

Abstract
The Mukaiyama aldol–Prins (MAP) cyclization of acetals stereoselectively provided substituted tetrahydropyrans. The scope of the reaction has been expanded to include other electrophiles, including ketals and α-acetoxy ethers. Finally, a double MAP cyclization with orthoformates is described.
Keywords: tandem reactions, aldol reactions, annulations, acetals, carbocations
The Prins reaction is a powerful transformation that generates a carbon-carbon bond through the coupling of an activated aldehyde or ketone to an olefin.1 The carbocation intermediate produced from this reaction has been utilized in subsequent processes to develop elaborate structures with high levels of stereoselectivity.2 The Mukaiyama aldol-Prins reaction is an alternative tandem process in which an oxocarbenium generated from the Mukaiyama aldol addition of an aldehyde to a homoallylic vinyl ether has been shown to undergo an intramolecular Prins reaction. The carbenium ion intermediate is trapped by addition of a nucleophile to generate cis-2,6-dialkyltetrahydropyrans (THPs) stereoselectively.3 The scope of the MAP reaction has been successfully extended to acetal-type electrophiles, and the preliminary results are described herein.
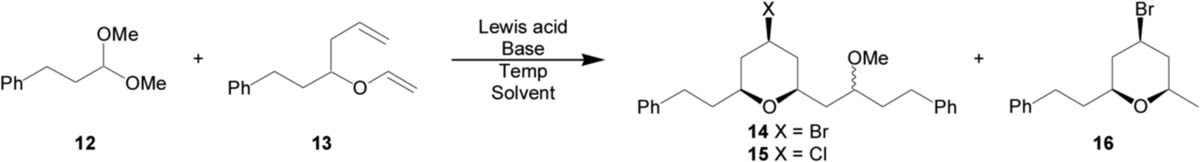
Recently, the MAP reaction has been applied to the total synthesis of the marine natural product leucascandrolide A.4 The reaction of aldehyde 1 with vinyl ether 2 produced THP 3 with high selectivity for the 1,3-anti product but only modest equatorial/axial selectivity at the bromide center (Scheme 1). The modest selectivity at the bromide center is a known issue with MAP cyclizations utilizing aldehydes with chelating groups.3 It has been hypothesized that an oxocarbenium ion electro-phile, which would not be subject to chelation, might avoid this issue. When dimethyl acetal 4 was employed as an electrophile the equatorial bromide was obtained in 8:1 selectivity. Unfortunately, the addition was not stereoselective at the methoxy center, but because of the high selectivity obtained at the bromide center, the MAP reaction with acetals warranted further investigation.
Scheme 1.

MAP cyclizations for leucascandrolide A
A proposed mechanism for the MAP reaction of acetals is shown in Scheme 2.4 Initial coordination of titanium tetrabromide to dimethyl acetal 6 provides oxocarbenium ion 7, which is activated toward nucleophilic addition. Mukaiyama aldol addition of vinyl ether 2 produces bromo ether 8 which is then activated to form ion pair 9. Direct collapse of this intermediate produces an axial bromide (10) while solvolysis produces an equatorial bromide (11).5,6 The selectivity of this last, partitioning step is not sensitive to the structure of the acetal and should provide high selectivities for the equatorial diastereomer. This variation of the MAP cyclization is expected to work with a variety of oxocarbenium ions.
Scheme 2.

Mechanism of the MAP reaction with acetals
Initial efforts were undertaken to optimize the MAP cyclization of dimethyl acetal 12 with homoallylic vinyl ether 13 (Table 1). The use of various bases to sequester adventitious protic acid were first explored. Without base, a considerable amount of proton mediated Prins cyclization (16) of vinyl ether 13 was observed (entry 1). Hünig's base proved detrimental to the reaction due to coordination to titanium (entry 2). Ultimately, the hindered pyridine base, 2,6-di-tert-butyl-4-methylpyridine (2,6-DBMP), proved ideal to buffer the system (entry 3). As expected, less polar solvent systems disfavored the proposed charged transition states and diminished yields were observed (entry 4). Additionally, various Lewis acid systems were explored to further enhance yields and selectivity. Both the attenuated Lewis acid system used for leucascandrolide A and titanium tetrachloride provided comparable selectivities, but reduced yields (entries 5 and 6). Bromotrimethylsilane has been shown to provide high selectivities for axial bromination in similar systems, but led only decomposition of the starting materials in this study (entry 7).6 Finally, an aluminum tri-bromide/trimethylaluminum mixed Lewis acid system had been demonstrated to promote very demanding Diels-Alder reactions,7 but the elevated temperatures required for the MAP acetal reaction with this catalyst furnished reduced yields and unremarkable selectivities (entry 8). Ultimately, the original conditions for the MAP cyclization of aldehydes proved to be optimal for acetal substrates.
