

Abstract
The Pd-catalyzed condensation of 2-bromostyrene and 2-chloroaniline derivatives yields stable diphenylamine intermediates, which are selectively converted to either 5-, 6-, or 7-membered heteroaromatics (indoles, carbazoles, acridines and dibenzazepines). The selectivity of these intramolecular transformations is uniquely ligand-controlled, and offers efficient routes to four important classes of heterocycles from a common precursor.

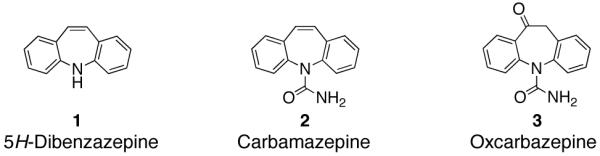
The biological activity manifested by tricyclic, nitrogen-containing heterocyclic compounds make them attractive targets for synthetic chemists.1 The 7-membered ring 5H-Dibenz[b,f]azepine nucleus (1) is a pharmaceutically important structure and constitutes the key subunit in tricyclic antidepressant drug substances as Carbamazepine (2) and Oxcarbazepine (3).2 These anticonvulsant and mood stabilizing drugs are primarily used for the treatment of epilepsy, bipolar disorder,3 trigeminal neuralgia4 and other neurological disorders.5 Currently, the most widely employed method for the construction of dibenzazepine analogs involves a gas-phase dehydrogenation of iminobibenzyls at high temperatures.6,7 The crude product in these processes is usually contaminated with 9-methylacridine.8 Thus, a general and efficient means for the synthesis of substituted dibenzazepines remains a challenging problem. In addition, the closely related acridine and carbazole tricyclic nuclei also feature prominently amongst natural products and drug substances.9 Various methods utilizing a number of synthetic platforms and starting materials are used for the construction of such heteroaromatic systems.1b,10 While there are many strategies available for the synthesis of carbazoles,2b,11 methods for the preparation of acridines12c,13 are limited and typically require harsh, functional group-intolerant conditions.
An examination of a series of tricyclic heteroaromatic compounds led us to the realization that 5H-dibenzazepines (4), 9-methylacridines (5) and vinylcarbazoles (6) might be derived from a common precursor via controlled intramolecular cyclizations . The key precursor (7) was prepared in 91% yield via a C-N coupling reaction14,15 of commercially available 2-bromostyrene and 2-chloroaniline, as shown in Scheme 1.
Scheme 1.
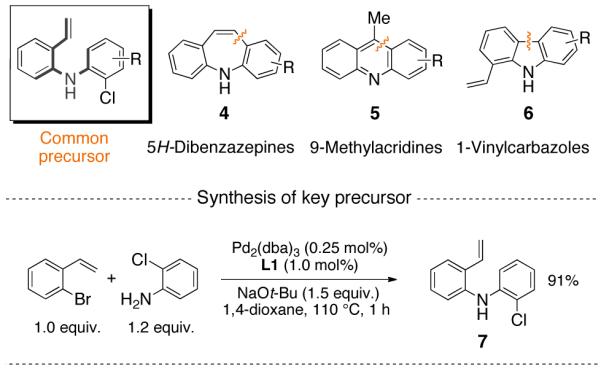
Proposed Common Precursor for the Synthesis of Tricyclic Nitrogen-Containing Heterocyclic Cores
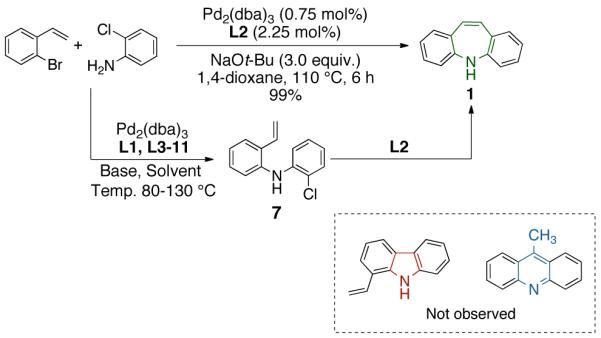
We reasoned that four major factors would control the regioselectivity of the Pd(0)-catalyzed transformation of 7: ligand, base, solvent and temperature. Further examination of these variables led us to discover that the mode of heterocyclization is almost exclusively controlled by the ligand employed. We examined a variety of phosphines (L1-L11)14a and found that DavePhos (L2) was highly effective ligand for the 7-endo cyclization of 7 to form 1. In contrast, TrixiePhos (L3) and L4,16 selectively furnished 1-vinylcarbazole (8) and 9-methylacridine (9), respectively (Scheme 2). An efficient catalyst for all transformations was formed from the combination of the appropriate ligand, Pd2dba3 and NaOt-Bu at 100-110 °C. Among the solvents screened, the use of 1,4-dioxane was superior in terms of conversion and selectivity for the formation of 5H-dibenzazepine and 1-vinylcarbazole. Regioselective 6-exo cyclization to form 9-methylacridine was achieved using L4 in toluene. It should be noted that no other heterocycles or side products were formed under the optimized conditions. Other combinations11b-d of catalyst, ligand, base and solvent led to the production of multiple products or gave low yields of the desired heterocycles.
