

Abstract
Cotranscriptional recruitment of pre-mRNA splicing factors to their genomic targets facilitates efficient and ordered assembly of a mature messenger ribonucleoprotein particle (mRNP). However, how the cotranscriptional recruitment of splicing factors is regulated remains largely unknown. Here, we demonstrate that protein arginine methylation plays a novel role in regulating this process in Saccharomyces cerevisiae. Our data show that Hmt1, the major type I arginine methyltransferase, methylates Snp1, a U1 small nuclear RNP (snRNP)-specific protein, and that the mammalian Snp1 homolog, U1-70K, is likewise arginine methylated. Genome-wide localization analysis reveals that the deletion of the HMT1 gene deregulates the recruitment of U1 snRNP and its associated components to intron-containing genes (ICGs). In the same context, splicing factors acting downstream of U1 snRNP addition bind to a reduced number of ICGs. Quantitative measurement of the abundance of spliced target transcripts shows that these changes in recruitment result in an increase in the splicing efficiency of developmentally regulated mRNAs. We also show that in the absence of either Hmt1 or of its catalytic activity, an association between Snp1 and the SR-like protein Npl3 is substantially increased. Together, these data support a model whereby arginine methylation modulates dynamic associations between SR-like protein and pre-mRNA splicing factor to promote target specificity in splicing.
In eukaryotic cells, pre-mRNA is processed and packaged into a mature messenger ribonucleoprotein particle (mRNP) prior to its export from the nucleus (reviewed in references 12, 25, and 44). The correct formation of an mRNP requires a web of physical interactions among RNA processing factors during transcription. An important step in the processing of eukaryotic RNA is pre-mRNA splicing, in which noncoding introns are removed to generate mature, translatable mRNAs. The splicing reaction is catalyzed by the spliceosome, which is composed of five small nuclear ribonucleoprotein particles (snRNPs) and many associated proteins (reviewed in references 62, 67, and 68). Like many other RNA processing factors that have been studied thus far, the components of the spliceosome are recruited cotranscriptionally (19, 36, 47). Chromatin immunoprecipitation (ChIP) experiments have shown that, in vivo, spliceosome components assemble on intron-containing genes (ICGs) in a stepwise manner, consistent with findings from in vitro studies of splicing complexes (19, 32, 47). Specifically, the U1 snRNP is recruited to the 5′ splice site (ss), and the branchpoint binding protein (BPP) and Mud2 (human U2AF65) are recruited to the intronic branch site and nearby sequences, respectively. Together, these factors define basic intron/exon consensus features and “commit” a pre-mRNA substrate to splicing. Subsequent assembly involves ordered recruitment of the U2 snRNP, the U5/U4/U6 tri-snRNP, and spliceosome activation factors such as the “nineteen complex” (NTC) (9). Posttranscriptional splicing can occur, both in vivo and in vitro (64), but the coupling of splicing to transcription is thought to maximize the fidelity and efficiency of the process (13, 26). Thus, differential cotranscriptional recruitment of splicing factors represents a mechanism by which splicing can be regulated.
Protein arginine methylation is a posttranslational modification that is common to many RNA-binding proteins (RBPs) (reviewed in references 3 and 4). The enzymes that catalyze this process are termed protein arginine methyltransferases (PRMTs). In both yeast and mammalian cells, heterogeneous nuclear ribonucleoproteins (hnRNPs)—which like the snRNPs are associated with pre-mRNAs and involved in mRNA biogenesis—are major substrates of the PRMTs. Methylated hnRNPs possess at least one N-terminal RNA recognition motif (RRM)-type RNA-binding motif in addition to C-terminal arginine-glycine-glycine (RGG)-rich repeats, where arginine methylation is often found (42).
Many studies have demonstrated that arginine methylation plays an important role in modulating protein-protein interactions. For example, arginine methylation of the mammalian transcriptional elongation factor Spt5 regulates its interaction with RNA polymerase II (Pol II), thereby affecting transcription at a global level (35). The loss of arginine methylation on the mammalian STAT1 protein prohibits its association with PIAS, the inhibitor of STAT1, resulting in a decreased STAT1-mediated interferon response (49). In some cases, arginine methylation of a specific factor can modulate subsequent posttranslational modification events. For example, arginine methylation of mammalian FOXO transcription factors inhibits their phosphorylation by Akt (70).
In Saccharomyces cerevisiae, the arginine methyltransferase that catalyzes the formation of most asymmetric dimethylarginines is called Hmt1 (also known as Rmt1) (17, 24). Hmt1 is important for the nuclear transport of several of the hnRNPs that participate in the mRNP biogenesis pathway (58) and for the biochemical association between Npl3 and Tho2 (71). Npl3, an Hmt1 substrate, is an RNA-binding protein that has characteristics of both SR family (15) and hnRNP-like family proteins (8, 57); it is important for mRNA export (29) and also plays a role in pre-mRNA splicing (33). Tho2 is a component of the evolutionarily conserved transcription/export) (TREX) complex and plays a role during the elongation phase of transcription (52). RNA in situ hybridization analysis has demonstrated that the loss of Hmt1 activity results in slowed release of the HSP104 mRNA from the site of transcription (71). Genome-wide localization analysis (also known as ChIP-chip) revealed that Hmt1 coordinates cotranscriptional assembly of the hnRNPs Nab2, Hrp1, and Yra1 (71). Together, these data demonstrated that Hmt1 promotes the dynamic interactions between RNA-binding proteins and pre-mRNAs that facilitate proper assembly of an mRNP during transcription. However, the previous studies left open the question of whether the catalytic activity of Hmt1 or some other feature of this protein is required for the regulation of the cotranscriptional recruitment of pre-mRNA splicing factors.
In this study, we demonstrate that Hmt1-catalyzed arginine methylation, specifically, facilitates the cotranscriptional recruitment of pre-mRNA splicing factors. Also, we identify Snp1, a component of the U1 snRNP, as a novel Hmt1 substrate. We further show that arginine methylation of Snp1 is evolutionarily conserved in higher eukaryotes. Using ChIP-chip, we determined the impact of protein arginine methylation on the cotranscriptional recruitment of splicing factors to ICGs; specifically, we show that in mutants lacking Hmt1 or its catalytic activity, the recruitment of splicing factors to genes that undergo regulated splicing is aberrant, resulting in the enhanced splicing of these transcripts. Finally, we show that arginine methylation regulates the association of Snp1 with the SR-like protein Npl3. Together, these results demonstrate that protein arginine methylation regulates the specificity of the splicing machinery targeting to particular pre-mRNAs.
MATERIALS AND METHODS
Yeast strains used in this study.
All yeast strains used are listed in Table S1 in the supplemental material. Cells were grown at 30°C on YEPD medium (1% yeast extract, 2% Bacto peptone, 2% [wt/vol] d-glucose) unless otherwise stated. Gene deletions and C-terminal epitope tags were generated as described previously (30, 43).
In vitro methylation of Snp1.
The in vitro methylation was performed as described previously (46) using recombinant Hmt1 and tandem affinity purification (TAP)-tagged Snp1 expressed in Δhmt1 cells. The TAP was carried out as described previously (55), with the exception of binding with IgG-Sepharose beads (GE Amersham) in 600 mM NaCl during the first affinity purification step to minimize the capturing of interactors. The entire in vitro methylation reaction was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a NuPAGE 4 to 12% Bis-Tris gel (Invitrogen), followed by fluorography as previously described (46). In parallel, the purified TAP-tagged Snp1 was subjected to immunoblotting using an anti-CBP (Open Biosystems) antibody.
Identification of in vivo methylation sites within U1-70K.
Monoclonal antibodies against U1-70K were used to immunoprecipitate endogenous U1-70K from HeLa cells grown in medium specifically designed for SILAC experiments (66). The immunoprecipitates were resolved using a NuPAGE 12% Bis-Tris gel (Invitrogen) and Coomassie blue stained. The major band slice at 70 kDa was excised and subjected to preparation for mass spectrometry analysis as previously described (63, 66). The liquid chromatography/tandem mass spectrometry (LC/MS-MS) analysis was carried out as previously described (66).