Table 1.
Optimization of the MAP reaction with acetals
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Lewis Acida | Base (equiv) | Temp | Solvent | Yield | dr (X) eq:axb | dr (OMe)c |
| 1d | TiBr4 | None | −78 °C | CH2Cl2 | 43% | 86:14 | 55:45 |
| 2d |
TiBr4 |
iPr2NEt (1.5) |
−78 °C |
CH2Cl2 |
29% |
82:18 |
55:45 |
| 3 |
TiBr4 |
2,6-DBMP (1.5) |
−78 °C |
CH2Cl2 |
67% |
92:8 |
55:45 |
| 4 | TiBr4 | 2,6-DBMP (1.5) | −78 °C | CH2Cl2/hexanes | 44% | 93:7 | 55:45 |
| 5 | TiBr4/Ti(OiPr)4e | 2,6-DBMP (1.5) | −78 °C | CH2Cl2 | 50% | 91:9 | 53:47 |
| 6 | TiCl4 | 2,6-DBMP (1.5) | −78 °C | CH2Cl2 | 44% | 90:10 | 57:43 |
| 7 | TMSBr | 2,6-DBMP (1.5) | 0 °C | CH2Cl2 | 0% | – | – |
| 8 | AlBr3f | AlMe3 (0.3) | −40 °C | CH2Cl2 | 40% | 80:20 | 56:44 |
Unless otherwise noted, 4 equiv of Lewis acid was used.
Determined by integration of the crude 1H NMR spectra.
Determined by GC analysis.
17% of THP 16 was also isolated.
Formed by pre-mixing an 8:1 solution of TiBr4 and Ti(OiPr)4.
1.5 equiv of AlBr3.
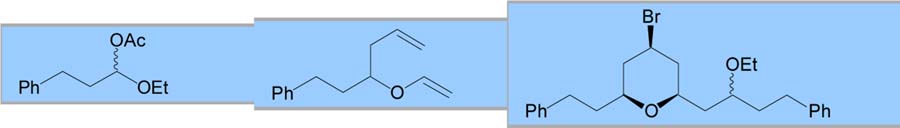
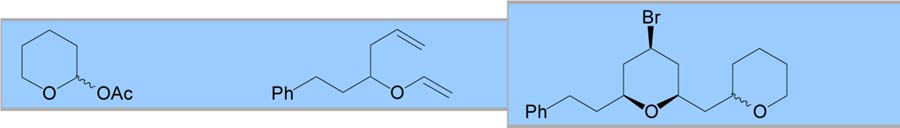
With an optimized procedure in hand, various acetals were explored as possible coupling partners (Table 2). A dibenzyl acetal was first employed to provide a benzyl ether that could easily be deprotected. This reaction proceeded with yields and selectivities comparable to the dimethyl acetal substrate (entry 1). Cyclic acetals were also employed as a possible method to control the newly formed ether stereocenter. Initial results with an achiral 5-membered cyclic acetal were discouraging (entry 2). Reduced selectivities were observed, possibly because the oxocarbenium ion of the cyclic acetal provided a manifold for chelation, which has been shown to decrease equatorial selectivity.3a C2-symmetric chiral cyclic acetals have shown enhanced selectivities for nucleophilic additions.8 When coupled with a chiral vinyl ether, a 71% yield was obtained with 89:11 bromide selectivity and 70:30 selectivity for the alkoxy stereocenter in favor of the expected diastereomer (entry 3).
Table 2.
