

Abstract
The human Rad51 protein requires ATP for the catalysis of DNA strand exchange, as do all Rad51 and RecA-like recombinases. However, understanding the specific mechanistic requirements for ATP binding and hydrolysis has been complicated by the fact that ATP appears to have distinctly different effects on the functional properties of human Rad51 versus yeast Rad51 and bacterial RecA. Here we use RNAi methods to test the function of two ATP binding site mutants, K133R and K133A, in human cells. Unexpectedly, we find that the K133A mutant is functional for repair of DNA double-strand breaks when endogenous Rad51 is depleted. We also find that the K133A protein maintains wild-type-like DNA binding activity and interactions with Brca2 and Xrcc3, properties that undoubtedly promote its DNA repair capability in the cell-based assay used here. Although a Lys to Ala substitution in the Walker A motif is commonly assumed to prevent ATP binding, we show that the K133A protein binds ATP, but with an affinity approximately 100-fold lower than that of wild-type Rad51. Our data suggest that ATP binding and release without hydrolysis by the K133A protein act as a mechanistic surrogate in a catalytic process that applies to all RecA-like recombinases. ATP binding promotes assembly and stabilization of a catalytically active nucleoprotein filament, while ATP hydrolysis promotes filament disassembly and release from DNA.
The human Rad51 protein (HsRad51) is the central catalytic component in the process of homologous genetic recombination and is essential for error-free repair of DNA double-strand breaks (DSBs)1 (1–4) and vertebrate cell survival (5, 6). Like its bacterial, yeast, and archaeal homologues (RecA, ScRad51, and RadA, respectively), the active form of HsRad51 is an extended nucleoprotein filament that catalyzes ATP-dependent DNA strand exchange between homologous single- and double-stranded DNA substrates (2, 7–9). While numerous studies suggest specific roles for ATP as an allosteric effector and energy source for RecA and ScRad51, it is currently not clear what step in the HsRad51 catalytic mechanism requires ATP binding, hydrolysis, or both. ATP and ATPγS induce a high-affinity binding state of RecA and ScRad51 for ssDNA (10–12), while for HsRad51, ATP has little effect on the equilibrium binding affinity for ssDNA (13–16).
HsRad51 contains a highly conserved 200-amino acid core domain that includes the Walker A and B motifs involved in ATP binding and hydrolysis (17–19). A universally conserved Lys residue in the Walker A motif consensus sequence GK(S/T) contacts the triphosphate group of bound ATP (20), and two mutations at this position have been widely used to dissect the mechanistic requirement for ATP binding and hydrolysis in recombinase function: Lys to Arg, which permits ATP binding but not hydrolysis, and Lys to Ala, which is assumed to eliminate ATP binding. For example, the RecA K72R protein catalyzes strand exchange for approximately 1.5 kb but cannot complete the reaction (21, 22), demonstrating that only ATP binding is required for formation of an active RecA nucleoprotein filament. This mutant is nonfunctional in vivo (23), and genetic studies support the idea that RecA K72A is nonfunctional as well (24). A yeast rad51 K191A mutant strain is as sensitive to DNA damage and as defective in spontaneous mitotic recombination as the rad51Δ strain (25). In contrast, the ScRad51 K191R mutant protein can complete strand exchange reactions and was found to complement the DNA repair defects of a rad51Δ strain (26). However, both results require a concentration of the K191R protein higher than that needed for wild-type ScRad51 (26, 27).
Attempts to gain insight into the effect of these substitutions on HsRad51 cellular function have thus far used heterologous cell types for expression of the human protein. When overexpressed in RAD51–/– chicken DT40 cells, the HsRad51 K133R mutant confers partial resistance to IR-induced DNA damage (28), suggesting that ATP hydrolysis is not essential for HsRad51 function. In contrast, Stark et al. (29) found that expression of HsRad51 K133R in mouse ES cells caused multiple types of DNA repair defects and hypersensitivity to IR (29), thus supporting a model in which ATP hydrolysis is important for HsRad51 function. In both studies, stable chicken and mouse cell lines expressing the HsRad51 K133A mutant could not be obtained, suggesting a lethal dominant-negative effect of this mutant. Therefore, the lethality of the K133A mutation has thus far prevented its investigation using a cell-based system.
In this study, we use a RNAi-based approach that has enabled us to investigate both the HsRad51 K133R and K133A mutants in human cells. Transgenes encoding the mutant proteins carry silent mutations protecting the mRNA from siRNAs that mediate degradation of the endogenous HsRad51 message. Thus, we can study mutant function independent of endogenous Rad51 as well as by a dominant-negative approach. We find that the K133A mutant supports DSB repair in cells depleted of endogenous Rad51 while the K133R mutant does not. Using a direct binding assay, we also show that the K133A protein binds ATP, but no hydrolysis is detected. These results suggest a model in which binding and release of nonhydrolyzed ATP by the K133A protein can mimic the ATP hydrolysis-dependent catalytic cycle of wild-type Rad51 filament assembly and disassembly required to drive DNA strand exchange in mammalian cells.
EXPERIMENTAL PROCEDURES
Cell Lines and Transfections
HEK293 cells were maintained in DMEM with 10% fetal bovine serum and 1% Pen/Strep. The GFP-RAD51SM construct (silent mutations) was made using pEGFP-C1 (Clontech, Palo Alto, CA), and several silent base changes were introduced into the siRNA-targeted region of the RAD51 structural gene (codons 65–70). The sequence defined by codons 65–70 is noted here followed by the same sequence with the silent mutations introduced to create the GFP-RAD51SM construct (wild-type RAD51, 5′-GGA ATT AGT GAA GCC AAA-3′; GFP-RAD-51SM, 5′-GGC ATC TCC GAG GCG AAG-3′). Top and bottom strands of the siRNA duplex used to knock down wild-type Rad51 are as follows: top, 5′-r(GGG AAU UAG UGA AGC CAA A)d(TT)-3′; bottom, 5′-r(UUU GGC UUC ACU AAU UCC C)d(TT)-3′. K133A and K133R mutations were introduced using the parental GFP-RAD51SM vector. The GFP-RAD51 construct without silent mutations was used in initial FACS experiments designed to optimize the efficiency of Rad51 knockdown. These involved testing various siRNA duplex sequences, concentrations of siRNA, and times post-transfection (see FACS Analysis). A construct carrying wild-type GFP-RAD51 in an N-terminal and C-terminal arrangement was created using pEGFP-N1 (Clontech) which was also used as a control in the cell-based DNA damage repair assays described below (see Comet Assays). A standard transfection protocol for most assays is as follows. Cells were seeded in a six-well plate (0.8 × 106 cells/mL) and transfected 24 h later with Rad51-specific siRNA duplex (final concentration of 5 nM; Qiagen, Studio City, CA) using a lipid transfection method (Lipofectamine 2000, Invitrogen, San Diego, CA). GFP-RAD51SM plasmids (4 μg) were transfected 12 h later.