Snp1 localization studies.
Wild-type (WT) or hmt1-null yeast strains containing green fluorescent protein (GFP)-tagged Snp1 from the GFP-tagged yeast library (27) were grown in synthetic complete (SC) medium until mid-log phase, at which time DAPI (4′,6-diamidino-2-phenylindole) stain was added to the medium for a final concentration of 1.2 μg/ml. Cells were then incubated for an additional 30 min. The differential interference contrast (DIC) and fluorescence microscopies were performed using an Axioplan 2 fluorescent microscope (Zeiss). Digital images were captured using the AxioCam MRm camera (Zeiss). Image acquisition and analysis were carried out using AxioVision 4.4 software (Zeiss).
Npl3-Snp1 interaction studies.
Yeast strains containing TAP-tagged Snp1 were grown in 500 ml of YEPD medium to mid-log phase and lysed with glass beads and a FastPrep machine (MP Biomedical) with a GigaPrep adapter five times at a setting of 6.5 for 30 s at 4°C. The cells were lysed in PBSMT buffer (phosphate-buffered saline with 0.5% Triton X-100, 3 mM KCl, and 2.5 mM MgCl2) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1.3 mM benzamide, and 2.5 μg/ml each leupeptin, chymostatin, antipain, pepstatin A, and aprotinin). Lysates were normalized for total protein, and TAP-tagged Snp1 fusion protein was purified by 2 h of incubation at 4°C with a 25-μl packed volume of IgG-Sepharose beads that had been prepared as previously described (71). Beads were washed three times with 1 ml of lysis buffer and twice with 1× TEV cleavage buffer (25 mM Tris-HCl [pH 8], 150 mM NaCl, 0.1% NP-40, 0.5 mM EDTA, 0.1 mM dithiothreitol [DTT]) prior to TEV cleavage with 1 μl of ProTEV protease (Promega) for 1 h at 16°C. The cleaved samples were eluted from the beads by a MolBicol column and precipitated with 12.5% trichloroacetic acid. Protein samples were dried, resuspended in protein sample buffer, resolved by SDS-PAGE using a 4 to 12% NuPAGE Bis-Tris gel (Invitrogen), and immunoblotted using anti-Npl3 antibody (gift from Pam Silver) (8). The immunoblot was then stripped and reprobed with an anti-CBP antibody (Open Biosystems). The immunoblot signals were digitally captured by a Bio-Rad ChemiDoc system.
Genome-wide localization (ChIP-chip) studies of splicing factors.
ChIP-chip studies were performed essentially as described previously (71). All of the experiments were performed with biological triplicates. The data obtained were analyzed using the Rosetta Resolver bioinformatics platform. Only genes with P values of <0.02 were considered for the final data analysis. Genes were considered bound if the immunoprecipitated fraction (IP)/background (whole-cell extract [WCE]) ratio was greater than 1. All of the raw output data from Rosetta Resolver and corrected data are available in Files S1 to S5 in the supplemental material.
Calculation of the intron distribution index (IDI).
For each ChIP-chip data set, a ranked list for total genes bound by a splicing factor was generated based on the IP/WCE ratio as described previously (71). Any ICG present within this ranked list is identified, and its distribution across the entire list is graphically plotted (see Fig. S2A to C in the supplemental material). Based on the total number of ICGs identified, a model distribution was generated to reflect a scenario in which the same numbers of ICGs are evenly distributed across a profile, yielding a straight line at a 45° angle (see Fig. S2A to C in the supplemental material). By plotting the actual experimental ICG ranking data as the x-axis coordinates with the model ICG ranking data as the y-axis coordinates, a nonlinear binding relationship was observed in which the line was skewed away from the assumed evenly distributed binding event. We then quantified the distance between this plot and the assumed model plot to measure the changes in the overall intron distribution between the experimental data and the model distribution data using the least-mean square distance (Drand) calculated by the following equation:
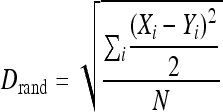 |
In this equation, Xi represents the ranking of each ICG in the data set being tested and Yi represents the corresponding ranking within the model reference. N is the number of ICGs shared between the two profiles compared (such as the WT versus Δhmt1 or the WT versus hmt1-G68R).
Directed ChIP experiments.
Directed ChIP experiments were performed as described previously (41, 71). For each immunoprecipitation, monoclonal anti-Myc (9E11; Fisher) antibody was precoupled to protein A-Sepharose beads. The oligonucleotides used in the ChIP experiments are listed in Table S2 in the supplemental material.
RNA isolation, cDNA preparation, and qPCR.
Total RNA was isolated by using the hot phenol method as described previously (71) followed by a DNase I (Invitrogen) treatment as per the manufacturer's instruction. The DNase I-treated RNA was then cleaned using an RNeasy kit (Qiagen). First-strand cDNA was synthesized from poly(A)-RNA using Superscript III reverse transcriptase (RT) (Invitrogen), oligo(dT), and 10 μg of RNA as described previously (47). cDNA dilution series were analyzed by PCR (24 to 26 cycles) to establish a quantitative, linear range of cDNA input for each set of quantitative PCR (qPCR) performed. The procedure for qPCR and the primer sets used were described previously (47), except [α-33P]dCTPs were used to radiolabel qPCR products. The radiolabeled PCR products were then resolved using an 8% Tris-borate-EDTA (TBE) gel, dried, and exposed to a PhosphorImager screen. The signals from spliced and unspliced PCR products were quantified using a PhosphorImager (Molecular Dynamics). The average ratios were calculated from triplicate sets of biological duplicates for HOP2 and triplicate sets of biological triplicates for REC107. Each RNA sample that had been treated by DNase I but was lacking the RT step was tested first by PCR using ADH3 primers to determine any potential genomic DNA contamination.
RESULTS
The U1 snRNP components Snp1 and U1-70K are arginine methylated.
Recent mammalian studies have implicated protein arginine methylation in pre-mRNA splicing (6, 11, 50). Snp1, a component of the S. cerevisiae U1 snRNP, has previously been shown to interact with the arginine methyltransferase Hmt1 in a comprehensive two-hybrid analysis (28). Analysis of the amino acid sequence of Snp1 revealed a number of arginine-glycine (RG) motifs that could potentially serve as sites for arginine methylation (see Fig. S1B, Snp1 sequence, in the supplemental material). We next set out to determine whether Snp1 is a substrate for Hmt1. We used a modified tandem affinity purification method to purify TAP-tagged Snp1 expressed from Δhmt1 cells under native, high-salt conditions. We then used this substrate, which has no endogenous methyl marks, in an in vitro methylation assay using recombinant Hmt1 as described previously (46).
The fluorograph from the in vitro methylation assay revealed a strong, positive signal corresponding to a band migrating at approximately 41 kDa (Fig. 1, left, arrow). Immunoblotting of the purified Snp1-CBP using an anti-CBP antibody (Fig. 1, right, arrow) confirmed that the 41-kDa band observed in the fluorograph is Snp1-CBP. Recombinant glutathione S-transferase (GST)-tagged Rps2 was used as a positive control (Fig. 1, asterisk), as described previously (51).
FIG. 1.