Exploration of various electrophiles for the MAP reaction
| Entry | Electrophile | Homoallylic Vinyl Ether | Major Product | Yield (%) | dr (Br) eq:axa | dr (OR) |
|---|---|---|---|---|---|---|
| 1 |
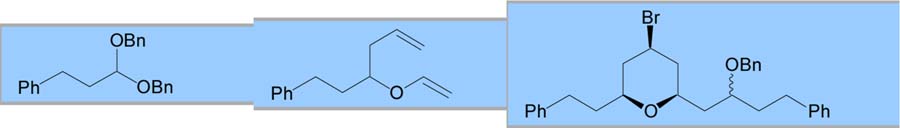
|
64 | >95:5 | 1:1 | ||
| 2 |

|
44 | 74:26 | 1:1 | ||
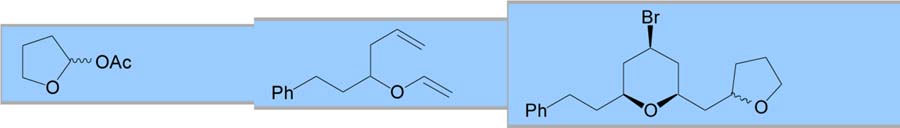
| 3 |
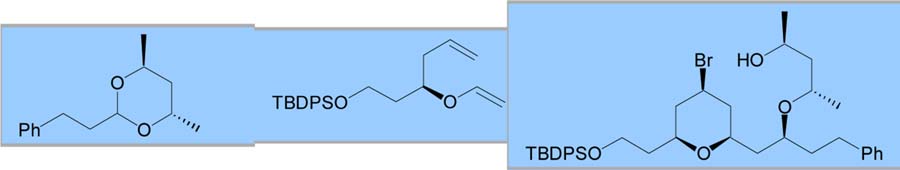
|
71 | 89:11 | 7:3b,c | ||
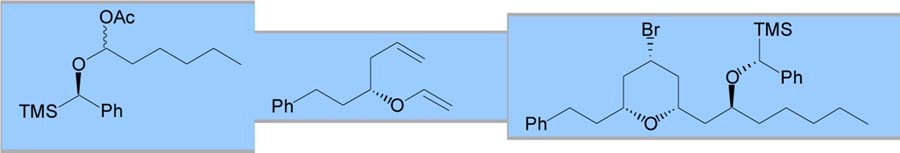
| 4 |
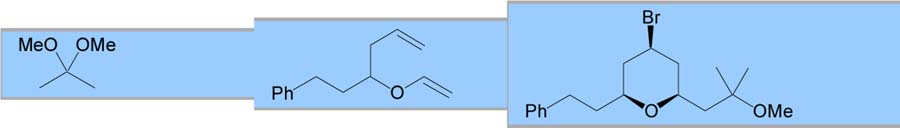
|
61 | 95:5 | – | ||
| 5 |
|
65 | 88:12 | 1:1 | ||
| 6 |
|
36 | 82:18 | 1:1 | ||
| 7 |
|
23 | 80:20 | 1:1 | ||
| 8 |
|
90 | 92:8 | 7:3d | ||
Ratios determined by integration of the crude 1H NMR spectra.
Ratio determined by HPLC analysis of the crude material.
Stereochemistry of the major isomer determined by removal of the chiral auxiliary and formation/analysis of both MPTA ester diastereomers.
Ratio of isolated products.
Various other electrophiles were also employed as coupling partners for the MAP cyclization. Ketals and α-acetoxy ethers were successful coupling partners, providing yields and selectivities similar to those observed for the acetals (entries 4 and 5).9 Cyclic α-acetoxy ethers were intriguing electrophiles since selective nucleophilic addition has been demonstrated depending on substitution.10 Unfortunately, neither simple five- or sixmembered α-acetoxy ethers provided useful yields of THP products (entries 6 and 7).11 Finally, chiral α-(trimethylsilyl)benzyl α-acetoxy ethers have been employed to enhance the selectivity of aldol additions.12 When this chiral auxiliary was utilized for the MAP cyclization, a 90% yield of the expected THP was produced with 7:3 selectivity for the aldol addition (entry 8).
Orthoesters were also examined as substrates for double MAP cyclizations (Scheme 3). Only orthoformates proved successful, providing interesting pseudo-C2-symmetric structures (18 and 19).13 Utilizing E homoallylic vinyl ether 20 as a nucleophile selectively produced bis-THP 21 with two equatorial methyl groups, in agreement with precedent.3c,14 Overall, six new stereo-centers were generated in this single reaction.
