remove nonspecifically bound material, proteins bound to the anti-V5 agarose beads were separated by SDS-PAGE and assayed by Western blotting using antibodies specific for each PP1c isoform or eIF2α. This showed that, as expected, the DP71L(s)-V5 protein was bound to the anti-V5 agarose beads. In addition, as shown in Fig. 3, eIF2α and all the three PP1c isoforms were precipitated with the anti-V5 antibody agarose beads from cells transduced with the lentiviral vector expressing DP71L(s)-V5 but not from the control cells. To rule out the possibility of nonspecific binding to the anti-V5 antibody agarose beads, Western blots were probed with an antibody against the NF-κB p50 subunit. Although the specific p50 band was detected in cell lysates, it was detected only very faintly in samples that were coprecipitated with the anti-SV5 agarose beads. These data indicate that DP71L(s) can be detected in complexes with all the isoforms of PP1c and eIF2α in cells, although they do not demonstrate that DP71L(s) is in a ternary complex including PP1c and eIF2α. Two alternatively spliced forms of PP1γ (39) have been reported. Since we used an antibody that recognizes both of these isoforms, we cannot distinguish if one or both isoforms of PP1γ precipitated with DP71L, although one of these splice variants is expressed predominantly in male germ cells.
FIG. 3.

eIF2α and isoforms α, β, and γ of PP1c are coprecipitated with DP71L(s). Lysates from 293FT cells and 293 cells expressing the DP71L(s)-V5 fusion protein (DP-V5) were immunoprecipitated with an antibody against the V5 tag, and the pulled-down immunocomplexes were washed to remove nonspecifically bound proteins. Proteins that specifically bound to the beads (top row) and the input lysates (bottom row) were then analyzed by Western blotting (WB) using antibodies specific to the three isoforms of PP1c, eIF2α, and the V5 tag (indicated on the top of the panel).
To test whether DP71L(s) can interact with the α, β, and γ isoforms of PP1c, we carried out yeast two-hybrid interaction trap experiments in which DP71L(s) or DP71L(l) forms were fused to the DNA binding domain of the yeast GAL4 protein (“bait”) and were cotransformed into yeast with a plasmid expressing the different isoforms of human PP1 (“prey”) fused to the GAL4 activation domain. Vectors expressing unfused GAL4 domains were included as negative controls. Figure 4 shows that both DP71L(s) and the longer form, DP71L(l), interact with all three isoforms of PP1c, although the interaction with the γ isoform seems to be the least avid. It has previously been reported that ICP34.5 and GADD34 recruit only PP1α to dephosphorylate eIF2α. When we investigated this, using the yeast two-hybrid system, we were able to show that both GADD34 and HSV-1 ICP34.5 interact with the α, β, and γ isoforms of PP1c (Fig. 4). We have previously demonstrated a direct interaction between DP71L and PP1c using purified recombinant proteins (32), and it is therefore extremely likely that the interactions that we observed between DP71L, GADD34, and HSV-1 ICP34.5 and the PP1c α, β, and γ isoforms are direct. Although it is theoretically possible that yeast proteins may be involved in bridging this interaction, the evolutionary distance of yeast from mammals makes this alternative explanation very unlikely.
FIG. 4.

DP71L(s), DP71L(l), GADD34, and HSV-1 ICP34.5 interact directly with the α, β, and γ isoforms of PP1, and DP71L(s) can recruit PP1α to eIF2α. (A) Plasmids expressing either DP71L(s), DP71L(l), GADD34, or HSV-1 ICP34.5 fused to the DNA binding domain of the yeast GAL4 protein (bait) were cotransformed into yeast with a plasmid expressing the indicated isoform of human PP1 (prey) fused to the GAL4 activation domain. Vectors expressing unfused GAL4 domains were included as negative controls. Primary transformants were selected on SD-LW (data not shown) and then streaked onto synthetic medium that also lacked histidine (SD-LWH) and contained 5 mM 3-aminotriazole to enhance stringency. Growth on this medium is indicative of protein-protein interaction. (B) A plasmid expressing eIF2α fused to the DNA binding domain of the yeast GAL4 protein (bait) was cotransformed into yeast with a plasmid expressing the indicated prey [DP71L(s), DP71L(l), GADD34, HSV-1 ICP34.5, or the α-isoform of human PP1]. Positive transformants were selected on SD-LW and then streaked onto SD-LWH with 5 mM 3-aminotriazole selective medium to assay for interactions. To study the ability of DP71L(s) to bridge an interaction between eIF2α and PP1, DP71L(s) was expressed as a nucleus-targeted nonfusion protein in cells also expressing eIF2α bait and PP1α prey; triple transformants were initially selected on SD-LWU and then streaked onto SD-LWUH with 5 mM 3-aminotriazole selective medium to assay for interactions.