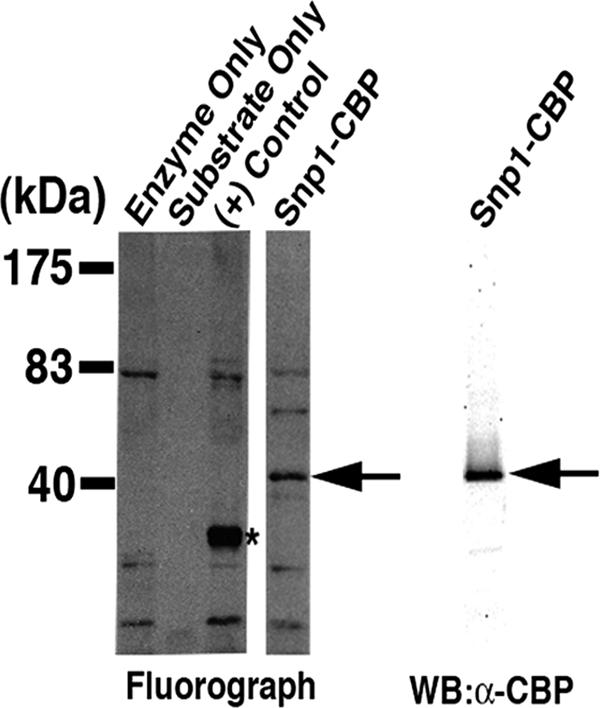
Arginine methylation is a feature of the yeast splicing factor Snp1. TAP-tagged Snp1 purified from the Δhmt1 strain was subjected to an in vitro methylation assay using recombinant Hmt1 and [methyl-3H]SAM. (Left) The full protein complement in each reaction was resolved on a 4 to 12% gel by SDS-PAGE and visualized by fluorography; (right) the substrate used in the in vitro methylation reaction was resolved in parallel, also on a 4 to 12% gel, and immunoblotted with an anti-CBP antibody. A band corresponding to methyl-3H-labeled Snp1-CBP was observed only in the presence of Hmt1 (Snp1-CBP lane, arrow [middle], compared to substrate only lane [left]). Recombinant GST-tagged Rps2 served as a control [left, (+) control lane, asterisk]. Immunoblotting carried out with an anti-CBP antibody (right) confirmed that the methylated band is Snp1-CBP (middle, arrow).
Hmt1 has been implicated in controlling nuclear localization of hnRNP-like proteins (20, 58). To determine if Hmt1 loss perturbs the cellular localization of Snp1, we chromosomally expressed a C-terminal GFP-tagged Snp1 in both wild-type and Δhmt1 cells and compared its in vivo cellular localization in these two cell types (see Fig. S1A in the supplemental material). In wild-type cells, Snp1-GFP was enriched in the nucleus (see Fig. S1A, WT panel, in the supplemental material), consistent with a previous report from a large-scale protein localization study in yeast (27). The same was true in the Δhmt1 cells (see Fig. S1A, Δhmt1 panel, in the supplemental material). Thus, Hmt1 loss has no effect on the cellular localization of Snp1.
The mammalian homolog of Snp1 is U1-70K (60), which was previously identified in a proteomic analysis of arginine-methylated protein complexes in HeLa cells (7). Although U1-70K was isolated using antibodies specific for symmetrical dimethylation (SYM10 and SYM11) (7), these antibodies strongly recognize core Sm proteins. Thus, the presence of U1-70K in those immunoprecipitates could have resulted from coisolation with native U1 snRNP particles. The presence of conserved RG motifs in the human U1-70K and the consensus sequences (see Fig. S1B, U1-70K and consensus sequence, in the supplemental material) suggests that these proteins may harbor modified arginine residues, which could account for their recognition by methylation-specific antibodies. We tested this possibility using a mass spectrometry approach and discovered that methylated arginines are present on U1-70K immunoprecipitated from HeLa cells (using a monoclonal anti-U1-70K antibody). Our mass spectrometry results indicated that 56% of the U1-70K protein was covered in the analysis, and the analysis of b and y ion series spectra indicated that 10 arginine residues (see Fig. S1B, asterisks, in the supplemental material) in the area covered were either mono- or dimethylated. Among the identified methylated arginines, many were found to match the RG and RXR motifs typical of most PRMT preferred sites (see Fig. S1B in the supplemental material). In summary, our data show that Snp1 is an in vitro substrate of Hmt1 and that its mammalian homolog, U1-70K, is likewise arginine methylated.
Hmt1 modulates the cotranscriptional recruitment of Snp1.
Since Hmt1 has been shown to signal the cotranscriptional recruitment of mRNP components (71) and U1 snRNP proteins are cotranscriptionally recruited (32, 36), we wanted to examine whether Hmt1 is necessary for cotranscriptional recruitment of Snp1 to its targets throughout the genome. To this end, we performed genome-wide occupancy analysis (ChIP-chip) of Snp1 in wild-type and Δhmt1 cells. A list of the relative rankings of all ICGs bound by Snp1 and other splicing factors examined in this study, in both wild-type and Δhmt1 cells, is presented in File S4 in the supplemental material. To probe for methylation-specific changes within the Snp1 ChIP-chip profiles, we used two complementary, quantitative analytical methods. First, we calculated the percentage of total Snp1-bound genes that were intron-containing genes in order to determine whether the Hmt1 loss leads to an increase or decrease in the percentage of ICGs bound within a profile. However, because a factor may bind to the same number and complement of ICGs yet the relative rankings of these genes may differ depending on the status of Hmt1, we also devised an intron distribution index (IDI) that assesses cumulative changes in the distribution of binding by Snp1 in a global manner (see Materials and Methods and Fig. S2 in the supplemental material). We calculated the IDI for each binding profile to distinguish changes in the relative rank of ICGs compared to that of other bound genes as a consequence of changes to Hmt1. A lower IDI in the Hmt1 mutant than in wild-type cells would reflect an overall decrease in the relative occupancy of the bound ICGs in the Hmt1 mutants. For example, although the same population of ICGs would be bound by a given factor, the factor's relative affinity for ICGs would be lower in Hmt1 mutants. Thus, using both approaches to analyze our Snp1 ChIP-chip data provides us with two separate but complementary methods to determine how methylation affects the cotranscriptional recruitment of Snp1.
Our Snp1 ChIP-chip data revealed that this protein binds a higher percentage of ICGs in Δhmt1 cells than in wild-type cells (Fig. 2 A, left). However, this is mostly based on the observation that Snp1 binding to non-ICGs was generally reduced in the Δhmt1 cells (Fig. 2A, middle); the total number of ICGs bound increased only slightly in the Δhmt1 cells (Fig. 2A, right). In sum, Snp1 displays less affinity for non-ICGs in the absence of Hmt1.
FIG. 2.

Hmt1 influences the cotranscriptional recruitment of U1 snRNP and its associated proteins. ChIP-chip was used to assess the effects of Hmt1 on the cotranscriptional recruitment of U1 snRNP and its associated proteins, which are Snp1 (A), Prp40 (B), and Mud2 (C). The impact of Hmt1 was assessed as the change in the percentage of intron-containing genes (ICGs) within the total number of genes bound by a splicing factor, as denoted by the bar graphs on the left. The total numbers of non-ICGs bound by each factor are shown by the middle bar graphs, and the total numbers of ICGs bound by each splicing factor are shown by the bar graphs on the right. The wild-type ChIP-chip data for Prp40 and Mud2 were obtained from Moore et al. (47).
In the absence of Hmt1, the recruitment of U1 snRNP-associated proteins to ICGs is increased.
To determine if Hmt1 affects the recruitment of other factors that associate with U1 snRNP, we performed ChIP-chip on the U1 snRNP-associated protein Prp40 and the pre-mRNA-U1 snRNP complex component Mud2. Prp40 interacts with the transcriptional machinery (48) and is considered one of the earliest splicing factors to be cotranscriptionally recruited. Thus, Prp40 has been proposed to sample transcripts for introns and to be specifically retained at ICGs. Similar to the trend observed for Snp1, Prp40 bound to a slightly higher percentage of ICGs in Δhmt1 cells than in wild-type cells (Fig. 2B, left) and to a slightly lower number of both non-ICGs and ICGs in total (Fig. 2B, middle).