Scheme 3.

MAP reaction of orthoformates
The MAP cyclization has proven to be a very effective method for the formation of a variety of substituted tetrahydropyrans. Utilization of acetals, α-acetoxy ethers, and orthoformates has expanded the scope and utility of the reaction. The use of various chiral auxiliaries led to enhanced stereoselectivity in the aldol addition step. The expanded scope of the MAP cyclization will be a powerful tool in natural product synthesis.
Acknowledgments
This work was supported by the National Cancer Institute (CA-081635) and a generous gift from Schering-Plough Research Institute.
References
- 1.a Arundale E, Mikeska LA. Chem. Rev. 1952;52:505–555. [Google Scholar]; b Adams DR, Bhatnagar SP. Synthesis. 1977:661–672. [Google Scholar]; c Snider BB. In: The Prins Reaction and Carbonyl Ene Reactions. Trost BM, Fleming I, Heathcock CH, editors. Vol. 2. Pergamon Press; New York: 1991. pp. 527–561. [Google Scholar]; d Pastor IM, Yus M. Curr. Org. Chem. 2007;11:925–957. [Google Scholar]
- 2.a Overman LE, Pennington LD. J. Org. Chem. 2003;68:7143–7157. doi: 10.1021/jo034982c. [DOI] [PubMed] [Google Scholar]; b Miles B, Davis CH, Coates RM. J. Org. Chem. 2006;71:1493–1501. doi: 10.1021/jo052142n. [DOI] [PubMed] [Google Scholar]
- 3.a Kopecky DJ, Rychnovsky SD. J. Am. Chem. Soc. 2001;123:8420–8421. doi: 10.1021/ja011377n. [DOI] [PubMed] [Google Scholar]; b Patterson B, Marumoto S, Rychnovsky SD. Org. Lett. 2003;5:3163–3166. doi: 10.1021/ol035303n. [DOI] [PubMed] [Google Scholar]; c Patterson B, Rychnovsky SD. Synlett. 2004:543–545. [Google Scholar]
- 4.Van Orden LJ, Patterson B, Rychnovsky SD. J. Org Chem. 2007;72:5784–5793. doi: 10.1021/jo070901r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alder RW, Harvey JN, Oakley MT. J. Am. Chem. Soc. 2002;124:4960–4961. doi: 10.1021/ja025902+. [DOI] [PubMed] [Google Scholar]
- 6.Jasti R, Vitale J, Rychnovsky SD. J. Am. Chem. Soc. 2004;126:9904–9905. doi: 10.1021/ja046972e. [DOI] [PubMed] [Google Scholar]
- 7.Jung ME, Ho D, Chu HV. Org. Lett. 2005;7:1649–1651. doi: 10.1021/ol050361p. [DOI] [PubMed] [Google Scholar]
- 8.Johnson WS, Edington C, Elliott JD, Silverman IR. J. Am. Chem. Soc. 1984;106:7588–7591. [Google Scholar]
- 9.Representative Experimental (Table 2, Entry 4). A solution of 2,6-di-tert-butyl-4-methylpyridine (77 mg, 0.38 mmol), homoallylic vinyl ether 13 (50 mg, 0.25 mmol), and 2,2-dimethoxypropane (0.022 mL, 0.25 mmol) in CH2Cl2 (2.5 mL) was cooled to −78 °C. Titanium tetrabromide (0.32 M in CH2Cl2, 3.1 mL, 1.0 mmol) was then added dropwise over 10 min. After 2 h, a 1:1 mixture of MeOH and Et3N (5 mL) was added and the reaction mixture was allowed to warm to room temperature. Saturated aqueous NaHCO3 (10 mL) was then added and the mixture was extracted with Et2O (2 × 10 mL). The combined organic layers were washed with brine (1 × 10 mL), dried over MgSO4, and concentrated under reduced pressure. Purification by flash chromatography (15:1 hexanes/Et2O) yielded 54 mg (61%) of the expected THP as a colorless oil: Rf = 0.31 (9:1 hexanes/Et2O); 1H NMR (500 MHz, CDCl3) δ 7.29 (t, J = 7.9, 2 H), 7.19 (t, J = 7.4, 3 H), 4.15 (tt, J = 12.0, 4.5, 1 H), 3.55−3.49 (m, 1 H), 3.32−3.26 (m, 1 H), 3.29 (s, 3 H), 2.83−2.77 (m, 1 H), 2.68−2.58 (m, 1 H), 2.24−2.16 (m, 2 H), 1.93−1.60 (m, 6 H), 1.26 (s, 3 H), 1.22 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 142.0, 128.53, 128.49, 126.0, 76.5, 74.3, 74.1, 49.3, 47.0, 45.9, 44.6, 43.4, 37.7, 32.0, 26.2, 25.3; IR (neat) 2925, 2860, 1603, 1454, 1080 cm−1; HRMS (ES/MeOH) m / z calcd for C18H27BrO2 [M + Na]+ 377.1092; found 377.1088.