Experiments to demonstrate an interaction between any of DP71L(s), DP71L(l), ICP34.5, GADD34, or PP1α and eIF2α using the yeast two-hybrid interaction system were unsuccessful. To determine if DP71L could bridge the interaction between eIF2α and PP1c, we expressed DP71L(s) as a nuclear-targeted nonfusion protein in yeast cells also expressing eIF2α bait and PP1α prey. Triple transformants were selected by growth on SD-LWU. Individual colonies were subsequently streaked onto SD-LWU medium also lacking histidine to assay for interactions (Fig. 4B). Growth of yeast colonies was detected under these selective conditions when DP71L(s) was expressed but not when the empty vector was used. This indicates that DP71L specifically bridges the interaction between eIF2α and PP1c.
Depletion of PP1c using siRNA increases the level of phosphorylated eIF2α in control cells and cells expressing DP71L.
Our data show that DP71L expression is associated with dephosphorylation of eIF2α, interacts directly with all three isoforms of PP1c, and is present in protein complexes with eIF2α. Since there is no evidence that DP71L has an independent phosphatase activity and PP1 is known to be a potent phosphatase of eIF2α, we hypothesize that DP71L-mediated dephosphorylation of eIF2α is dependent on PP1c, as is observed for the cellular proteins GADD34 and CreP (13). To investigate this, we knocked down PP1c expression in both cells transduced with the lentivirus vector expressing DP71L(s)-V5 and control cells using a mixture of siRNAs targeting all the three PP1c isoforms and examined the effect on the eIF2α phosphorylation by Western blotting. As shown in Fig. 5, transfection of scrambled control RNA had no effect on the expression levels of any of the proteins, PP1α, PP1β, PP1γ, total eIF2α, P-eIF2α, or DP71L(s)-V5. In both control HEp2 cells and HEp2 cells transduced with lentiviral vector expressing DP71L(s)-V5, PP1c-specific siRNAs decreased the protein levels of PP1α and PP1β significantly compared to those in cells transfected with control scrambled siRNAs or mock-transfected cells. Interestingly, PP1c depletion led to a significant increase in P-eIF2α levels in the control HEp2 cells, indicating that PP1c plays a homeostatic role in determining the level of P-eIF2α in cells. Consistent with our hypothesis that DP71L acts through PP1c to dephosphorylate eIF2α, PP1c depletion also caused a dramatic increase in the level of eIF2α phosphorylation in DP71L(s)-V5-expressing cells; the level of P-eIF2α under these conditions was at least as high as that seen in the control cells. In contrast, if DP71L acted via a phosphatase other than PP1c, knockdown of PP1c by siRNAs would not result in an increase in P-eIF2α. Interestingly, the increase in the level of P-eIF2α was coincidental with the decrease of the steady-state level of DP71L(s)-V5 (Fig. 5). This may indicate that DP71L(s)-V5 is a short-lived protein and is rapidly degraded following the translation arrest caused by phosphorylation of eIF2α, although other explanations are possible, including alteration in the level of the mRNA for DP71L(s)-V5 mediated by transcriptional or posttranscriptional regulation.
FIG. 5.