Scheme 2.

Pd/Ligand Controlled Selective Cyclizations
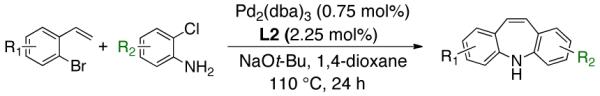
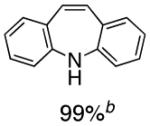
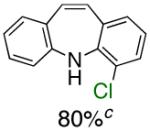
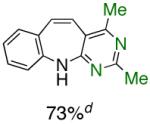
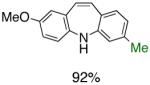
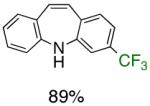
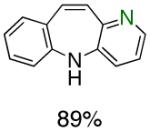
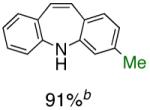
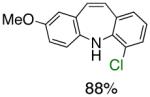
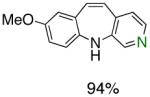
To the best of our knowledge, our results represent the first cases of such intramolecular reactions that incorporate a 7-endo cyclization. Control experiments were performed and demonstrated that no reactions occurred in the absence of ligand. Interestingly, of all the ligands screened, L2 was unique in promoting the cascade synthesis17 of 5H-dibenzazepine (Scheme 3). This direct transformation was achieved via a tandem reaction of 2-bromostyrene with 2-chloroaniline, which proceeded smoothly in the presence of Pd2(dba)3, L2 and NaOt-Bu, to provide 1 in 99% isolated yield. Other ligands (L1, L3-L11) afforded diarylamine intermediate as the only observed product. As shown in Table 1, our protocol for the tandem synthesis of 5H-dibenzazepine derivates is quite general.
Scheme 3.
One-Pot Direct Synthesis of 5H-Dibenzazepine
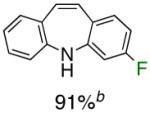
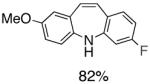
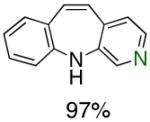
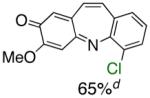
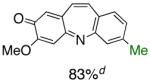
Table 1.
Pd-Catalyzed Tandem Formation of Dibenzazepines
 | |||
|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Reaction conditions: a Isolated yields (average of two runs). 1.0 mmol of styrene, 1.2 mmol of amine, 1 mL of dry 1,4-dioxane, 3.0 mmol of NaOt-Bu, 0.0075 mmol Pd2(dba)3, 0.023 mmol L2; Ar atmosphere; 110°C, 24h. b Reaction time - 6h. c Reaction proceeded to 85% conversion (GC). d 2.5 mol% Pd2(dba)3 and 8 mol% of L2 are required.
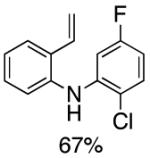
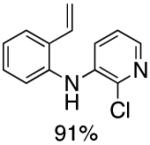
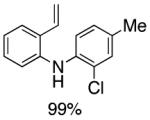
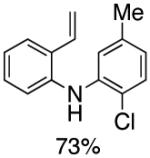
Following the initial survey of ligands (L3 and L4 for the formation of carbazoles and acridines respectively) and optimization of Pd sources, we next prepared a range of vinyldiarylamines (as shown in Table 2). The intermediates so prepared, were then subjected to Pd-catalyzed cyclization conditions to provide a range of vinylcarbazoles and acridines in a highly regioselective manner and in good to excellent yields (Table 3). Notably, both electron-rich and and electron-deficient vinyldiarylamines were transformed in an efficient manner.