FACS Analysis
Initial experiments using RNAi-mediated depletion of the GFP signal from cells transfected with GFP-RAD51 carrying the wild-type RAD51 gene sequence were performed to optimize the efficiency of Rad51 knockdown. HEK293 cells were transfected as described above and analyzed for loss of GFP signal at various times following transfection with the GFP-RAD51 construct. Cells were trypsinized, pelleted, and suspended in 0.5% PBS for analysis using a Becton-Dickenson FACScan flow cytometer and quantified using Cell Quest (Becton-Dickenson).
Western Blots
HEK293 cells transfected as described above were harvested 12 h after transfection of the GFP-RAD51SM vectors, washed with PBS, and lysed with RIPA buffer [25 mM Tris (pH 7.4), 0.5% Triton X-100, 0.5% sodium deoxycholate, 0.05% sodium dodecyl sulfate, 0.05 mM EDTA, and 75 mM NaCl], and the total amount of protein was determined with the BCA protein assay kit (Pierce, Rockford, IL). SDS–polyacrylamide gels (10%) were run with 80 μg of total protein in each lane and transferred to PVDF membranes at 25 V for 30 min in transfer buffer [192 mM glycine, 25 mM Tris (pH 7.4), and 20% methanol]. Membranes were incubated in blocking buffer [10 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 0.25% Tween 20] containing 15% instant nonfat dry milk overnight at 4 °C. A Rad51 monoclonal antibody (Upstate Biotechnology, Lake Placid, NY) was added (1:2000 dilution) in blocking buffer containing 2% instant nonfat dry milk for 1 h. Parallel blots stained for actin (monoclonal antibody at a 1:2000 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) served as loading controls. Membranes were washed for 5 × 5 min in blocking buffer, and HRP-conjugated secondary antibodies at a 1:12000 dilution (Pierce) were added for 1 h. Membranes were washed as described above, incubated with LumiGLO chemiluminescent substrate (KPL, Gaithersburg, MD) for 1 min, exposed to X-Omat Blue XB-1 X-ray film (Eastman Kodak, Rochester, NY), and developed (Kodak 2000A XOMAT processor).
Comet Assays
HEK293 cells were transfected as described above and 12 h following transfection with the GFP-RAD51SM vectors were exposed to 7.5 Gy IR using a Gammacell 40 instrument (MDS Nordion). Cells were pulled before irradiation, 30 min and 10 h after damage, and mixed with 0.5% low-melt agarose. Approximately 10 000 cells (60 μL) were spotted on a glass slide coated with 1.5% agarose, and a coverslip was placed over the cells. Once the agarose had solidified, coverslips were removed, 50 μL of 0.5% low-melt agarose was added, and coverslips were replaced. Slides were immersed in lysing solution [2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 1% Triton X-100, and 10% DMSO (pH 10.0)] at 4 °C overnight, rinsed for 3 × 5 min in neutralization buffer [0.4 M Tris (pH 7.5)], and subjected to alkaline electrophoresis [300 mM NaOH and 1 mM EDTA (pH 13.2)] at a current of 300 mA (25 V) for 20 min. Slides were washed with neutralization buffer, covered with 100% ethanol (4 °C), and dried overnight. Slides were stained with EtBr, pictures taken with a fluorescence microscope (Axioskop2 plus, Zeiss), and images analyzed with Cometscore (Tritek software) using “tail moment” (tail length × the percent of total DNA in the tail) as the measure of×DNA damage and repair. The use of GFP fusion proteins in this assay allowed the assessment of transfection efficiency (see Results), and controls using nonfused wild-type Rad51 gave results similar to those seen for the GFP–Rad51 fusion (not shown).
DNA Damage-Induced Protein Focus Formation
Cells transfected as described above were grown on coverslips, exposed to 7.5 Gy IR, allowed to recover for 1 h at 37 °C (5% CO2), washed once with PBS, immersed in 100% methanol (−20 °C) for 5 min, and blocked in PBS with 4% BSA overnight at 4 °C. Coverslips were mounted using Vectashield with DAPI, sealed with polyurethane, and stored in the dark at 4 °C. GFP was visualized by confocal microscopy (Leica TCS SP2 AOBS), and image processing was performed using the accompanying Leica Confocal Software TCS SP2.
Co-Immunoprecipitation Assays
HEK293 cells were seeded into six-well plates, allowed to grow overnight to approximately 90% confluence, and transfected in triplicate as described above. Twenty hours post-transfection, cells were collected and washed with PBS, and cells from three wells were combined, lysed with 150 μL of lysis buffer [10 mM HEPES (pH 7.4), 150 mM KCl, 10 mM MgCl2, and 50 μg/mL digitonin] containing protease inhibitors, and incubated on ice for 10 min. Samples were centrifuged for 10 min (13000g), and the supernatant was assayed for total protein (BCA assay, Pierce). For preclearing, 250 μg of total protein was mixed with 25 μL of protein G magnetic beads (New England Biolabs, Beverly, MA) and incubated for 1 h at 4 °C, tubes were placed in a magnetic separation rack (New England Biolabs), and the supernatant was transferred to a clean tube. Anti-GFP mouse monoclonal antibody (5 μg, Roche) was added to the precleared supernatant, and the mixture was incubated for 1 h at 4 °C followed by addition of protein G magnetic beads (25 μL) and incubation for 1 h at 4 °C. Beads were washed three times with 500 μL of lysis buffer and resuspended in Laemmli loading buffer (30 μL) at 70 °C for 5 min; samples were placed in the magnetic separation rack, and the supernatant was loaded onto a SDS–polyacrylamide gel (10%). Western blots were performed as described above using either HsRad51 polyclonal antibodies diluted 1:3000 (Oncogene) or Xrcc3 monoclonal antibodies diluted 1:5000 (Novus) and goat HRP conjugate secondary antibodies diluted 1:12000 (Pierce).
Yeast Two-Hybrid Assays
DNA sequences encoding Xrcc3, the BRC3 repeat of BRCA2, and wild-type HsRad51 were fused to the LexA DNA binding domain in plasmid pBTM116. Wild-type and mutant HsRAD51 genes were fused to the GAL-4 activation domain in plasmid pGAD10. Yeast strain L40 was transformed with each plasmid, incubated overnight, plated onto SD-Leu-Trp medium, and incubated at 30 °C for 72 h. Colonies were grown overnight at 30 °C in SD-Leu-Trp medium, and at an A600 of ≈0.7, 1 mL of culture was centrifuged. Cells were washed in 1 mL of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, and 1 mM MgSO4), repelleted, and suspended in 150 μL of Z buffer plus [35 mM βME, 23% (v/v) chloroform, and 0.01% SDS], vortexed for 15 s followed by addition of Z buffer (700 μL) with βME and ONPG (1 mg/mL). Reaction mixtures were incubated for 30 min (30 °C), reactions stopped via addition of 1 M NaCO3 (500 μL), and mixtures centrifuged for 10 min (13000g). A420 was measured using a Beckman Coulter DU 7400 spectrophotometer, and Miller units were determined using (A420 × 1000)/(A600 × minutes × milliliter).