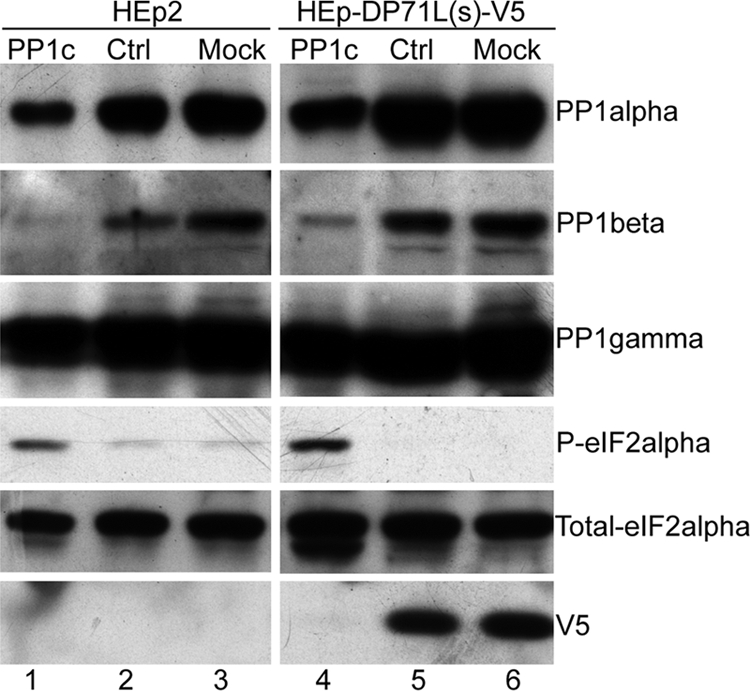
DP71L-mediated dephosphorylation of eIF2α is dependent on PP1c. HEp2 (lanes 1 to 3) and HEp-DP71L(s)-V5 cells (lanes 4 to 6) were transfected with a mixture of siRNAs targeting all PP1c isoforms (lanes 1 and 4) or control (Ctrl) scrambled siRNAs (lanes 2 and 5) or were mock transfected (lanes 3 and 6) for 24 h. Cell lysates were separated by SDS-PAGE and analyzed by Western blotting using antibodies against PP1α, PP1β, PP1γ, total and P-eIF2α, and V5-tagged DP71L(s) (V5), as indicated at the right. The lane numbers are indicated at the bottom.
DP71L inhibits the P-eIF2α-ATF4-CHOP signaling pathway in the unfolded protein response.
Translation of some mRNAs, including that for the transcription factor ATF4, continues in the presence of P-eIF2α. ATF4 activates transcription of a number of downstream targets, including the proapoptotic protein CHOP (35, 40). We predicted that by reducing the levels of P-eIF2α, DP71L would also reduce ATF4 translation and induction of CHOP. To investigate the effect of DP71L on expression of ATF4 and CHOP, we treated cells with an N-glycosylation inhibitor, tunicamycin (Tm), at 10 μg/ml for 4 h or a reducing agent, dithiothreitol (DTT), at 2 mM for 30 min to cause stress and compared the levels of the P-eIF2α, ATF4, and CHOP proteins in control cells and cells transduced with lentiviral vector expressing DP71L(s)-V5. Western blotting results showed that both tunicamycin and DTT treatments caused a dramatic increase in the levels of P-eIF2α in control PK15 cells but only a very slight increase in the cells transduced with lentiviral vector expressing DP71L(s)-V5. Tunicamycin treatment also caused a significant increase in expression of CHOP and ATF4 in control PK15 cells, but this was only very faintly detected in cells transduced with the lentiviral vector expressing DP71L(s)-V5 (Fig. 6 A). DTT treatment did not increase the level of ATF4 or CHOP expression in either control cells or cells transduced with lentiviral vectors expressing DP71L(s)-V5. This was probably due to the short treatment period. The lentivirus-transduced PK15 cells were not cloned but consisted of a pool of cells expressing DP71L(s)-V5 protein at different levels. We also observed the induction of CHOP by confocal microscopy following treatment with tunicamycin for 4 h. As shown in Fig. 6B, all PK15 control cells treated with tunicamycin for 4 h expressed CHOP with a distinct nuclear localization, whereas only a small group of cells that did not express DP71L(s)-V5 were stained positive for CHOP in the cells transduced with lentivirus expressing DP71L(s)-V5. No induction of CHOP was observed in cells which were not treated with drugs (data not shown). Taken together, these results demonstrate that DP71L(s) expression inhibits ATF4 and CHOP expression following induction of UPR caused by ER stress. This is most likely due to the ability of DP71L(s) to reduce the levels of eIF2α phosphorylation, but we cannot rule out other eIF2α phosphorylation-independent mechanisms.
FIG. 6.