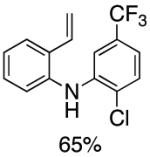
Table 2.
Synthesis of Diarylamine Intermediates
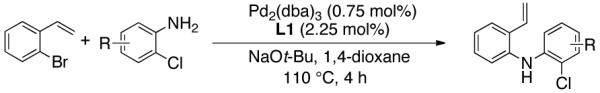 | ||
|---|---|---|
|
|
|
|
|
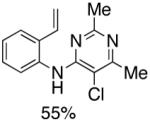
|
Reaction conditions: 1.0 mmol of 2-bromostyrene, 1.2 mmol of amine, 0.0075 mmol of Pd2(dba)3, 0.023 mmol of L1 (BrettPhos), 1.0 mL of dry 1,4-dioxane, 1.5 mmol of NaOt-Bu; Ar atmosphere; 110°C, 4h. Isolated yields.
Table 3.
Selective Formation of Acridines and Carbazoles
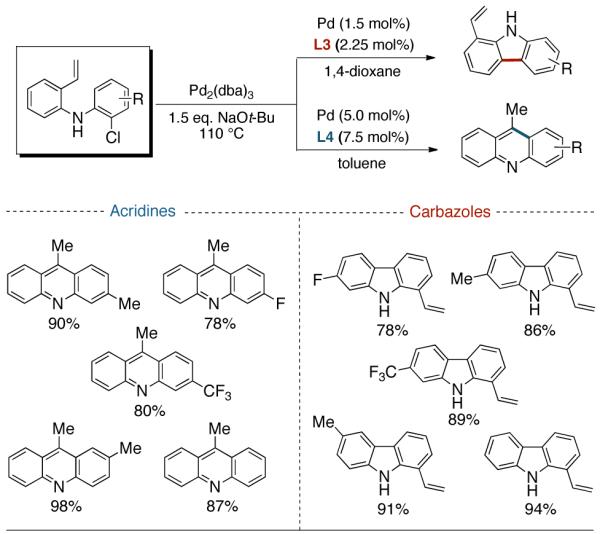
|
Reaction conditions: Isolated yields (average of two runs). 1.0 mmol of intermediate, 1.5 mmol of NaOt-Bu, 110 °C, 24 h. For acridines: 1 mL of dry toluene, 0.025 mmol Pd2(dba)3, 0.075 mmol L3. For vinylcarbazoles: 1 mL of dry 1,4-dioxane, 0.0075 mmol Pd2(dba)3, 0.023 mmol L4.
As an expansion of this study, we explored the preparation of N-arylindoles through the use of an oxidative cyclization.1b,18 Using the same precursors as above, the construction of arylindoles was achieved via intramolecular C-N bond formation. These reactions proceed in the presence of Pd(OAc)2, Cu(OAc)2 and acetic acid in DMF at 100°C, to provide 2-chloro-N-arylindoles in excellent yields (Table 4).
Table 4.
Pd(II)-Catalyzed Synthesis of N-Arylindoles
 | ||
|---|---|---|
|
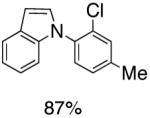
|
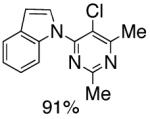
|
Reaction conditions: Isolated yields (average of two runs). 1.0 mmol of intermediate, 1.5 mmol of Cu(OAc)2, 0.1 mmol of Pd(OAc)2, 1 mL of AcOH, 3 mL DMF. 110 °C, 24h.
In Scheme 4 we suggest plausible mechanisms for the transformations described above. The dibenzazepine, vinylcarbazole, and acridine syntheses are likely initiated by the oxidative addition of Pd(0) to 7. Carbon-carbon bond formation, in the construction of 5H-dibenzazepine, may proceed via intermediate A to form eight-membered palladacycle B, which then undergoes reductive elimination to afford 1 (pathway I).19 With L3, the oxidative addition complex may undergo intramolecular C-H activation to give a six-membered palladacycle C, which yields 1-vinylcarbazole 8 after reductive elimination11b-d (pathway II). Acridine formation may proceed via a “normal” Heck pathway,20 producing intermediate C, from which β-hydride elimination can take place to generate 9 (pathway III). Finally, the formation of N-arylindole 10 presumably results from a Pd(II)-mediated intramolecular amination of olefin (Wacker-type transformation21), as shown in pathway IV. Transformations of this type were reported in the pioneering work of Hegedus.18b
Scheme 4.