Mud2 associates with the 3′ end of an intron (2) and interacts with the U2 snRNP by recognizing the branchpoint sequence (1). Our ChIP-chip data show that Mud2 is bound to a higher percentage of ICGs in Δhmt1 cells than in wild-type cells (Fig. 2C, left). As is the case for Snp1, this is based on the fact that a significantly lower overall number of non-ICGs bound (Fig. 2C, middle), despite only a slight decrease in the total number of ICGs bound (Fig. 2C, right). This overall decrease in the number of non-ICGs bound suggests that Mud2 has a significantly lower relative affinity for non-ICGs in the absence of Hmt1.
Together, the Prp40 and Mud2 data suggest that Hmt1 prevents Mud2 from being recruited to additional spurious non-ICGs and that this is also the case for Prp40, but to a lesser degree.
In the absence of Hmt1 activity, the cotranscriptional recruitment of splicing factors after U1 snRNP addition is decreased.
Our data show that Hmt1 affects the cotranscriptional recruitment of U1 snRNP and its associated components, and we wanted to determine if Hmt1 also affects splicing factors involved in other major steps of the canonical spliceosome assembly pathway. Therefore, we extended this approach to investigation of the splicing factors Prp11, Prp5, Prp2, and Brr2. Prp11 is a component of the U2 snRNP and is required for binding of the U2 snRNP to pre-mRNA during spliceosome assembly (56). Prp5 bridges the U1 and U2 snRNPs to allow U2 snRNP to stably associate with introns (31). Prp2 is an RNA-dependent ATPase of the DEAH-box family and is required for the first transesterification reaction of splicing (59). Brr2 is an RNA helicase; it associates with the U5 snRNP (23) and is required for activating the spliceosome for catalysis by disrupting U4/U6 base pairing in native snRNPs (39, 53).
In comparison to the U1 snRNP and the associated components that have been examined, all four of the U2 and U4/U5/U6 components and their associated factors bound to a lower percentage of ICGs in the absence of Hmt1 (Fig. 3, left panels). In the case of Prp11, this result was particularly striking, with ICG binding reduced to only 13% in the Δhmt1 mutant (Fig. 3A, left). Moreover, for each of these proteins other than Prp2, the total number of non-ICGs bound was increased (Fig. 3, middle panels), whereas the total number of ICGs bound was generally decreased, with the only exception being Prp5 (Fig. 3, right panels). Nevertheless, we observed reduced IDIs for all four components in the context of Δhmt1 cells, with the reduction being particularly drastic in the cases of both Prp11 and Brr2 (see Fig. S2D, panels Prp11, Prp2, Prp5, and Brr2, in the supplemental material). These findings further support an overall loss of relative affinity for ICGs, an overall increase of relative affinity for non-ICGs, or both. Together, these data reveal that the absence of Hmt1 significantly increases the relative affinity of post-U1 snRNP addition splicing factors for non-ICGs and, to a lesser degree, decreases their relative affinities for ICGs.
FIG. 3.

Arginine methylation negatively affects the recruitment of U2 and U4/U5/U6 tri-snRNP components and their associated proteins. The genome-wide cotranscriptional recruitment of Prp11 (A), Prp5 (B), Prp2 (C), and Brr2 (D) in the wild-type and Δhmt1 strains is mapped as described in the legend to Fig. 2. The requirement for Hmt1 activity (versus Hmt1 presence) in cotranscriptional recruitment was tested for Prp11 (E) and Mud2 (F). The wild-type ChIP-chip data for Prp11, Prp5, Prp2, and Brr2 were obtained from Moore et al. (47).
To establish the role of the catalytic activity of Hmt1 in the cotranscriptional recruitment of splicing factors, we took advantage of a previously published Hmt1 catalytic mutant, hmt1-G68R (46). We focused on Mud2 and Prp11, since these proteins play key roles in the formation of the commitment complex yet display opposite responses in recruitment to ICGs in the context of HMT1 deletion. Our data with the hmt1-G68R mutants reveal that arginine methylation affects the cotranscriptional recruitment of both Mud2 and Prp11 in a manner that is consistent with the loss of the entire gene (Fig. 3E and F). Thus, we attribute the changes observed in our ChIP-chip analysis to the catalytic activity of Hmt1.
Directed ChIP validation of genome-wide occupancy analysis.
To validate the results of our genome-wide binding studies, we performed directed ChIP on genes bound by Snp1, Mud2, Prp11, Prp2, and Prp5 in both wild-type and Δhmt1 cells (Fig. 4). In each case, we chose genes that the ChIP-chip data suggest have either different or similar binding based on the ranked list created with each profile. We also chose genes with different expression levels, including a highly expressed one (RPS13), a moderately expressed one (ARP9), and ones expressed at low levels (meiotic ICGs REC107 and HOP2). In the case of RPS13, a relatively high level of binding was detected for all five splicing factors in both wild-type and Δhmt1 cells, consistent with the data from genome-binding studies (Fig. 4A to E, RPS13 graphs). In the case of ARP9, we did not detect any difference in binding by Snp1 and observed only a slight change in Mud2 binding in Δhmt1 cells compared to that in wild-type cells (Fig. 4A and B, ARP9 graphs). Nevertheless, we did observe a significant enrichment for Prp11, Prp2, and Prp5 binding to ARP9 in Δhmt1 cells (Fig. 4C to E, ARP9 graphs). Overall, these trends support the ChIP-chip data obtained for both RPS13 and ARP9.
FIG. 4.

Validation of genome-wide occupancy results. Directed ChIPs were performed on either wild-type or hmt1-null cells using anti-Myc. The splicing factors examined are Snp1 (A), Mud2 (B), Prp11 (C), Prp5 (D), and Prp2 (E). The choice of genes tested is based on the ChIP-chip data obtained. The bar graph depicts the normalized ratio of experimental signal to the intergenic region.
In the case of both REC107 and HOP2, our ChIP results were also generally consistent with our ChIP-chip data (Fig. 4A to E, REC107 and HOP2 graphs). The one exception had to do with Prp11 recruitment to HOP2 in Δhmt1 cells. In this instance, a positive ChIP result was obtained (Fig. 4C, HOP2 graph). Upon reanalysis of the original Prp11 ChIP-chip data for HOP2 in Δhmt1 cells, we discovered that binding had been observed, but its P value was only 0.16 and thus did not meet the stringent P value cutoff of 0.02 (see File S5 in the supplemental material). As such, these data points were excluded from the data set that was further evaluated, despite the fact that it had an IP/input ratio that reflected a bound state (i.e., greater than 1). Notably, our ChIP-chip binding data for Prp11 in hmt1-G68R cells indicate that Prp11 is bound to HOP2 (see File S6 in the supplemental material). Thus, it is likely that Prp11 is also bound to HOP2 in the Δhmt1 cells and that this interaction was reflected in our directed ChIP data. As a whole, our directed ChIP results are in agreement with the ChIP-chip data.
Hmt1-dependent arginine methylation is required for regulated splicing of meiosis-related genes.