ASFV DP71L(s) inhibits AFT4 and CHOP induction. (A) PK15 (lanes 1 to 3) and PK-DP71L(s)-V5 (lanes 4 to 6) cells were mock treated (lanes 1 and 4) or were treated with DTT for 30 min (lanes 2 and 5) or tunicamycin at 10 μg/ml for 4 h (lanes 3 and 6). Western blotting was used to analyze the cell lysates for the levels of total and phosphorylated eIF2α, ATF4, and CHOP proteins (indicated at the right). The total eIF2α was also used as the loading control. The lane numbers are indicated at the bottom. (B) Confocal microscopy was used to analyze the induction of CHOP in PK15 (top row) and PK-DP71(s)-V5 (bottom row) cells. The cells were treated with tunicamycin at 10 μg/ml for 4 h, and then they were permeabilized and costained with antibodies against V5 tag and CHOP, respectively, followed by appropriate Alexa Fluor 568- or 488-conjugated secondary antibodies. CHOP was stained green, and the V5 tag was stained red. The nucleus was stained blue with DAPI.
ASFV-mediated inhibition of eIF2α phosphorylation and CHOP induction is observed when the DP71L gene is deleted.
It has been shown that deletion of the DP71L gene does not reduce ASFV replication in vitro in Vero cells or porcine macrophages. It was demonstrated previously that infection of Vero cells with the BA71V isolate of ASFV resulted in an approximately 2-fold reduction of the levels of P-eIF2α at late times postinfection (32). Furthermore, ASFV infection inhibited UPR, including CHOP induction (28). To determine the role of the DP71L protein in reducing eIF2α phosphorylation and CHOP induction during ASFV infection, we infected primary porcine macrophages with two wild-type ASFV isolates, Malawi Lil20/1 and Benin 97/1, and two DP71L-knockout mutant viruses, MalΔNL, which has a deletion of the DP71L(l) from the Malawi Lil 20/1 isolate genome, and E70ΔN, which has a deletion of the DP71L(s) gene from the E70 isolate. We compared the phosphorylation of eIF2α and CHOP expression in infected cells by using confocal microscopy at 24 h postinfection, including tunicamycin treatment for the final 4 h. Cells were costained for P-eIF2α or CHOP (red) and the late viral protein VP72 (green), and DNA was counterstained blue with DAPI. As shown in Fig. 7, in mock-infected cells strong cytoplasmic staining of P-eIF2α and distinct nuclear staining of CHOP were observed, whereas in the cells infected with all of the four viruses described above, only a weak and scattered background staining pattern was observed for both P-eIF2α and CHOP, and there was no obvious difference between the four viruses. All the infected cells showed the characteristic staining pattern of VP72 in viral factories (Fig. 7). These data confirm that ASFV infection inhibits phosphorylation of eIF2α and induction of CHOP and indicate that ASFV may have other proteins as well as DP71L to carry out these functions.
FIG. 7.

ASFV inhibits eIF2α phosphorylation and CHOP induction independently of DP71L. Primary porcine macrophages were infected for 24 h with four viruses, as indicated on the top of the panel (Mal, ASFV Malawi Lil20/1; MalΔNL, DP71L knock-out mutant of ASFV Malawi Lil20/1; Ben, ASFV Benin isolate; and E70ΔNL, DP71L knock-out mutant of ASFV E70 isolate), with tunicamycin (10 μg/ml) being added for the final 4 h before fixation. Cells were permeabilized and stained with a rabbit antibody against P-eIF2α (top row) or a rabbit antibody against CHOP (bottom row), followed by an Alexa Fluor 568-conjugated secondary antibody (red). The cells were also costained for the viral late protein VP72 with a mouse monoclonal antibody, followed by an Alexa Fluor 488-conjugated secondary antibody (green). DNA was counterstained blue with DAPI. Only the merged images were shown.