Protein Purification
Wild-type RAD51 and mutants were expressed from pET15B vectors (13) in Escherichia coli strain BLR(DE3) carrying a plasmid encoding the chaperone proteins GroEL and GroES (pGro7, Takara, Japan). The K133A and K133R mutations were constructed from the parental pET-HsRAD51 vectors (QuikChange, Stratagene). Cells were grown in ½× superbroth (1.8 L) containing 100 μg/mL ampicilin, 20 μg/mL chloramphenicol, and 1 mg/mL L-arabinose at 37 °C. At an A600 of 0.75, the temperature was reduced to 18 °C, and cultures were induced with 0.1 mM IPTG for approximately 16 h. Cells were resuspended in 40 mL of buffer [250 mM Tris (pH 7.5) and 25% (w/v) sucrose] and protease inhibitors (Sigma). Cells were lysed, and the HsRad51 protein was purified as described previously (13).
DNA Binding Assays
Single-stranded DNA cellulose (Sigma, St. Louis, MO) was swelled in R buffer [20 mM Tris-HCl (pH 7.5), 5% (w/v) glycerol, 100 μM EDTA, and 5 mM βME] and 50 mM (NH4)2SO4 and loaded into a column with a final bed volume of 1.4 mL. Using a protocol developed previously for screening the DNA binding properties of proteins in treated cell extracts (30), extracts containing wild-type or mutant HsRad51 proteins in R buffer with 50 mM (NH4)2SO4 were loaded and allowed to bind for 30 min at 4 °C. The column was washed with 2 × 5 column volumes of R buffer and 50 mM (NH4)2SO4 buffer, and bound protein was eluted with R buffer and 1 M (NH4)2SO4. Fractions were analyzed by SDS gel electrophoresis and Coomassie staining. Gel shift assays were performed following incubation of purified proteins with a 5′-fluorescein-labeled 54-base oligonucleotide (50 nM) in buffer containing 50 mM TEA, 5 mM MgCl2, 75 mM KCl, 1 mM DTT, 100 μg/mL BSA, and 2 mM ATPγS for 10 min (22 °C), followed by addition of glutaraldehyde to 1% and incubation for 10 min. Reactions were analyzed by electrophoresis on 1.0% agarose gels (1× TBE with 5 mM MgCl2), and DNA was visualized at 473 nm using a Fuji FLA Multi-functional Imagining System (Fuji) and Image Reader software (Fuji).
8-N3-ATP Cross-Linking
Cross-linking was performed in a buffer containing 10 mM MgOAc, 20 mM Tris (pH 7.5), 100 mM KCl, 10 μM HsRad51, and 0.5 mM 8-N3-ATP (Sigma) with 1–5 μCi of [α-32P]-8-N3-ATP (MP Biomedicals). Reaction mixtures (50 μL) were incubated on ice (10 min) prior to UV irradiation using a Rayonet photochemical reactor. Samples (10 μL) were removed at the indicated times, mixed with Laemmli sample buffer containing 10 mM DTT, and analyzed by SDS gel electrophoresis (10% acrylamide). Incorporation of radiolabeled N3-ATP was assessed by analyzing gels using an FX molecular imager and QuantityOne (Bio-Rad).
RESULTS
Rad51 K133A Is Functional for DNA Double-Strand Break Repair in Human Cells
To examine the ability of Rad51 mutant proteins to function in HEK293 cells in both the presence and absence of endogenous wild-type Rad51, we created GFP fusion vectors in which the HsRAD51 gene carries silent mutations (RAD51SM), making it resistant to the siRNA used to knock down endogenous Rad51 (Figure 1). DNA repair proficiency was determined by comet assays (Figure 2) using tail moment as a measure of DNA damage and repair. In each experiment, a minimum of 50 cells were analyzed, and all experiments were performed in triplicate. DNA repair is inhibited by transfection with Rad51-specific siRNA but not a negative control siRNA (Figure 2B). Transfection of Rad51-specific siRNA followed by the GFP-RAD51SM vector results in complete recovery of DNA repair, while the Rad51 K133R mutant shows little to no DNA repair. Unexpectedly, cells carrying the K133A mutant exhibit DNA repair activity similar to that of wild-type GFP-Rad51 or nontransfected cells. Assays were repeated in triplicate several times, and plasmids were resequenced on three separate occasions to ensure correct mutant identity. In all trials, the K133A mutant exhibited DNA repair capacity similar to that of wild-type Rad51, and the K133R mutant was inactive. Student’s t-tests comparing the 0 h with the 10 h time points and the 0.5 h with the 10 h time points (150 measurements) within each data set confirmed the statistical significance of DNA repair by wild-type Rad51 (p < 0.49 and p < 1 × 10−26, respectively) and the K133A mutant (p < 0.2 and p < 4 × 10−22, respectively), and the lack of DNA repair by the K133R mutant (p < 1 × 10−17 and p < 0.42, respectively) and in cells treated with×Rad51 siRNA (p < 4 × 10−14 and p < 0.38, respectively). We also found that both proteins behaved as dominant-negative mutants (Figure 2C); i.e., no DNA repair was detected above the level seen in cells transfected only with Rad51-specific siRNA (Figure 2B,C). We were also unable to make stable cell lines expressing the K133A mutant, a result consistent with previous studies using chicken and mouse cells (28, 29). Also, as previously reported (31), we found that GFP fused to the C-terminus of Rad51 renders the protein nonfunctional (Figure 2C).
Figure 1.
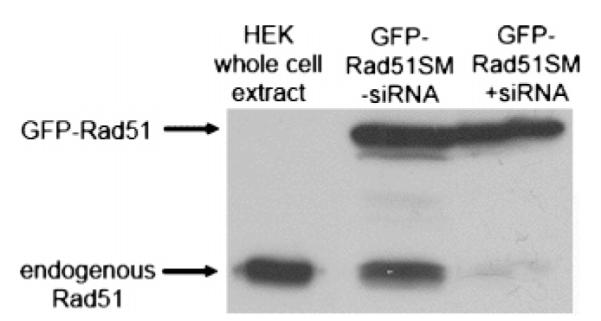
GFP-RAD51SM transgene expression is protected against siRNAs used to knock down endogenous Rad51. Extracts of HEK293 cells transfected with a Rad51-specific siRNA duplex and the HsRAD51SM vector were analyzed with Western blots (80 μg of total protein/lane). Blots were developed using a HsRad51 monoclonal antibody and show endogenous wild-type HsRad51 (≈37 kDa) and the GFP-Rad51 fusion protein (≈65 kDa). The right-most lane shows that the siRNA targets only endogenous Rad51.