We previously demonstrated that differential spliceosome recruitment predicts regulated splicing and found that this was most striking in the case of meiosis-related genes (47). The genes that encode these regulated transcripts are transcribed in the vegetative growth state but are not spliced until meiosis (14). Since our ChIP-chip data indicated that the recruitment and distribution of splicing factors to ICGs change in Hmt1 mutants, we examined whether these changes affect regulated splicing in Hmt1 mutants. To this end, we monitored the changes in pre-mRNA splicing for these meiosis-related transcripts. We analyzed our ChIP-chip data and evaluated changes in recruitment of the seven splicing factors assayed to meiosis-related genes in the Hmt1-null mutants (see File S6 in the supplemental material). We found that both Snp1 and Mud2 bound a significant fraction of meiosis-related ICGs in wild-type cells and that the loss of Hmt1 had some effect on the recruitment of Snp1, but not Mud2, to this population of ICGs (Fig. 5 A, compare WT and Hmt1 mutant). All other splicing factors examined, with the exception of Prp40, showed increased association with meiosis-related ICGs in Δhmt1 cells, with little or no binding to these genes in wild-type cells (Fig. 5B, compare WT and Hmt1 mutant). These data indicated that Hmt1 contributes to the recruitment of specific splicing factors to meiosis-related ICGs. To further test this possibility, we performed a gene ontology (GO) test by using the GO Slim Mapper feature of the Saccharomyces Genome Database (SGD) to determine the effect of Hmt1 on the occupancy of meiotic ICGs for each of the splicing factors examined in this study (Fig. 5C). As a reference, the genome frequency for “meiosis” as a GO slim term is 155 genes out of 6,310 genes. Of the 294 annotated ICGs in the SGD, 17 map to the “meiosis” GO slim term, as indicated in the bar graph (Fig. 5C, SGD reference bar). Using the GO test, we found that in the cases of U1 snRNP and its associated factors Prp40, Snp1, and Mud2, the number of meiotic ICGs bound in Δhmt1 cells either did not change or decreased with respect to the number bound in wild-type cells (Fig. 5C, Prp40, Snp1, and Mud2 bars). In the case of splicing factors that are added after U1 snRNP is added, however, the number of meiotic ICGs bound was consistently higher in Δhmt1 cells than in their wild-type counterparts (Fig. 5C, Prp11, Prp2, and Prp5 bars). This trend also holds true if one calculates the “cluster frequency” by dividing the number of meiotic ICGs bound by the number of total ICGs bound for each splicing factor. Thus, Hmt1 loss leads to an increase in the impact of occupancy of meiotic ICGs by Prp11, Prp2, Prp5, and Brr2.
FIG. 5.

Changes in arginine methylation-dependent cotranscriptional recruitment influences regulated splicing. (A) The percentages of meiosis-related genes bound by the U1 snRNP components and associated factors Prp40, Snp1, and Mud2 in wild-type and Δhmt1 cells are shown. Where a specific meiosis-related gene is bound by a particular factor in both wild-type and Δhmt1 cells, the percentage of these bound genes within the total number of meiosis-related genes is calculated and denoted as “Both” on the bar graph. (B) The percentages of meiosis-related genes bound by U2 and the U4/U5/U6 tri-snRNP components and associated proteins Prp11, Prp5, Prp2, and Brr2 in wild-type and Hmt1 mutants are shown (representation is as described for panel A). (C) The effects of Hmt1 on the occupancy of meiotic ICGs relative to all ICGs bound by a splicing factor. A gene ontology (GO) test using the “Yeast GO-Slim: Process” feature of the SGD was performed, testing the total number of ICGs bound by each splicing factor in either wild-type or Δhmt1 cells. Of the entire 294 ICGs annotated in the yeast genome (SGD reference, gray bar), 17 map to the “meiosis” GO slim term (SGD reference, black bar). The results for each splicing factor are plotted as bars. (D) The splicing of meiotically regulated transcripts REC107 and HOP2 in wild-type, Δhmt1, and hmt1-G68R cells was measured by quantitative PCR using [α-33P]dCTP as described in Materials and Methods. The products were resolved on an 8% TBE gel, dried, exposed to a phosphorimager screen, and quantified by a PhosphorImager. The unspliced (U) and predicted spliced (S) products are indicated on the left. ECM33, a constitutively spliced transcript, and ADH3, an unspliced transcript, were used as controls in the quantitative PCR to demonstrate similarity in the levels of input cDNA used in the quantitative PCR. (E) The ratios of spliced to unspliced transcript levels from REC107 and HOP2 in wild-type, Δhmt1, and hmt1-G68R cells are shown.
The enhanced recruitment of spliceosome components to meiosis-related genes in Hmt1 mutants suggests that splicing of these transcripts may be enhanced in Hmt1 mutants. To test this possibility, we examined splicing of the meiosis-related gene HOP2, which encodes a meiosis-specific protein that prevents synapsis between nonhomologous chromosomes while ensuring synapsis between homologs (10). In Δhmt1 cells, five of the seven splicing factors examined bound the HOP2 locus, whereas only two of them bound this locus in wild-type cells (see File S6 in the supplemental material). For comparison, we examined REC107, another meiosis-related gene, which is known to undergo Mer1-dependent meiosis-specific splicing (61). Our data indicate that REC107 is not bound by any splicing factors in Δhmt1 cells and that it is bound by only Prp40 in the wild-type cells (see File S6 in the supplemental material).
To examine how arginine methylation affects the splicing of REC107 and HOP2, we performed quantitative PCR (qPCR) on cDNA produced from wild-type, Δhmt1, and hmt1-G68R cells (Fig. 5D). The signal corresponding to unspliced and predicted spliced transcripts in each background was quantified to calculate an internal ratio for comparison (Fig. 5E). Mutations in Hmt1 had little effect on the ratio of spliced to unspliced transcripts for REC107 (Fig. 5E, REC107 graph), consistent with the observation that the loss of Hmt1 did not affect the recruitment of splicing factors to this locus. In contrast, the level of splicing increased 30% and 50% for the HOP2 transcript in the cases of the Δhmt1 and hmt1-G68R mutants, respectively (Fig. 5E, HOP2 graph), supporting our prediction that increased recruitment of splicing factors to the HOP2 gene enhances the splicing of HOP2 transcripts in vegetative cells. As a control for our qPCRs and isolation of total RNA, we examined the level of a constitutively spliced transcript encoded by ECM33 and the level of non-intron-containing transcript encoded by ADH3. Levels of these RNAs were similar in the wild-type and mutant strains (Fig. 5D, ECM33 and ADH3). A control in which a DNase I-treated, non-reverse-transcribed total RNA template was used produced no products, indicating that our RNA sample was not contaminated with genomic DNA (data not shown). Thus, our data demonstrate that arginine methylation can affect cotranscriptional recruitment of pre-mRNA splicing factors, leading to increased pre-mRNA splicing of the affected target.
Hmt1-dependent arginine methylation modulates the biochemical interaction between Npl3 and Snp1.
Protein arginine methylation was previously shown to be critical in modulating protein complexes early in mRNP formation (71). Therefore, arginine methylation of Snp1 may regulate its interactions with nascent mRNPs. Npl3, a yeast SR-like protein (15) with known roles in mRNA export (29), is recruited during the early stages of transcription (40). Recently, Npl3 has been demonstrated to promote pre-mRNA splicing for a large subset of ICGs and to facilitate cotranscriptional binding of early splicing factors to their genomic targets (33). Since Npl3 and Snp1 have been shown to interact with each other as part of the cap-binding protein (Cbc2) complex (18) and since both proteins are substrates of Hmt1, we hypothesized that arginine methylation may modulate their biochemical interaction. To test this hypothesis, we expressed a TAP-tagged Snp1 fusion protein (Snp1-TAP) in wild-type, Δhmt1, and hmt1-G68R backgrounds and then copurified Snp1 and its associated proteins. Immunoblotting using an anti-Npl3 antibody was then performed to probe for changes in the amount of Npl3 that copurified with Snp1 (Fig. 6, left). Immunoblotting revealed an intense band migrating at approximately 55 kDa in both the Δhmt1 and hmt1-G68R lanes (Fig. 6, left); this molecular mass corresponds to the size of Npl3 in its unmethylated form (46). In wild-type cells, a fainter band of slightly higher molecular mass corresponding to that expected for methylated Npl3 was also observed (Fig. 6, left, WT lane). The difference in the migration between the methylated and unmethylated forms of Npl3 is more readily visible in a control immunoblot, using a fraction of the input lysate (Fig. 6, input panel). The drastic contrast in the amount of Npl3 associated with Snp1 in the Hmt1 mutants indicates that the biochemical interaction between Npl3 and Snp1 was increased in the absence of Hmt1 or of its catalytic activity. The immunoblot was stripped and reprobed with an anti-CBP antibody to show that similar amounts of Snp1 were purified and loaded onto the gel (Fig. 6, right). A negative control was generated by purifying proteins from a yeast strain that does not express any TAP-tagged proteins [Fig. 6, (-) control lane].
FIG. 6.