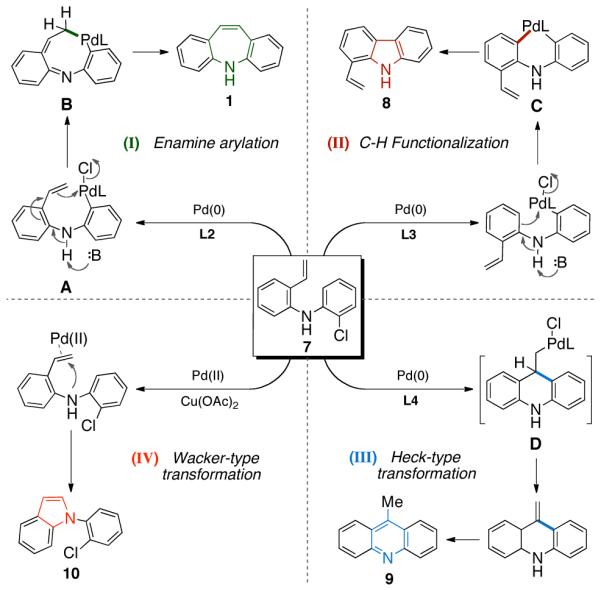
Possible Mechanistic Pathways
In conclusion, the Pd-catalyzed condensation of 2-bromostyrene and 2-chloroaniline derivatives yields stable diphenylamine intermediates, which are selectively converted to form, either 5-, 6-, or 7-membered heterocycle systems (indoles, carbazoles, acridines and dibenzazepines). The selectivity of these intramolecular transformations appears to be completely ligand controlled, and offers a unique opportunity for efficient routes to four important heterocyclic derivates from a common precursor. The novel Pd-catalyzed synthesis of dibenzazepines and 9-methylacridines developed in this study is highly efficient and should provide an access to a range of other tricyclic derivates. Our future efforts will be devoted to obtaining a clearer understanding of the mechanism of these highly ligand-dependent transformations.
Supplementary Material
Acknowledgment
This research was financially supported by the National Institutes of Health (NIH) (GM-58160) and Novartis AG.
Footnotes
Supporting information: Experimental details for the synthesis of all new compounds and spectral data. This information is available free via the Internet at http://pubs.acs.org.
References
- (1).(a) Lednicer D. Strategies for Organic Drugs Synthesis and Design. 2 ed. Wiley; Hoboken: 2009. [Google Scholar]; (b) Thansandote P, Lautens M. Chem. Eur. J. 2009;15:5874. doi: 10.1002/chem.200900281. [DOI] [PubMed] [Google Scholar]
- (2).For a recent review on the synthesis of carbamazepine and oxcarbazepine see: Singh H, Gupta N, Kumar P, Dubey SK, Sharma PK. Org. Process Res. Dev. 2009;13:870. and references therein.
- (3).(a) Ambrosio AF, Soares-da-Silva P. Neurochem Res. 2002;27:121. doi: 10.1023/a:1014814924965. [DOI] [PubMed] [Google Scholar]; (b) Hirschfeld RMA, Kasper S. Int. J. Neuropsychoph. 2004;7:507. doi: 10.1017/S1461145704004651. [DOI] [PubMed] [Google Scholar]; (c) Okuma T, Kishimoto A. Psychiatry Clin. Neurosci. 1998;52:3. doi: 10.1111/j.1440-1819.1998.tb00966.x. [DOI] [PubMed] [Google Scholar]
- (4).Gomez-Arguelles JM, Dorado R, Sepulveda JM, Huet R, Arrojo FG, Aragon E, Herrera A, Trron C, Anciones B. J. Clin. Neurosci. 2008;15:516. doi: 10.1016/j.jocn.2007.04.010. [DOI] [PubMed] [Google Scholar]
- (5).Albani F, Riva R, Baruzzi A. Pharmacopsychiat. 1995;28:235. doi: 10.1055/s-2007-979609. [DOI] [PubMed] [Google Scholar]
- (6).Kricka LJ, Ledwith A. Chem. Rev. 1974;74:101. and references therein. Tokmakov GP, Grandberg II. Tetrahedron. 1995;51:2091.