Figure 2.

HsRad51 K133A (KA) mutant that repairs DNA breaks in vivo. Comet assays were performed to analyze Rad51-mediated repair of DNA double-strand breaks. (A) DNA from single HEK293 cells is visualized by EtBr staining following electrophoresis. Cells were exposed to IR as indicated and grown for 0.5 or 10 h. Results for cells expressing GFP-HsRAD51 transgenes in the presence (B) or absence (C) of Rad51-specific siRNA as indicated. At least 50 cells were analyzed for each time point, and all assays were performed in triplicate a minimum of four separate times. Student’s t-tests were performed to ensure the statistical significance of the data (see the text).
Given that the efficiencies of both transfection with the GFP-RAD51 constructs and knockdown of endogenous Rad51 are critical technical elements of analysis of transgene function in the absence of endogenous Rad51, both procedures were optimized prior to the performance of comet assays. Transfection efficiency was determined by confocal microscopy. Analysis of cells using the transfection procedure described above showed that HEK293 cells transfected with GFP-RAD51 or GFP-RAD51SM constructs resulted in a transfection efficiency of 94.4% (238 cells expressing GFP of a total 252 cells observed in nine different bright field images). Similar efficiencies were observed using samples of cells transfected for comet assays. The efficiency of RNAi-mediated knockdown was initially determined by FACS analyses of cells expressing a transgene carrying the wild-type RAD51 sequence (GFP-RAD51) that had also been transfected with a RAD51-specific siRNA duplex. Of the four original siRNA duplexes targeting different regions of the RAD51 structural gene, one showed a knockdown efficiency approximating 93–95% as judged by depletion of the GFP signal. These FACS data are consistent with Western blot analyses (for example, Figure 1) showing a similar level of knockdown of endogenous wild-type Rad51 in cells expressing the RNAi-resistant RAD51 gene in the GFP-RAD51SM constructs. Therefore, for the conditions used in comet assays, both the tranfection efficiency of the GFP-RAD51 vectors and the efficiency of RNAi-mediated knockdown of endogenous Rad51 are approximately 95%.
Rad51 K133A and K133R Proteins Form DNA Damage-Dependent Nuclear Foci
In normal cycling cells that have not been exposed to DNA damage, Rad51 is seen to form occasional and small nuclear foci (31, 32) that have been called “S-phase foci” (32). These are indicated by the white arrows in the left-hand panel of Figure 3, which shows cells expressing wild-type GFP-Rad51 that have not been exposed to IR. These background foci are also formed by the K133R and K133A mutant proteins (not shown). Formation of nuclear Rad51 foci that are larger in number and size is a hallmark of the initial phase of the cellular response to DNA damage. As demonstrated previously (31, 33, 34), we show in Figure 3 that wild-type GFP-Rad51 forms distinct nuclear foci in response to IR treatment. Additionally, both the K133R and K133A mutant proteins form DNA damage-induced nuclear foci (Figure 3), and each formed similar nuclear foci whether endogenous Rad51 was RNAi-depleted. Therefore, neither ATP site mutation weakens the ability of Rad51 to form foci in either the absence or presence of DNA damage. Additionally, as suggested by the images in Figure 3, cell counts in confocal images with 10 or more cells in a given field revealed that the vast majority of transfected cells exhibited DNA damage-induced foci [wild-type GFP-Rad51, 87.3% (103 of 120); GFP-Rad51 K133R, 92.9% (104 of 112); and GFP-Rad51 K133A, 95.9% (94 of 98)].
Figure 3.

K133A and K133R proteins form DNA damage-induced nuclear foci. HEK293 cells transfected with the wild-type GFP-RAD51SM vector and not exposed to DNA damage (left-most panel) show expression of GFP-Rad51 in the cytoplasm (C) and background nuclear foci that have been termed S-phase foci (white arrows). HEK293 cells expressing the indicated fusion protein were exposed to 7.5 Gy IR, grown for 1 h at 37 °C, and fixed. GFP fusion proteins were visualized by confocal microscopy.
Rad51 K133A and K133R Proteins Maintain Normal Protein–Protein Interactions
Rad51 protein–protein interactions, e.g., self-association, Xrcc3 and Brca2, were assessed using co-ip and yeast two-hybrid methods. HEK293 cells expressing wild-type GFP-Rad51, GFP K133R, or GFP K133A were lysed 14 h following transfection. Fusion proteins were precipitated using GFP antibodies, and SDS gels were analyzed for the presence of either endogenous Rad51 or Xrcc3. Western blots (Figure 4) show that equivalent levels of endogenous Rad51 and Xrcc3 co-ip with each fusion protein, and neither endogenous protein is detected when GFP alone is immunoprecipitated. This demonstrates that interactions of Rad51 with itself and Xrcc3 are not disrupted by either of the mutations. In yeast two-hybrid analyses, wild-type and mutant Rad51 proteins exhibit equivalent levels of interaction with wild-type Rad51, Xrcc3, and the BRC3 peptide repeat of Brca2 (Figure 5). Together, these data show that normal Rad51 protein interactions are not disrupted by either of the ATP binding site mutations.
Figure 4.

Wild-type and mutant GFP-HsRad51 proteins interact with endogenous Rad51 and Xrcc3. Cells expressing the indicated fusion protein were lysed, precleared, and incubated with an anti-GFP monoclonal antibody. Following recovery of the antibody using protein G magnetic beads, Western blots were performed with antibodies specific for either HsRad51 or HsXrcc3. Controls marked GFP are from cells transfected with the pEGFP-C1 parental vector.
Figure 5.
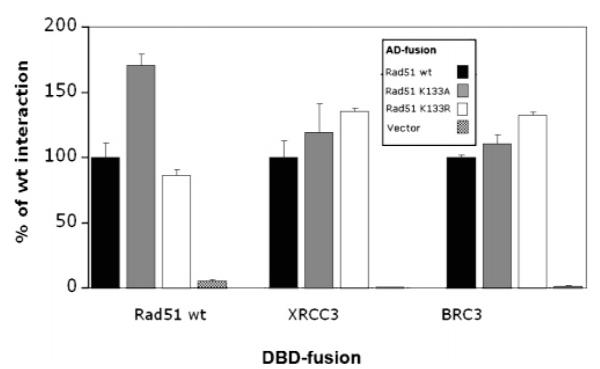
Wild-type and mutant GFP-HsRad51 proteins interact with wild-type HsRad51, HsXrcc3, and the BRC3 peptide from HsBRCA2. Yeast two-hybrid assays were performed using the indicated LexA-DNA binding domain (DBD) and Gal4 activation domain (AD) fusions. Results of β-galactosidase assays are expressed as the percentage activity relative to that of the wild-type HsRad51 activation domain fusion. Experiments were repeated four times.