- 10.a Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. J. Am. Chem. Soc. 1999;121:12208–12209. [Google Scholar]; b Ayala L, Lucero CG, Romero JAC, Tabacco SA, Woerpel KA. J. Am. Chem. Soc. 2003;125:15521–15528. doi: 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]; c Lewis MD, Cha JK, Kishi Y. J. Am. Chem. Soc. 1982;104:4976–4978. [Google Scholar]
- 11.Cyclic α-methoxy ethers and more complicated sugar moieties provided no THP products when employed as electrophiles.
- 12.Rychnovsky SD, Cossrow J. Org. Lett. 2003;5:2367–2370. doi: 10.1021/ol0347904. [DOI] [PubMed] [Google Scholar]
- 13.Neither orthoacetates nor orthocarbonates were reactive towards nucleophilic addition.
- 14.bis-THP (21). A solution of 2,6-di-tert-butyl-4-methylpyridine (35 mg, 0.58 mmol), homoallylic vinyl ether 20 (48 mg, 0.22 mmol), and triethyl orthoformate (0.018 mL, 0.11 mmol) in CH2Cl2 (1.1 mL) was cooled to −78 °C. Titanium tetrabromide (0.32 M in CH2Cl2, 1.4 mL, 0.44 mmol) was then added dropwise over 10 min. After 2 h, a 1:1 mixture of MeOH and Et3N (5 mL) was added and the reaction mixture was allowed to warm to room temperature. Saturated aqueous NaHCO3 (5 mL) was then added and the mixture was extracted with Et2O (2 × 5 mL). The combined organic layers were washed with brine (1 × 5 mL), dried over MgSO4, and concentrated under reduced pressure. Purification by flash chromatography (20:1 to 15:1 hexanes/Et2O) yielded 37 mg (52%) of the title compound as a colorless oil: [α]24D +1.1 (c 1.9, CHCl3); Rf = 0.33 (9:1 hexanes/Et2O); 1H NMR (500 MHz, CDCl3) δ 7.32−7.06 (m, 10 H), 4.07−3.99 (m, 1 H), 3.93 (td, J = 11.6, 4.6, 1 H), 3.84 (td, J = 11.3, 4.5, 1 H), 3.75−3.65 (m, 1 H), 3.55−3.43 (m, 1 H), 3.38−3.29 (m, 2 H), 3.21 (t, J = 9.1, 1 H), 3.04 (t, J = 9.5, 1 H), 2.85−2.60 (m, 4 H), 2.35−2.22 (m, 2 H), 2.00−1.58 (m, 12 H), 1.23 (t, J = 6.9, 3 H), 1.13 (d, J = 6.5, 3 H), 1.10 (d, J = 6.4, 3 H); 13C NMR (125 MHz, CDCl3) δ 141.9, 128.8, 128.5, 126.0, 79.3, 78.4, 76.4, 75.8, 72.3, 64.9, 57.7, 57.2, 45.8, 45.6, 44.6, 44.5, 39.2, 38.8, 37.5, 32.1, 31.5, 16.4, 15.8; IR (neat) 2927, 2854, 1456, 1086 cm−1; HRMS (ES/MeOH) m / z calcd for C33H46Br2O3 [M + Na]+ 671.1711; found 671.1719.