DISCUSSION
In eukaryotic cells, the reversible phosphorylation of eIF2α is a key rate-limiting step in the control of protein synthesis. A number of stimuli, including virus infection, amino acid starvation, and ER stress, activate pathways leading to phosphorylation of eIF2α and reduction in global protein synthesis. Four protein kinases have been characterized to be involved in this process. One of these, PKR, is activated by double-stranded RNA produced during virus infection, and this contributes to the shutoff of host protein synthesis, which can reduce virus replication by limiting translation of virus proteins. So, it is not surprising that many viruses reduce phosphorylation of eIF2α by various mechanisms to enhance their replication and that virus proteins involved in this process can influence virulence. Here we have demonstrated that ASFV DP71L protein expression in cells transduced with lentiviral vectors expressing DP71L(s) reduces the level of phosphorylated eIF2α to undetectable levels. One predicted effect of this is to enhance translation, and we demonstrated that cotransfection into cells of a plasmid expressing DP71L(s) with plasmids expressing reporter genes increased the expression of the reporter genes. We provided strong evidence to support the hypothesis that DP71L acts to reduce the amount of phosphorylated eIF2α by recruiting PP1c to dephosphorylate eIF2α. We showed previously that purified recombinant DP71L protein binds directly to purified PP1, as was predicted, since DP71L contains a putative PP1 docking site. Here we showed, using the yeast two-hybrid system, that DP71L(s) interacts with all three isoforms of PP1c (α, β, γ) and is coprecipitated from mammalian cells with all the three PP1c isoforms as well as eIF2α. However, the exact composition of the ternary protein complexes in cells is unknown. Using the yeast two-hybrid system, we demonstrated that eIF2α did not interact directly with either PP1c or DP71L but that DP71L could bridge an interaction between eIF2α and PP1α. This supports the hypothesis that DP71L is involved in recruiting PP1c to dephosphorylate eIF2α. The mechanism by which this is mediated is not yet defined for DP71L or ICP34.5 and GADD34.
The ability of DP71L to reduce the levels of phosphorylated eIF2α, most likely by recruitment of PP1c to dephosphorylate eIF2α, is shared with the herpes simplex virus-encoded ICP34.5 protein and with the host DNA damage response protein GADD34 and its constitutively expressed homologue, CreP. Our data strongly suggest that DP71L should also be classified as a member of PP1 regulatory family 15 (PP1R15). Interestingly, the DP71L(s) protein is only 70 to 72 amino acids long, whereas ICP34.5 is about 250 amino acids long and GADD34 is about 670 amino acids long. Thus, our data help to define the minimal domain required to cause dephosphorylation of eIF2α. Previous studies (18) have shown that the ICP34.5 protein binds to the α isoform of PP1c. We show here that both ICP34.5 and GADD34 bind to all three isoforms of PP1c, and this extends knowledge about how these two proteins may function. It is not clear if complexes between the different PP1c isoforms and DP71L have different functions in cells or if all three isoforms are involved in dephosphorylation of eIF2α. We also demonstrated that expression of DP71L(s) in cell lines inhibited induction of the ATF4-CHOP signaling branch of the UPR, most likely as the consequence of causing eIF2α dephosphorylation, but we cannot rule out P-eIF2α-independent mechanisms. Using cells expressing a form of eIF2α mutated at Ser51 which cannot be phosphorylated would help clarify this.
Previous results (11) show that the DP71L(s) and DP71L(l) forms can be localized in the nucleus as well as the cytoplasm, suggesting that the protein has a role in the nucleus, in addition to the cytoplasmic function in controlling eIF2α dephosphorylation.
DP71L is not required for viral replication in vitro. Deleting DP71L from ASFV E70 resulted in attenuation in vivo, but no attenuation was achieved by deleting the long-form gene from the Malawi Lil20/1 isolate or the short-form gene from the Pr4 isolate. The authors speculated that E70 virus might have lost other genes which are complementary in function to DP71L(l). In the present study, we compared the levels of phosphorylated eIF2α and CHOP in primary porcine macrophages infected with wild-type ASFV and two DP71L gene deletion mutants: MalΔNL, which has a deletion of the DP71L(l) from the Malawi Lil 20/1 isolate genome, and E70ΔN, which has a deletion of the DP71L(s) gene from the E70 isolate. Infection with all these viruses inhibited phosphorylation of eIF2α and induction of CHOP, suggesting that the viruses have genes in addition to DP71L which target these pathways. The data presented above also suggest that another as yet defined function of DP71L may be responsible for the reduction in virulence caused by deleting the DP71L gene from the E70 genome. This is supported by previous findings which showed that CHOP induction was inhibited at an early stage of ASFV infection (28), whereas DP71L is expressed late in infection (11). Inhibition of eIF2α phosphorylation and of CHOP induction is advantageous for virus replication, since preventing both inhibition of global protein synthesis and induction of apoptosis would increase virus production. ASFV has been reported to have other mechanisms to inhibit apoptosis, including virus-encoded Bcl2 and IAP homologues. As yet no other mechanisms by which ASFV prevents shutdown of translation have been described. It has been well documented that viral gene products inhibit eIF2α kinases. For example, the poxvirus E3L protein (33) and the HSV-1 Us11 protein (31) inhibit PKR, and HSV-1 glycoprotein gB binds to PERK to prevent its activation (27). It would be an interesting and logical step forward to identify the ASFV inhibitors of eIF2α kinases.