DNA Binding by the K133R and K133A Mutant Proteins
Using a ssDNA cellulose binding screen designed for qualitative assessment of the DNA binding properties of proteins in cell extracts (30), we find that the wild-type Rad51, K133R, and K133A proteins bind ssDNA with similar efficiency (Figure 6A). Additionally, gel shift assays were performed using purified proteins, and consistent with recent studies (16), these show that the ssDNA binding affinity of the K133R and K133A protein approximates that of wild-type Rad51 (Figure 6B).
Figure 6.

Wild-type Rad51 and the K133R and K133A mutant proteins bind ssDNA with equivalent affinities. (A) A ssDNA cellulose binding screen was used to assess the relative ability of non-GFP-fused wild-type and mutant proteins to bind DNA. Treated cell extracts were loaded onto a ssDNA column (1.5 mL) and the load (L), flow-through (FT), wash (W1 and W2), and high-salt bump (B) fractions analyzed on SDS gels. (B) Gel shift assays were performed using purified proteins in the absence or presence of ATP. Following incubation of the indicated proteins with a 5′-fluorescein-labeled oligonucleotide, samples were analyzed on a 1.0% agarose gel.
The K133A Mutant Protein Binds ATP
ATP is a required cofactor for the recombinational activity of Rad51, and the fact that the K133A protein is functional in cell-based DNA repair assays suggests that it interacts productively with ATP. Although the Walker A motif Lys to Ala mutation is commonly assumed to inhibit ATP binding, we were unable to find a report in which a direct binding assay had been performed for any mutant ATPase that included this substitution. Therefore, we performed a direct ATP binding assay using [α-32P]-8-N3-ATP photolabeling. In reaction mixtures containing 0.5 mM 8-N3-ATP and no competing cold ATP, we found that the K133A protein incorporated N3-ATP approximately 20% as well as either wild-type Rad51 or the K133R protein (Figure 7A). A decreased level of labeling with [α-32P]-N3-ATP using UV exposures of ≥60 s resulted from protein cross-linking (Figure 7A and data not shown). Inclusion of ssDNA in the photolabeling experiments showed no significant differences in N3-ATP binding (data not shown).
Figure 7.

HsRad51 K133A protein that binds ATP. Purified proteins were incubated with [α-32P]-8-azido ATP on ice (10 min) and UV-irradiated for the indicated times. Samples were analyzed on SDS gels, and the histogram (A) shows quantification of radiolabel incorporation following UV irradiation for 30 and 60 s. Increasing amounts of cold ATP were added to reaction mixtures containing the indicated protein, and incorporation of [α-32P]-8-azido ATP following UV irradiation for 30 s was assessed by analysis of SDS gels (B and C). Data for wild-type Rad51 and the K133R mutant protein are averaged over four separate experiments with an estimated standard error of ≤10%. Data for the K133A mutant protein are averaged over four separate experiments using two different preparations of the protein, and standard errors are given.
Competition experiments using cold ATP were designed assuming a KD for ATP approximating 30 μM, as determined by measurements of the ATP dependence of hydrolysis (35). Titration of cold ATP from 0.05 to 1.0 mM decreased the level of N3-ATP labeling of wild-type Rad51 and the K133R mutant protein (Figure 7B,C), and a Ki of 20 μM was calculated for both proteins by nonlinear regression analysis using the formula y = 1 − (ATP/ATP- Ki). Interestingly, for the K133A protein, we found that increasing the level of cold ATP over this same range resulted in enhanced N3-ATP incorporation (Figure 7B,C). Addition of cold ATP to a concentration of 5 mM resulted in a decreased level of N3-ATP incorporation (Figure 7B,C). The fact the an increase in the level of N3-ATP incorporation peaked when the overall concentration of N3-ATP and ATP approached 3 mM and decreased from 5 mM and beyond provides an estimated KD in the range of 2–4 mM. Although this substitution is assumed to prevent ATP binding, no published studies have used an ATP concentration as high as this when analyzing various ATP-dependent activities for mutant Rad51 (16) or RecA proteins (reviewed in refs 8 and 9). The specificity of ATP binding is supported by the competition studies for wild-type Rad51 and the K133R protein and by the fact that although the K133A protein has an estimated KD close to 3 mM, binding is in competition at higher concentrations (Figure 7B,C). Additionally, when each of the three proteins is denatured, no N3-ATP incorporation is observed (not shown). The increased level of N3-ATP labeling for the K133A protein with an increasing cold ATP concentration up to 3 mM also suggests a cooperativity not observed for wild-type Rad51 (35; see Discussion). Thus, despite the frequent assumption that a Walker A motif Lys to Ala substitution prevents ATP binding, we show that this mutation in HsRad51 still allows 20% binding under standard conditions (0.5 mM N3-ATP) and a significantly higher degree of binding at more elevated ATP concentrations. ATPase assays showed wild-type Rad51 to have a turnover of approximately 0.2 min−1, consistent with other studies (2, 15), while the K133A and K133R proteins exhibited no activity above background.
DISCUSSION
We have used a RNAi-based approach in human cells to study the DNA repair activity of two HsRad51 ATP site mutants, K133A and K133R. An important feature of this approach is that it allowed analysis of the K133A mutant, which has been refractory to study in other cell systems due to its lethal dominant-negative effect. While the K133R mutant is nonfunctional in this system, we have made the surprising observation that the K133A mutant is functional for DSB repair in HEK293 cells depleted of endogenous Rad51. We show that both mutant proteins maintain certain functions indicative of normal Rad51 activity, e.g., formation of DNA damage-induced nuclear foci in response to DNA damage, protein self-association, and heterologous protein interactions with Xrcc3 and Brca2. Additionally, their DNA binding properties are similar to those of wild-type HsRad51. Given the common assumption that a Lys to Ala substitution in the Walker A motif inhibits ATP binding, a particularly important result is our demonstration that the K133A protein binds ATP, although with an affinity approximately 100-fold lower than that of wild-type HsRad51.
Until now, studies of HsRad51 ATP site mutants have used heterologous cell systems. Overexpression of the HsRad51 K133R mutant partially complements the IR sensitivity of RAD51–/– chicken DT40 cells (28), and because the purified K133R protein showed no evidence of ATP turnover, the authors suggested that ATP hydrolysis was not essential for HsRad51 function in vivo. In contrast, mouse ES cells expressing the same HsRad51 mutant showed a hypersensitivity to IR and significant defects in homology-directed DSB repair, thus supporting the idea that ATP hydrolysis is important to HsRad51 function in vivo (29). In both of these studies, cell lines carrying the HsRAD51 K133A mutant could not be made, suggesting that it behaves as a lethal dominant-negative mutation (28, 29). It is important to note that in our study both the K133A and K133R mutants display a dominant-negative phenotype in human cells expressing endogenous Rad51. We were also unable to create stable cell lines expressing the K133A mutant, a result consistent with studies of the K133A mutant in chicken and mouse cells. However, unlike our RNAi-based procedures, no determination of possible K133A activity could be undertaken in the absence of endogenous Rad51 in the chicken and mouse cell work (28, 29).