Arginine methylation modulates the biochemical association between Npl3 and Snp1. TAP-tagged Snp1 expressed in wild-type, Δhmt1, and hmt1-G68R cells was purified and resolved on a 4 to 12% gel by SDS-PAGE. (Left) The amount of Npl3 that copurified with Snp1 was determined using an anti-Npl3 antibody. The arrow (Npl3-CH3) denotes a band corresponding to Npl3 lacking methylated arginines (lanes Δhmt1 and hmt1-G68R). A faint, slightly higher-molecular-mass band for the wild-type cells corresponds to arginine-methylated Npl3 (WT lane, arrow with Npl3 plus CH3). (Input panel) The degree of change is readily visualized from the input of each lysate, probed with anti-Npl3 antibody (arrows). (Right) The same immunoblot was stripped and reprobed with anti-CBP antibody to detect the level of Snp1-CBP (arrow denotes Snp1-CBP). A control using a strain that does not express any TAP-tagged protein was run in parallel [(-) control lane].
DISCUSSION
During mRNP biogenesis, a network of cross-stimulatory connections and physical interdependencies is crucial for the formation of an export-competent mRNP (reviewed in references 12 and 44). Cotranscriptional recruitment of splicing factors constitutes an integral part of mRNP formation. In yeast, the ordered association of spliceosomal components during transcription has been shown to facilitate efficient cotranscriptional mRNA processing (19, 47). In the current study, we define a novel role for protein arginine methylation in the cotranscriptional recruitment of pre-mRNA splicing factors. We have identified Snp1, a component of the canonical U1 snRNP (which participates in the early steps of spliceosomal complex assembly during transcription [60]), as a novel substrate of Hmt1 in vitro. Using ChIP-chip, we uncovered a role for protein arginine methylation in modulating the recruitment of splicing factors to ICGs. A number of these data were validated by directed ChIP experiments. Changes in the cotranscriptional recruitment of splicing factors due to the loss of arginine methylation affects regulated splicing. We show that the absence of arginine methylation leads to an aberrant interaction between Snp1 and the SR-like protein Npl3. Overall, our data demonstrate that protein arginine methylation plays a critical role in defining proper cotranscriptional recruitment of pre-mRNA splicing factors.
To date, studies characterizing the recruitment of pre-mRNA splicing factors on a genome-wide scale have focused on measuring changes in the absolute number or percentage of ICGs bound by a splicing factor (32, 47, 64). This kind of measurement does not reveal how the distribution of bound ICGs changes in the context of a given condition or mutation. It is possible, for example, that a splicing factor is bound to the identical number and complement of ICGs under different conditions but that the ICGs bound in one case occupy the highly bound portion of the total bound population and in the other occupy the least-bound portion. Thus, the IDI serves as a novel method to assay the relative changes in ICG association when comparing two ChIP-chip profiles. In the study here, it reveals that the loss of Hmt1 or of its activity perturbs the relative degree of occupancy of ICGs for all U snRNPs examined, with the exception of U1 snRNP and its associated proteins.
Since the dynamics of U1 and U2 snRNP recruitment are intimately linked (19, 37, 65), it is likely that a change in the recruitment dynamics of the U1 snRNP would produce a downstream effect on the recruitment of U2 and other U snRNPs. This could be achieved by perturbing the formation of the commitment complex. Our ChIP-chip data are consistent with such an effect, as they demonstrate that recruitment of the U2 snRNP and the U4/U5/U6 snRNP, as well as of their associated proteins, is reduced in Hmt1 mutants relative to that in wild-type cells. Since the maximal U1 snRNP recruitment occurs near the 5′ ss (64), it is likely that the loss of arginine methylation in Hmt1 mutants results in increased retention of the U1 snRNP and its associated proteins at ICGs. This may be reflected by the observed decrease in binding of U1 snRNP and its associated proteins at non-ICGs, since fewer U1 snRNPs would be available to “scan” the non-ICGs as they are effectively “titrated away” as a consequence of longer ICG retention. Disruption of this in vivo recruitment dynamic could theoretically prevent recognition of the other components of the spliceosomal complex (such as proteins from the U2 snRNP and the U4/U5/U6 tri-snRNP) and thus their efficient recruitment to genomic targets. This could give rise to a scenario in which formation of the commitment complex is compromised. Indeed, our ChIP-chip data—according to which the U2 snRNP, the U4/U5/U6 tri-snRNP, and their associated proteins all exhibited lower relative affinities for ICGs in Hmt1 mutants—support such a scenario.
Our genome-wide assays led to the identification of a meiosis-related gene whose transcripts display enhanced splicing during vegetative growth in Hmt1 mutants. The observation that the loss of arginine methylation leads to aberrant recruitment of splicing factors and misregulated splicing of these targets supports our previous study, in which we showed that differential spliceosome recruitment predicts regulated splicing in precisely the same population of genes, i.e., meiosis-related ICGs (47). The relatively small increases in the pre-mRNA splicing of HOP2 may be due to the nuclear mRNA surveillance machinery monitoring sites of regulated splicing, since increased levels of unspliced meiotic mRNAs have been observed in Δrrp6 cells, which lack a subunit of the nuclear exosome (47). Testing a mutant in which both HMT1 and RRP6 are deleted may reveal more dramatic changes. Notably, Δhmt1 cells display decreased sporulation frequency (16). Whether this abnormality results from altered splicing of meiosis-related transcripts remains to be investigated. Nevertheless, our present study has established arginine methylation as a requirement for regulated recruitment of the spliceosome.
Arginine methylation has been demonstrated to affect the function of protein substrates by modulating their interactions with other proteins (5). For example, the interaction of mammalian PRMT1 and transcription factor Ying Yang 1 (YY1) is necessary for the recruitment of histone H4-specific methyltransferase activity (54). Hmt1 has been demonstrated to control the biochemical association between mRNA export factor Npl3 and transcriptional elongation factor Tho2, as well as the self-association of Npl3 (71). While the predominant role of Npl3 is in the export of mRNAs (29), it was recently revealed that Npl3 also promotes pre-mRNA splicing, as it is required for the cotranscriptional recruitment of early splicing factors such as the U1 snRNP protein Prp40 and the U2 snRNP protein Lea1 (33). Furthermore, Npl3 is a yeast SR-like protein (15), and SR proteins in mammalian cells are known to help stabilize the U1 snRNP during the spliceosome assembly (69). These facts suggested that the loss of Hmt1 or its activity would impact how Npl3 interacts with the early splicing factors and that this interaction might account for the decreased recruitment of U2 and U4/U5/U6 tri-snRNP proteins in our ChIP-chip experiment. In support of this hypothesis, we observed an aberrant biochemical association between Npl3 and Snp1 in both the Δhmt1 and hmt1-G68R mutants (Fig. 5). Since Hmt1 also methylates Npl3, it is possible that arginine methylation regulates the cotranscriptional recruitment of splicing factors by altering Npl3 recruitment. However, our previous analysis shows that the loss of Hmt1 does not alter the recruitment of Npl3 to either ICGs or non-ICGs (see Fig. S4 in the supplemental material). Rather, the dynamics of biochemical association between Npl3 and Snp1 are most likely to control how the rest of the splicing factors are recruited in the context of mRNP biogenesis. New studies, using methylation-specific mutants of both Npl3 (45) and Snp1, are under way to address the influence of methylation on the ability of each factor to promote this association.
A recent study demonstrated that the chromatin-modifying activity of the histone acetyltransferase Gcn5 is functionally linked to the cotranscriptional recruitment of pre-mRNA splicing factors (21). Specifically, Gcn5 was found to acetylate the ICG-bound histone H3 (21). Notably, a screen used to identify protein-protein interactions that are triggered by posttranslational modifications identified Hmt1 as a binding partner for acetylated histones (22). Hmt1 binds both acetylated histones H3 and H4 but methylates only H4 (34). Given that Hmt1 methylates histone H4 at position 3 (H4R3) in a chromatin-specific context (72) and that Hmt1 loss does not abolish bulk changes in H4R3 methylation (38), it would be interesting to determine whether Hmt1 plays a role in the status of H4R3 methylation within ICG-bound histones. Such a study would determine whether the chromatin-modifying activity of Hmt1 is linked to its role in optimizing the cotranscriptional recruitment of splicing factors.