PP1 is ubiquitously expressed and regulates many physiological processes. Its phosphatase activity and substrate specificity are dictated by different regulatory subunits, and so far at least 100 of these have been identified (34). Given the importance of protein phosphorylation, it is widely accepted that there should be no free PP1c in the cell. They are associated with either inhibitory subunits or activating subunits that target certain substrates. In the present study, we have generated evidence to support that the ASFV DP71L protein recruits PP1c to dephosphorylate eIF2α. However, it is still unclear whether the DP71L protein displaces the inhibitory subunits or cooperates with other regulatory subunits to realize this activity. In comparison with GADD34 and ICP34.5, DP71L has unique features; notably, it is a very compact protein composed of only 70 to 72 amino acids in the short form. It has the conserved PP1c docking motif and a 38-amino-acid motif [Vx(7,8)Rx3Wx5DRxRFxRRx11L] common to GADD34 and ICP34.5, which are required for PP1 binding and eIF2α phosphatase activity. However, DP71L lacks other domains present in GADD34 and ICP34.5, including the N-terminal domain in GADD34 involved in promoting apoptosis, the PCNA binding domain, and the nuclear export signal of 3-amino-acid PAT repeats in ICP34.5. At the C terminus, DP71L protein lacks the RARA-AR motif found in both GADD34 and ICP34.5. This motif was critical for them to bind to PP1c and/or form an active eIF2α phosphatase (42), suggesting that DP71L may use a mechanism different from that used by GADD34 or ICP34.5 (4, 17). Moreover, ICP34.5 has other functions independent of eIF2α phosphorylation, such as regulating autophagy via the interaction with beclin 1 (30), modulating viral glycoprotein processing (7), and targeting TBK1-mediated signaling (38). Although dephosphorylation of eIF2α is likely to be a key function of DP71L, we predict that it also has other functions since both the DP71L(s) and DP71L(l) forms are also localized to the nucleus. In addition, the extra residues present in the DP71L(l) form are likely to be involved in other functions.
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council (grants BB/E021239 and BB/E019781/1).
We thank Pippa Hawes for assistance with confocal microscopy and Laszlo Zsak and Dan Rock for providing ASFV mutant viruses.
Footnotes
Published ahead of print on 11 August 2010.
REFERENCES
- 1.Afonso, C. L., L. Zsak, C. Carrillo, M. V. Borca, and D. L. Rock. 1998. African swine fever virus NL gene is not required for virus virulence. J. Gen. Virol. 79:2543-2547. [DOI] [PubMed] [Google Scholar]
- 2.Barber, G. N., M. Wambach, M. L. Wong, T. E. Dever, A. G. Hinnebusch, and M. G. Katze. 1993. Translational regulation by the interferon-induced double-stranded-RNA-activated 68-kDa protein-kinase. Proc. Natl. Acad. Sci. U. S. A. 90:4621-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berlanga, J. J., J. Santoyo, and C. de Haro. 1999. Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2 alpha kinase. Eur. J. Biochem. 265:754-762. [DOI] [PubMed] [Google Scholar]
- 4.Brush, M. H., D. C. Weiser, and S. Shenolikar. 2003. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 23:1292-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castello, A., A. Quintas, E. G. Sanchez, P. Sabina, M. Nogal, L. Carrasco, and Y. Revilla. 2009. Regulation of host translational machinery by African swine fever virus. PLoS Pathog. 5:e1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman, D. A., V. Tcherepanov, C. Upton, and L. K. Dixon. 2008. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 89:397-408. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee, S., J. W. Wang, M. J. Cismowski, J. R. Bower, and K. S. Rosenthal. 2009. HSV-2 ICP34.5 protein modulates herpes simplex virus glycoprotein processing. Arch. Virol. 154:661-663. [DOI] [PubMed] [Google Scholar]