- (7).(a) Craig PN, Lester BM, Saggiomo AJ, Kaiser C, Zirkle CL. J. Org. Chem. 1961;26:135. [Google Scholar]; (b) Monti KD, Maciejewski AB. Appl. Catal., A. 1995;121:139. [Google Scholar]
- (8).Knell A, Monti D, Baiker A. Cat. Lett. 1995;31:197. and references therein.
- (9).(a) Denny WA. Med. Chem. Rev. 2004;1:257. [Google Scholar]; (b) Knölker H-J, Reddy KR. Chem. Rev. 2002;102:4303. doi: 10.1021/cr020059j. [DOI] [PubMed] [Google Scholar]
- (10).For the review that incorporate this topic see: Negishi E.-i., Coperet C, Ma S, Liou S-Y, Liu F. Chem. Rev. 1996;96:365. doi: 10.1021/cr950020x.
- (11).(a) Tsang WCP, Zheng N, Buchwald SL. J. Am. Chem. Soc. 2005;127:14560. doi: 10.1021/ja055353i. [DOI] [PubMed] [Google Scholar]; (b) Bedford RB, Betham M. J. Org. Chem. 2006;71:9403. doi: 10.1021/jo061749g. [DOI] [PubMed] [Google Scholar]; (c) Bedford RB, Betham M, Charmant JPH, Weeks AL. Tetrahedron. 2008;64:6038. [Google Scholar]; (d) Bedford RB, Cazin CSJ. Chem. Commun. 2002:2310. doi: 10.1039/b207712b. [DOI] [PubMed] [Google Scholar]; (e) Zeni G, Larock RC. Chem. Rev. 2006;107:303. doi: 10.1021/cr0683966. [DOI] [PubMed] [Google Scholar]
- (12).(a) Bergmann ED, Rabinovitz M, Bromberg A. Tetrahedron. 1968;24:1289. [Google Scholar]; (b) Bergmann ED, Rabinovitz M. J. Org. Chem. 1960;25:827. [Google Scholar]; (c) Kricka LJ, Ledwith A. Chem. Rev. 1974;74:101. [Google Scholar]
- (13).Rogness DC, Larock RC. J. Org. Chem. 2010;75:2289. doi: 10.1021/jo1000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For selected reviews see: Surry DS, Buchwald SL. Angew. Chem. Int. Ed. 2008;47:6338. doi: 10.1002/anie.200800497. Hartwig JF. Angew. Chem. Int. Ed. 1998;37:2046. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L.
- (15).Fors BP, Watson DA, Biscoe MR, Buchwald SL. J. Am. Chem. Soc. 2008;130:13552. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Netherton MR, Fu GC. Org. Lett. 2001;3:4295. doi: 10.1021/ol016971g. [DOI] [PubMed] [Google Scholar]
- (17).For selected reviews that incorporate this topic, see: Parsons PJ, Penkett CS, Shell AJ. Chem. Rev. 1996;96:195. doi: 10.1021/cr950023+. and references therein. Topczewski JJ, Callahan MP, Jeffrey D, Neighbors JD, Wiemer DF. J. Am. Chem. Soc. 2009;131:14630. doi: 10.1021/ja906468v.
- (18).(a) Cacchi S, Fabrizi G. Chem. Rev. 2005;105:2873. doi: 10.1021/cr040639b. [DOI] [PubMed] [Google Scholar]; (b) Harrington PJ, Hegedus LS. J. Org. Chem. 1984;49:2657. [Google Scholar]; (c) Humphrey GR, Kuethe JT. Chem. Rev. 2006;106:2875. doi: 10.1021/cr0505270. [DOI] [PubMed] [Google Scholar]
- (19).We believe that the vinylogous enamine nature of the terminal alkene is primarily responsible for the observed regioselectivity of this transformation.
- (20).Beletskaya IR, Cheprakov AV. Chem. Rev. 2000;100:3009. doi: 10.1021/cr9903048. and references therein.
- (21).For recent reviews see: Balme G, Bouyssi D, Lomberget T, Monteiro N. Synthesis. 2003:2115.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.