Overall, our data support a model in which Hmt1-catalyzed arginine methylation controls the cotranscriptional recruitment of splicing factors by promoting proper Npl3-Snp1 interaction. Npl3 is cotranscriptionally recruited during the early stages of transcription as part of mRNP biogenesis. As a yeast SR-like protein, it may stabilize the U1 snRNP, much as SR family proteins do in mammalian cells. During the early phase of the transcription process, the Snp1-containing U1 snRNP samples transcribed genes to identify any introns. The detection of an intron leads to the formation of a commitment complex, followed by subsequent recruitment of the rest of the spliceosome. Given that mRNP assembly involves a multitude of associations and dissociations of its components, aberrant interactions between Npl3 and Snp1 in the Hmt1 mutants may cause the U1 snRNP proteins to be retained at their genomic targets for longer times than optimal. This disruption in the dynamics of U1 snRNP recruitment, in turn, would likely affect the formation of the commitment complex, judging by the ChIP-chip data for Prp11 and Mud2. Alternatively, it is possible that a change in the dynamics of U1 snRNP recruitment due to Hmt1 activity results in a rearrangement within the spliceosome that subsequently prevents proper recruitment of the U2 and U4/U5/U6 tri-snRNP components. In summary, our study establishes a novel and key regulatory role for protein arginine methylation in controlling the dynamics of cotranscriptional recruitment of pre-mRNA splicing factors, an important facet in the assembly of an mRNP. The fact that the arginine methylation of the Snp1 homolog U1-70K is conserved in higher eukaryotes suggests that the role of arginine methylation in regulating the spliceosome may also be conserved.
Supplementary Material
Acknowledgments
We thank M. Ares, S. Komili, E. P. Lei, A. McBride, M. Moore, and S. Roberts for critical reading of the manuscript, the Cullen lab members for microscopy assistance, and members of the Yu laboratory for helpful discussions. We also thank Pam Silver for antibodies and yeast strains and Manny Ares for research reagents and technical advice.
This work was supported by a Scientist Development grant (0830279N) from the American Heart Association to M.C.Y.
Footnotes
Published ahead of print on 7 September 2010.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Abovich, N., X. C. Liao, and M. Rosbash. 1994. The yeast MUD2 protein: an interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev. 8:843-854. [DOI] [PubMed] [Google Scholar]
- 2.Abovich, N., and M. Rosbash. 1997. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89:403-412. [DOI] [PubMed] [Google Scholar]
- 3.Bachand, F. 2007. Protein arginine methyltransferases: from unicellular eukaryotes to humans. Eukaryot. Cell 6:889-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedford, M. T., and S. G. Clarke. 2009. Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bedford, M. T., A. Frankel, M. B. Yaffe, S. Clarke, P. Leder, and S. Richard. 2000. Arginine methylation inhibits the binding of proline-rich ligands to Src homology 3, but not WW, domains. J. Biol. Chem. 275:16030-16036. [DOI] [PubMed] [Google Scholar]
- 6.Boisvert, F. M., J. Cote, M. C. Boulanger, P. Cleroux, F. Bachand, C. Autexier, and S. Richard. 2002. Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. J. Cell Biol. 159:957-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boisvert, F. M., J. Cote, M. C. Boulanger, and S. Richard. 2003. A proteomic analysis of arginine-methylated protein complexes. Mol. Cell. Proteomics 2:1319-1330. [DOI] [PubMed] [Google Scholar]
- 8.Bossie, M. A., C. DeHoratius, G. Barcelo, and P. Silver. 1992. A mutant nuclear protein with similarity to RNA binding proteins interferes with nuclear import in yeast. Mol. Biol. Cell 3:875-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan, S. P., D. I. Kao, W. Y. Tsai, and S. C. Cheng. 2003. The Prp19p-associated complex in spliceosome activation. Science 302:279-282. [DOI] [PubMed] [Google Scholar]
- 10.Chen, Y. K., C. H. Leng, H. Olivares, M. H. Lee, Y. C. Chang, W. M. Kung, S. C. Ti, Y. H. Lo, A. H. Wang, C. S. Chang, D. K. Bishop, Y. P. Hsueh, and T. F. Wang. 2004. Heterodimeric complexes of Hop2 and Mnd1 function with Dmc1 to promote meiotic homolog juxtaposition and strand assimilation. Proc. Natl. Acad. Sci. U. S. A. 101:10572-10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng, D., J. Cote, S. Shaaban, and M. T. Bedford. 2007. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell 25:71-83. [DOI] [PubMed] [Google Scholar]
- 12.Cole, C. N., and J. J. Scarcelli. 2006. Transport of messenger RNA from the nucleus to the cytoplasm. Curr. Opin. Cell Biol. 18:299-306. [DOI] [PubMed] [Google Scholar]
- 13.Das, R., K. Dufu, B. Romney, M. Feldt, M. Elenko, and R. Reed. 2006. Functional coupling of RNAP II transcription to spliceosome assembly. Genes Dev. 20:1100-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis, C. A., L. Grate, M. Spingola, and M. Ares, Jr. 2000. Test of intron predictions reveals novel splice sites, alternatively spliced mRNAs and new introns in meiotically regulated genes of yeast. Nucleic Acids Res. 28:1700-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dreyfuss, G., V. N. Kim, and N. Kataoka. 2002. Messenger RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 3:195-205. [DOI] [PubMed] [Google Scholar]
- 16.Enyenihi, A. H., and W. S. Saunders. 2003. Large-scale functional genomic analysis of sporulation and meiosis in Saccharomyces cerevisiae. Genetics 163:47-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gary, J. D., W. J. Lin, M. C. Yang, H. R. Herschman, and S. Clarke. 1996. The predominant protein-arginine methyltransferase from Saccharomyces cerevisiae. J. Biol. Chem. 271:12585-12594. [DOI] [PubMed] [Google Scholar]
- 18.Gavin, A. C., M. Bosche, R. Krause, P. Grandi, M. Marzioch, A. Bauer, J. Schultz, J. M. Rick, A. M. Michon, C. M. Cruciat, M. Remor, C. Hofert, M. Schelder, M. Brajenovic, H. Ruffner, A. Merino, K. Klein, M. Hudak, D. Dickson, T. Rudi, V. Gnau, A. Bauch, S. Bastuck, B. Huhse, C. Leutwein, M. A. Heurtier, R. R. Copley, A. Edelmann, E. Querfurth, V. Rybin, G. Drewes, M. Raida, T. Bouwmeester, P. Bork, B. Seraphin, B. Kuster, G. Neubauer, and G. Superti-Furga. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415:141-147. [DOI] [PubMed] [Google Scholar]
- 19.Gornemann, J., K. M. Kotovic, K. Hujer, and K. M. Neugebauer. 2005. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol. Cell 19:53-63. [DOI] [PubMed] [Google Scholar]
- 20.Green, D. M., K. A. Marfatia, E. B. Crafton, X. Zhang, X. Cheng, and A. H. Corbett. 2002. Nab2p is required for poly(A) RNA export in Saccharomyces cerevisiae and is regulated by arginine methylation via Hmt1p. J. Biol. Chem. 277:7752-7760. [DOI] [PubMed] [Google Scholar]
- 21.Gunderson, F. Q., and T. L. Johnson. 2009. Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet. 5:e1000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo, D., T. R. Hazbun, X. J. Xu, S. L. Ng, S. Fields, and M. H. Kuo. 2004. A tethered catalysis, two-hybrid system to identify protein-protein interactions requiring post-translational modifications. Nat. Biotechnol. 22:888-892. [DOI] [PubMed] [Google Scholar]