- 8.Childs, K. S., J. Andrejeva, R. E. Randall, and S. Goodbourn. 2009. Mechanism of mda-5 inhibition by paramyxovirus V proteins. J. Virol. 83:1465-1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cobbold, C., and T. Wileman. 1998. The major structural protein of African swine fever virus, p73, is packaged into large structures, indicative of viral capsid or matrix precursors, on the endoplasmic reticulum. J. Virol. 72:5215-5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demaison, C., K. Parsley, G. Brouns, M. Scherr, K. Battmer, C. Kinnon, M. Grez, and A. J. Thrasher. 2002. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency [correction of imunodeficiency] virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum. Gene Ther. 13:803-813. [DOI] [PubMed] [Google Scholar]
- 11.Goatley, L. C., M. B. Marron, S. C. Jacobs, J. M. Hammond, J. E. Miskin, C. C. Abrams, G. L. Smith, and L. K. Dixon. 1999. Nuclear and nucleolar localization of an African swine fever virus protein, I14L, that is similar to the herpes simplex virus-encoded virulence factor ICP34.5. J. Gen. Virol. 80:525-535. [DOI] [PubMed] [Google Scholar]
- 12.Han, A. P., C. Yu, L. Lu, Y. Fujiwara, C. Browne, G. Chin, M. Fleming, P. Leboulch, S. H. Orkin, and J. J. Chen. 2001. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 20:6909-6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harding, H. P., Y. Zhang, D. Scheuner, J. J. Chen, R. J. Kaufman, and D. Ron. 2009. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc. Natl. Acad. Sci. U. S. A. 106:1832-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harding, H. P., Y. H. Zhang, and D. Ron. 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271-274. [DOI] [PubMed] [Google Scholar]
- 15.Haresnape, J. M., P. J. Wilkinson, and P. S. Mellor. 1988. Isolation of African swine fever virus from ticks of the Ornithodoros moubata complex (Ixodoidea:Argasidae) collected within the African swine fever enzootic area of Malawi. Epidemiol. Infect. 101:173-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He, B., J. Chou, D. A. Liebermann, B. Hoffman, and B. Roizman. 1996. The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the gamma(1)34.5 gene of herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. J. Virol. 70:84-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He, B., M. Gross, and B. Roizman. 1998. The gamma(1)34.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase I regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 273:20737-20743. [DOI] [PubMed] [Google Scholar]
- 18.He, B., M. Gross, and B. Roizman. 1997. The gamma(1)34.5 protein of herpes simplex virus I complexes with protein phosphatase 1 alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 94:843-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiss, D. C., and G. A. Gabriels. 2009. Implications of endoplasmic reticulum stress, the unfolded protein response and apoptosis for molecular cancer therapy. Part I. Targeting p53, Mdm2, GADD153/CHOP, GRP78/BiP and heat shock proteins. Expert Opin. Drug Discov. 4:799-821. [DOI] [PubMed] [Google Scholar]
- 20.Jousse, C., A. Bruhat, V. Carraro, F. Urano, M. Ferrara, D. Ron, and P. Fafournoux. 2001. Inhibition of CHOP translation by a peptide encoded by an open reading frame localized in the chop 5′ UTR. Nucleic Acids Res. 29:4341-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King, P., and S. Goodbourn. 1994. The interferon-beta promoter responds to priming through multiple independent regulatory elements. J. Biol. Chem. 269:30609-30615. [PubMed] [Google Scholar]
- 22.Krishnamoorthy, T., G. D. Pavitt, F. Zhang, T. E. Dever, and A. G. Hinnebusch. 2001. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 21:5018-5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langland, J. O., and B. L. Jacobs. 2002. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology 299:133-141. [DOI] [PubMed] [Google Scholar]