In our study, the expression level of transiently transfected plasmid-borne HsRad51 proteins is between 2- and 10-fold higher than the normal level of endogenous Rad51, and both mutant proteins maintain wild-type-like protein–protein interactions. Both considerations are undoubtedly important to promoting the function of the K133A protein, and each has been shown to positively affect the function of Rad51 mutant proteins in other studies. For example, the ScRad51 K191R mutant is functional both in vivo and in vitro but only when the protein is present at concentrations higher than that required for wild-type ScRad51 (26, 27). Additionally, the IR sensitivity of the ScRad51 K191R haploid strain is suppressed by high-copy expression of RAD54 that is dependent on RAD57, suggesting that the mediator function of the Rad57 (36) and Rad54 proteins (37–39) compensates for the weakly active Rad51 K191R protein (27). Recent studies show that HsRad51–ssDNA nucleoprotein filament structures are irregular in shape and unstable (40), supporting the idea that filament stability requires the assistance of other factors. Brca2 promotes HsRad51 filament assembly via coordination of its Rad51 and DNA binding activities (41, 42), and Xrcc3 interacts directly with HsRad51 and promotes assembly of Rad51 complexes in response to DNA damage (43, 44). We show that both the K133A and K133R mutants maintain the ability to form homo- and hetero-oligomeric complexes similar to wild-type HsRad51. Therefore, we propose that the K133A protein interacts productively with mediator proteins needed for appropriate initiation and completion of DNA strand exchange and that these interactions are especially important for the activity of a recombinase with a significant reduction in ATP binding affinity.
Our findings are supported by recent biochemical studies of the HsRad51 K133R and K133A proteins (16). As in our work, Chi et al. (16) found that both mutant proteins bind to ss- and dsDNA in a manner independent of ATP and with affinities similar to that of wild-type HsRad51. However, significant differences were observed in the ATP-mediated stabilization of the different nucleoprotein filaments. ATP induced a stable complex between wild-type Rad51 and ssDNA that dissociated upon ATP hydrolysis. However, the K133R–ATP–ssDNA complex was so stable that little to no dissociation was observed (16). This property could explain our observations that the K133R protein is nonfunctional on its own and behaves as a dominant-negative mutant in human cells (this study) and mouse cells (29). The fact that the HsRad51 K133R protein shows partial function in chicken cells (28) could be related to the presence of a helicase activity in those cells that is more robust than that in mammalian cells which aids in the disassembly of Rad51 nucleoprotein filaments, similar to the Saccharomyces cerevisiae srs2 or Schizosaccharomyces pombe fbh1 proteins (45–48) or possibly S. cerevisiae Rad54 (37).
Chi et al. (16) also showed that ATP did not induce stable complex formation between the K133A protein and ssDNA. However, the concentration of ATP was 0.1 mM in each of the assays that was used, and given our N3-ATP labeling data, it may be possible that some amount of stable K133A–ssDNA complex would be observed at ATP concentrations in the range of 1–3 mM. In our studies, we find that the level of N3-ATP labeling of the K133A protein increases as the ATP concentration is increased from 0 to 3 mM, a result expected for an allosteric protein in which ATP binding to one subunit increases the affinity of neighboring subunits for ATP. A decrease in the level of labeling as the ATP concentration is increased beyond 3 mM likely results from a combination of direct competition, as seen for wild-type Rad51 and the K133R mutant, as well as the inner filter effect (absorption of exciting wavelength responsible for photoactivation of N3-ATP). Although cooperative ATP binding is well-established for the homologous bacterial RecA protein (49), the human Rad51 protein binds ATP noncooperatively (35). If cooperative ATP binding is an inherently subtle process for Rad51, it must occur at very low ATP concentrations for wild-type Rad51 and the K133R mutant protein, and thus under the conditions of our experiments, only inhibition by unlabeled ATP is observed for these two proteins. The significantly reduced ATP binding affinity of the K133A protein, however, may permit detection of this phenomenon.
Our data support the idea that ATP serves very related functions for all recombinase enzymes. At a minimum, ATP binding promotes assembly of a stable, functional nucleoprotein filament competent for catalysis of DNA strand exchange while ATP hydrolysis (or simple ATP release in the case of the HsRad51 K133A protein) promotes disassembly of the nucleoprotein filament by destabilizing the oligomeric complex and lowering its DNA binding affinity. The idea that formation of an active recombinase filament is coupled directly to ATP binding and does not require the energy derived from ATP hydrolysis was originally proposed by Menetski et al. (11) through studies of the RecA protein. This was supported by work showing that the RecA K72R protein carries out DNA strand exchange in vitro for approximately 1.5 kb without ATP hydrolysis (21, 22) but cannot complete the reaction. Completion of DNA strand exchange requires disassembly of the RecA nucleoprotein filament, and this step depends on ATP hydrolysis (50). An additional complexity of the recombination mechanism involves how ATP binding and hydrolysis are coupled to the processing of DNA substrates during ongoing strand exchange reactions. This was addressed by studies of RecA that led to a model of “facilitated rotation” mediated by cycling ATP binding and hydrolysis events (50). Given the fundamental similarities in recombinase function and structures (see below), a related model likely applies to HsRad51, and in the case of the K133A protein, this probably occurs via binding and simple release of ATP with assistance from mediator proteins (51, 52). With regard to the dominant-negative activity of the K133A protein, it is possible that mixed filaments of wild-type and mutant Rad51 are incapable of coordinating ATP binding and hydrolysis and/or release with nucleoprotein filament assembly and disassembly, although further studies will be required to test this idea.
The idea that assembly and disassembly of recombinase filaments are coupled to ATP binding and hydrolysis, respectively, is now supported by recent modeling and crystallographic studies of bacterial RecA (53), ScRad51 (54), and archaeal MvRadA (55). These structures show that ATP binds directly between subunits in the active filament. Additionally, each structure contains a newly identified motif termed the ATP cap, highlighted by a conserved Pro residue that stacks against the adenine ring of ATP in the neighboring subunit (55).
Data presented in this study support a model in which the weak ATP binding properties of the HsRad51 K133A mutant protein, when overexpressed in human cells depleted of endogenous Rad51, allow it to mimic the ATP hydrolysis cycle of wild-type HsRad51 by simple binding and release of ATP. Together with its DNA binding properties and its ability to interact productively with mediator proteins, it can therefore exist transiently in an active state competent for catalysis of DSB repair. Clearly, studies of Walker A motif recombinase mutants from different species show disparities in recombination and DNA repair defects, which undoubtedly reflect specific mechanistic differences between the enzymes as well as differences in the cell types used for DNA repair assays. Although ATP may regulate functions specific to a given recombinase, there is now strong mechanistic and structural evidence that ATP binding and hydrolysis regulate properties common to all RecA and Rad51-like recombinases: formation and stabilization of an active nucleoprotein filament and its ultimate destabilization and disassembly.
ACKNOWLEDGMENT
We are grateful to John Latino for technical support, Dr. Nick Rhind for assistance with FACS analysis, Annamarie Piermarini for help with the figures, and Drs. Otto Gildemeister, William Kobertz, and Anthony Carruthers for critical reading of the manuscript.
Footnotes
This work was supported by NIH Grant GM44772 (K.L.K.) and DOD Breast Cancer Research Program Predoctoral Fellowship W81XWH-04-1-0601 (A.L.F.).
- ssDNA
- single-stranded DNA
- dsDNA
- double-stranded DNA
- IR
- ionizing radiation
- co-ip
- co-immunoprecipitation
- DSB
- double-strand break
- MMS
- methylmethane sulfonate
- UV
- ultraviolet
REFERENCES
- 1.Benson FE, Stasiak A, West SC. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J. 1994;13:5764–5771. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumann P, Benson FE, West SC. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–766. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- 3.Gupta RC, Bazemore LR, Golub EI, Radding CM. Activities of human recombination protein Rad51. Proc. Natl. Acad. Sci. U.S.A. 1997;94:463–468. doi: 10.1073/pnas.94.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benson FE, Baumann P, West SC. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature. 1998;391:401–404. doi: 10.1038/34937. [DOI] [PubMed] [Google Scholar]
- 5.Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol. Cell. Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radding CM. Helical RecA nucleoprotein filaments mediate homologous pairing and strand exchange. Biochim. Biophys. Acta. 1989;1008:131–145. doi: 10.1016/0167-4781(80)90001-9. [DOI] [PubMed] [Google Scholar]
- 8.Roca AI, Cox MM. RecA protein: Structure, function, and role in recombinational DNA repair. Prog. Nucleic Acid Res. Mol. Biol. 1997;56:129–223. doi: 10.1016/s0079-6603(08)61005-3. [DOI] [PubMed] [Google Scholar]
- 9.McGrew DA, Knight KL. Molecular design and functional organization of the RecA protein. Crit. Rev. Biochem. Mol. Biol. 2003;38:385–432. doi: 10.1080/10409230390242489. [DOI] [PubMed] [Google Scholar]
- 10.Silver MS, Fersht AR. Direct observation of complexes formed between recA protein and a fluorescent single-stranded deoxyribonucleic acid derivative. Biochemistry. 1982;21:6066–6072. doi: 10.1021/bi00267a007. [DOI] [PubMed] [Google Scholar]
- 11.Menetski JP, Kowalczykowski SC. Interaction of recA protein with single-stranded DNA. Quantitative aspects of binding affinity modulation by nucleotide cofactors. J. Mol. Biol. 1985;181:281–295. doi: 10.1016/0022-2836(85)90092-0. [DOI] [PubMed] [Google Scholar]
- 12.Namsaraev EA, Berg P. Binding of Rad51p to DNA. Interaction of Rad51p with single- and double-stranded DNA. J. Biol. Chem. 1998;273:6177–6182. doi: 10.1074/jbc.273.11.6177. [DOI] [PubMed] [Google Scholar]
- 13.De Zutter JK, Knight KL. The hRad51 and RecA proteins show significant differences in cooperative binding to single-stranded DNA. J. Mol. Biol. 1999;293:769–780. doi: 10.1006/jmbi.1999.3200. [DOI] [PubMed] [Google Scholar]
- 14.Tombline G, Heinen CD, Shim KS, Fishel R. Biochemical characterization of the human RAD51 protein. III. Modulation of DNA binding by adenosine nucleotides. J. Biol. Chem. 2002;277:14434–14442. doi: 10.1074/jbc.M109917200. [DOI] [PubMed] [Google Scholar]
- 15.Shim KS, Schmutte C, Tombline G, Heinen CD, Fishel R. hXRCC2 enhances ADP/ATP processing and strand exchange by hRAD51. J. Biol. Chem. 2004;279:30385–30394. doi: 10.1074/jbc.M306066200. [DOI] [PubMed] [Google Scholar]
- 16.Chi P, Van Komen S, Sehorn MG, Sigurdsson S, Sung P. Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair. 2006;5:381–391. doi: 10.1016/j.dnarep.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshimura Y, Morita T, Yamamoto A, Matsushiro A. Cloning and sequence of the human RecA-like gene cDNA. Nucleic Acids Res. 1993;21:1665. doi: 10.1093/nar/21.7.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shinohara A, Ogawa H, Matsuda Y, Ushio N, Ikeo K, Ogawa T. Cloning of human, mouse and fission yeast recombination genes homologous to RAD51 and recA. Nat. Genet. 1993;4:239–243. doi: 10.1038/ng0793-239. [DOI] [PubMed] [Google Scholar]
- 20.Saraste M, Sibbald PR, Wittinghofer A. The P-loop: A common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- 21.Rehrauer WM, Kowalczykowski SC. Alteration of the nucleoside triphosphate (NTP) catalytic domain within Escherichia coli recA protein attenuates NTP hydrolysis but not joint molecule formation. J. Biol. Chem. 1993;268:1292–1297. [PubMed] [Google Scholar]
- 22.Shan Q, Cox MM, Inman RB. DNA strand exchange promoted by RecA K72R. Two reaction phases with different Mg2+ requirements. J. Biol. Chem. 1996;271:5712–5724. doi: 10.1074/jbc.271.10.5712. [DOI] [PubMed] [Google Scholar]
- 23.Konola JT, Logan KM, Knight KL. Functional characterization of residues in the P-loop motif of the RecA protein ATP binding site. J. Mol. Biol. 1994;237:20–34. doi: 10.1006/jmbi.1994.1206. [DOI] [PubMed] [Google Scholar]
- 24.Logan KM, Knight KL. Mutagenesis of the P-loop motif in the ATP binding site of the RecA protein from Escherichia coli. J. Mol. Biol. 1993;232:1048–1059. doi: 10.1006/jmbi.1993.1459. [DOI] [PubMed] [Google Scholar]