- 24.Lee, Y. Y., R. C. Cevallos, and E. Jan. 2009. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2 alpha phosphorylation. J. Biol. Chem. 284:6661-6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marais, R., Y. Light, H. F. Paterson, and C. J. Marshall. 1995. RAS recruits RAF-1 to the plasma-membrane for activation by tyrosine phosphorylation. EMBO J. 14:3136-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masson, N., M. Ellis, S. Goodbourn, and K. A. W. Lee. 1992. Cyclic AMP response element-binding protein and the catalytic subunit of protein kinase A are present in F9 embryonal carcinoma-cells but are unable to activate the somatostatin promoter. Mol. Cell. Biol. 12:1096-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulvey, M., C. Arias, and I. Mohr. 2007. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 81:3377-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Netherton, C. L., J. C. Parsley, and T. Wileman. 2004. African swine fever virus inhibits induction of the stress-induced proapoptotic transcription factor CHOP/GADD153. J. Virol. 78:10825-10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novoa, I., H. Q. Zeng, H. P. Harding, and D. Ron. 2001. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2 alpha. J. Cell Biol. 153:1011-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orvedahl, A., D. Alexander, Z. Talloczy, Q. H. Sun, Y. J. Wei, W. Zhang, D. Burns, D. A. Leib, and B. Levine. 2007. HSV-1ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23-35. [DOI] [PubMed] [Google Scholar]
- 31.Poppers, J., M. Mulvey, D. Khoo, and I. Mohr. 2000. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 74:11215-11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivera, J., C. Abrams, B. Hernaez, A. Alcazar, J. M. Escribano, L. Dixon, and C. Alonso. 2007. The MyD116 African swine fever virus homologue interacts with the catalytic subunit of protein phosphatase 1 and activates its phosphatase activity. J. Virol. 81:2923-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharp, T. V., F. Moonan, A. Romashko, B. Joshi, G. N. Barber, and R. Jagus. 1998. The vaccinia virus E3L gene product interacts with both the regulatory and the substrate binding regions of PKR: implications for PKR autoregulation. Virology 250:302-315. [DOI] [PubMed] [Google Scholar]
- 34.Shi, Y. G. 2009. Serine/threonine phosphatases: mechanism through structure. Cell 139:468-484. [DOI] [PubMed] [Google Scholar]
- 35.Todd, D. J., A. H. Lee, and L. H. Glimcher. 2008. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 8:663-674. [DOI] [PubMed] [Google Scholar]
- 36.Tulman, E. R., G. A. Delhon, B. K. Ku, and D. L. Rock. 2009. African swine fever virus. Curr. Top. Microbiol. Immunol. 328:43-87. [DOI] [PubMed] [Google Scholar]
- 37.Vattem, K. M., and R. C. Wek. 2004. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 101:11269-11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verpooten, D., Y. J. Ma, S. W. Hou, Z. P. Yan, and B. He. 2009. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 284:1097-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Virshup, D. M., and S. Shenolikar. 2009. From promiscuity to precision: protein phosphatases get a makeover. Mol. Cell 33:537-545. [DOI] [PubMed] [Google Scholar]
- 40.Wek, R. C., H. Y. Jiang, and T. G. Anthony. 2006. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 34:7-11. [DOI] [PubMed] [Google Scholar]
- 41.Zeenko, V. V., C. Wang, M. Majumder, A. A. Komar, M. D. Snider, W. C. Merrick, R. J. Kaufman, and M. Hatzoglou. 2008. An efficient in vitro translation system from mammalian cells lacking the translational inhibition caused by eIF2 phosphorylation. RNA 14:593-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang, C. Z., J. Tang, J. Xie, H. K. Zhang, Y. P. Li, J. Zhang, D. Verpooten, B. He, and Y. J. Cao. 2008. A conserved domain of herpes simplex virus ICP34.5 regulates protein phosphatase complex in mammalian cells. FEBS Lett. 582:171-176. [DOI] [PubMed] [Google Scholar]
- 43.Zhou, D. H., L. R. Palam, L. Jiang, J. Narasimhan, K. A. Staschke, and R. C. Wek. 2008. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J. Biol. Chem. 283:7064-7073. [DOI] [PubMed] [Google Scholar]
- 44.Zsak, L., Z. Lu, G. F. Kutish, J. G. Neilan, and D. L. Rock. 1996. An African swine fever virus virulence-associated gene NL-S with similarity to the herpes simplex virus ICP34.5 gene. J. Virol. 70:8865-8871. [DOI] [PMC free article] [PubMed] [Google Scholar]