- 25.Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 26.Sung P, Stratton SA. Yeast Rad51 recombinase mediates polar DNA strand exchange in the absence of ATP hydrolysis. J. Biol. Chem. 1996;271:27983–27986. doi: 10.1074/jbc.271.45.27983. [DOI] [PubMed] [Google Scholar]
- 27.Morgan EA, Shah N, Symington LS. The requirement for ATP hydrolysis by Saccharomyces cerevisiae Rad51 is bypassed by mating-type heterozygosity or RAD54 in high copy. Mol. Cell. Biol. 2002;22:6336–6343. doi: 10.1128/MCB.22.18.6336-6343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morrison C, Shinohara A, Sonoda E, Yamaguchi-Iwai Y, Takata M, Weichselbaum RR, Takeda S. The essential functions of human Rad51 are independent of ATP hydrolysis. Mol. Cell. Biol. 1999;19:6891–6897. doi: 10.1128/mcb.19.10.6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stark JM, Hu P, Pierce AJ, Moynahan ME, Ellis N, Jasin M. ATP hydrolysis by mammalian RAD51 has a key role during homology-directed DNA repair. J. Biol. Chem. 2002;277:20185–20194. doi: 10.1074/jbc.M112132200. [DOI] [PubMed] [Google Scholar]
- 30.Logan KM, Skiba MC, Eldin S, Knight KL. Mutant RecA proteins which form hexamer-sized oligomers. J. Mol. Biol. 1997;266:306–316. doi: 10.1006/jmbi.1996.0751. [DOI] [PubMed] [Google Scholar]
- 31.Essers J, Houtsmuller AB, van Veelen L, Paulusma C, Nigg AL, Pastink A, Vermeulen W, Hoeijmakers JH, Kanaar R. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J. 2002;21:2030–2037. doi: 10.1093/emboj/21.8.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarsounas M, Davies D, West SC. BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene. 2003;22:1115–1123. doi: 10.1038/sj.onc.1206263. [DOI] [PubMed] [Google Scholar]
- 33.Yu DS, Sonoda E, Takeda S, Huang CL, Pellegrini L, Blundell TL, Venkitaraman AR. Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2. Mol. Cell. 2003;12:1029–1041. doi: 10.1016/s1097-2765(03)00394-0. [DOI] [PubMed] [Google Scholar]
- 34.Forget AL, Bennett BT, Knight KL. Xrcc3 is recruited to DNA double strand breaks early and independent of Rad51. J. Cell. Biochem. 2004;93:429–436. doi: 10.1002/jcb.20232. [DOI] [PubMed] [Google Scholar]
- 35.Tombline G, Fishel R. Biochemical characterization of the human Rad51 protein. I. ATP hydrolysis. J. Biol. Chem. 2002;277:14417–14425. doi: 10.1074/jbc.M109915200. [DOI] [PubMed] [Google Scholar]
- 36.Sung P. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev. 1997;11:1111–1121. doi: 10.1101/gad.11.9.1111. [DOI] [PubMed] [Google Scholar]
- 37.Solinger JA, Kiianitsa K, Heyer WD. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles Rad51: dsDNA filaments. Mol. Cell. 2002;10:1175–1188. doi: 10.1016/s1097-2765(02)00743-8. [DOI] [PubMed] [Google Scholar]
- 38.Wolner B, van Komen S, Sung P, Peterson CL. Recruitment of the recombinational repair machinery to a DNA double-strand break in yeast. Mol. Cell. 2003;12:221–232. doi: 10.1016/s1097-2765(03)00242-9. [DOI] [PubMed] [Google Scholar]
- 39.Sugawara N, Wang X, Haber JE. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 40.Ristic D, Modesti M, van der Heijden T, van Noort J, Dekker C, Kanaar R, Wyman C. Human Rad51 filaments on double- and single-stranded DNA: Correlating regular and irregular forms with recombination function. Nucleic Acids Res. 2005;33:3292–3302. doi: 10.1093/nar/gki640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- 42.Yang H, Li Q, Fan J, Holloman WK, Pavletich NP. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005;433:653–657. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- 43.Bishop DK, Ear U, Bhattacharyya A, Calderone C, Beckett M, Weichselbaum RR, Shinohara A. Xrcc3 is required for assembly of Rad51 complexes in vivo. J. Biol. Chem. 1998;273:21482–21488. doi: 10.1074/jbc.273.34.21482. [DOI] [PubMed] [Google Scholar]
- 44.Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell. Biol. 2001;21:2858–2866. doi: 10.1128/MCB.21.8.2858-2866.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, Sung P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003;423:305–309. doi: 10.1038/nature01577. [DOI] [PubMed] [Google Scholar]
- 46.Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- 47.Osman F, Dixon J, Barr AR, Whitby MC. The F-Box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Mol. Cell. Biol. 2005;25:8084–8096. doi: 10.1128/MCB.25.18.8084-8096.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morishita T, Furukawa F, Sakaguchi C, Toda T, Carr AM, Iwasaki H, Shinagawa H. Role of the Schizosaccharomyces pombe F-Box DNA helicase in processing recombination intermediates. Mol. Cell. Biol. 2005;25:8074–8083. doi: 10.1128/MCB.25.18.8074-8083.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mikawa T, Masui R, Kuramitsu S. RecA protein has extremely high cooperativity for substrate in its ATPase activity. J. Biochem. 1998;123:450–457. doi: 10.1093/oxfordjournals.jbchem.a021958. [DOI] [PubMed] [Google Scholar]
- 50.Cox MM. The bacterial RecA protein as a motor protein. Annu. Rev. Microbiol. 2003;57:551–577. doi: 10.1146/annurev.micro.57.030502.090953. [DOI] [PubMed] [Google Scholar]
- 51.Beernink HT, Morrical SW. RMPs: Recombination/replication mediator proteins. Trends Biochem. Sci. 1999;24:385–389. doi: 10.1016/s0968-0004(99)01451-6. [DOI] [PubMed] [Google Scholar]
- 52.Sung P, Krejci L, Van Komen S, Sehorn MG. Rad51 recombinase and recombination mediators. J. Biol. Chem. 2003;278:42729–42732. doi: 10.1074/jbc.R300027200. [DOI] [PubMed] [Google Scholar]
- 53.VanLoock MS, Yu X, Yang S, Lai AL, Low C, Campbell MJ, Egelman EH. ATP-Mediated Conformational Changes in the RecA Filament. Structure. 2003;11:187–196. doi: 10.1016/s0969-2126(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 54.Conway AB, Lynch TW, Zhang Y, Fortin GS, Fung CW, Symington LS, Rice PA. Crystal structure of a Rad51 filament. Nat. Struct. Mol. Biol. 2004;11:791–796. doi: 10.1038/nsmb795. [DOI] [PubMed] [Google Scholar]
- 55.Wu Y, He Y, Moya IA, Qian X, Luo Y. Crystal structure of archaeal recombinase RADA: A snapshot of its extended conformation. Mol. Cell. 2004;15:423–435. doi: 10.1016/j.molcel.2004.07.014. [DOI] [PubMed] [Google Scholar]