- 23.Hacker, I., B. Sander, M. M. Golas, E. Wolf, E. Karagoz, B. Kastner, H. Stark, P. Fabrizio, and R. Luhrmann. 2008. Localization of Prp8, Brr2, Snu114 and U4/U6 proteins in the yeast tri-snRNP by electron microscopy. Nat. Struct. Mol. Biol. 15:1206-1212. [DOI] [PubMed] [Google Scholar]
- 24.Henry, M. F., and P. A. Silver. 1996. A novel methyltransferase (Hmt1p) modifies poly(A)+-RNA-binding proteins. Mol. Cell. Biol. 16:3668-3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hieronymus, H., and P. A. Silver. 2004. A systems view of mRNP biology. Genes Dev. 18:2845-2860. [DOI] [PubMed] [Google Scholar]
- 26.Howe, K. J., C. M. Kane, and M. Ares, Jr. 2003. Perturbation of transcription elongation influences the fidelity of internal exon inclusion in Saccharomyces cerevisiae. RNA 9:993-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huh, W. K., J. V. Falvo, L. C. Gerke, A. S. Carroll, R. W. Howson, J. S. Weissman, and E. K. O'Shea. 2003. Global analysis of protein localization in budding yeast. Nature 425:686-691. [DOI] [PubMed] [Google Scholar]
- 28.Ito, T., T. Chiba, R. Ozawa, M. Yoshida, M. Hattori, and Y. Sakaki. 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. U. S. A. 98:4569-4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kadowaki, T., S. Chen, M. Hitomi, E. Jacobs, C. Kumagai, S. Liang, R. Schneiter, D. Singleton, J. Wisniewska, and A. M. Tartakoff. 1994. Isolation and characterization of Saccharomyces cerevisiae mRNA transport-defective (mtr) mutants. J. Cell Biol. 126:649-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knop, M., K. Siegers, G. Pereira, W. Zachariae, B. Winsor, K. Nasmyth, and E. Schiebel. 1999. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15:963-972. [DOI] [PubMed] [Google Scholar]
- 31.Kosowski, T. R., H. R. Keys, T. K. Quan, and S. W. Ruby. 2009. DExD/H-box Prp5 protein is in the spliceosome during most of the splicing cycle. RNA 15:1345-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kotovic, K. M., D. Lockshon, L. Boric, and K. M. Neugebauer. 2003. Cotranscriptional recruitment of the U1 snRNP to intron-containing genes in yeast. Mol. Cell. Biol. 23:5768-5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kress, T. L., N. J. Krogan, and C. Guthrie. 2008. A single SR-like protein, Npl3, promotes pre-mRNA splicing in budding yeast. Mol. Cell 32:727-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo, M. H., X. J. Xu, H. A. Bolck, and D. Guo. 2009. Functional connection between histone acetyltransferase Gcn5p and methyltransferase Hmt1p. Biochim. Biophys. Acta 1789:395-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwak, Y. T., J. Guo, S. Prajapati, K. J. Park, R. M. Surabhi, B. Miller, P. Gehrig, and R. B. Gaynor. 2003. Methylation of SPT5 regulates its interaction with RNA polymerase II and transcriptional elongation properties. Mol. Cell 11:1055-1066. [DOI] [PubMed] [Google Scholar]
- 36.Lacadie, S. A., and M. Rosbash. 2005. Cotranscriptional spliceosome assembly dynamics and the role of U1 snRNA:5′ss base pairing in yeast. Mol. Cell 19:65-75. [DOI] [PubMed] [Google Scholar]
- 37.Lacadie, S. A., D. F. Tardiff, S. Kadener, and M. Rosbash. 2006. In vivo commitment to yeast cotranscriptional splicing is sensitive to transcription elongation mutants. Genes Dev. 20:2055-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lacoste, N., R. T. Utley, J. M. Hunter, G. G. Poirier, and J. Cote. 2002. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J. Biol. Chem. 277:30421-30424. [DOI] [PubMed] [Google Scholar]
- 39.Laggerbauer, B., T. Achsel, and R. Luhrmann. 1998. The human U5-200kD DEXH-box protein unwinds U4/U6 RNA duplices in vitro. Proc. Natl. Acad. Sci. U. S. A. 95:4188-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lei, E. P., H. Krebber, and P. A. Silver. 2001. Messenger RNAs are recruited for nuclear export during transcription. Genes Dev. 15:1771-1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lei, E. P., and P. A. Silver. 2002. Intron status and 3′-end formation control cotranscriptional export of mRNA. Genes Dev. 16:2761-2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu, Q., and G. Dreyfuss. 1995. In vivo and in vitro arginine methylation of RNA-binding proteins. Mol. Cell. Biol. 15:2800-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longtine, M. S., A. McKenzie III, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953-961. [DOI] [PubMed] [Google Scholar]
- 44.Luna, R., H. Gaillard, C. Gonzalez-Aguilera, and A. Aguilera. 2008. Biogenesis of mRNPs: integrating different processes in the eukaryotic nucleus. Chromosoma 117:319-331. [DOI] [PubMed] [Google Scholar]
- 45.McBride, A. E., J. T. Cook, E. A. Stemmler, K. L. Rutledge, K. A. McGrath, and J. A. Rubens. 2005. Arginine methylation of yeast mRNA-binding protein Npl3 directly affects its function, nuclear export, and intranuclear protein interactions. J. Biol. Chem. 280:30888-30898. [DOI] [PubMed] [Google Scholar]
- 46.McBride, A. E., V. H. Weiss, H. K. Kim, J. M. Hogle, and P. A. Silver. 2000. Analysis of the yeast arginine methyltransferase Hmt1p/Rmt1p and its in vivo function. Cofactor binding and substrate interactions. J. Biol. Chem. 275:3128-3136. [DOI] [PubMed] [Google Scholar]
- 47.Moore, M. J., E. M. Schwartzfarb, P. A. Silver, and M. C. Yu. 2006. Differential recruitment of the splicing machinery during transcription predicts genome-wide patterns of mRNA splicing. Mol. Cell 24:903-915. [DOI] [PubMed] [Google Scholar]
- 48.Morris, D. P., and A. L. Greenleaf. 2000. The splicing factor, Prp40, binds the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 275:39935-39943. [DOI] [PubMed] [Google Scholar]
- 49.Mowen, K. A., J. Tang, W. Zhu, B. T. Schurter, K. Shuai, H. R. Herschman, and M. David. 2001. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell 104:731-741. [DOI] [PubMed] [Google Scholar]
- 50.Ohkura, N., M. Takahashi, H. Yaguchi, Y. Nagamura, and T. Tsukada. 2005. Coactivator-associated arginine methyltransferase 1, CARM1, affects pre-mRNA splicing in an isoform-specific manner. J. Biol. Chem. 280:28927-28935. [DOI] [PubMed] [Google Scholar]
- 51.Perreault, A., C. Lemieux, and F. Bachand. 2007. Regulation of the nuclear poly(A)-binding protein by arginine methylation in fission yeast. J. Biol. Chem. 282:7552-7562. [DOI] [PubMed] [Google Scholar]
- 52.Piruat, J. I., and A. Aguilera. 1998. A novel yeast gene, THO2, is involved in RNA Pol II transcription and provides new evidence for transcriptional elongation-associated recombination. EMBO J. 17:4859-4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raghunathan, P. L., and C. Guthrie. 1998. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr. Biol. 8:847-855. [DOI] [PubMed] [Google Scholar]
- 54.Rezai-Zadeh, N., X. Zhang, F. Namour, G. Fejer, Y. D. Wen, Y. L. Yao, I. Gyory, K. Wright, and E. Seto. 2003. Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes Dev. 17:1019-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rigaut, G., A. Shevchenko, B. Rutz, M. Wilm, M. Mann, and B. Seraphin. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030-1032. [DOI] [PubMed] [Google Scholar]
- 56.Ruby, S. W., T. H. Chang, and J. Abelson. 1993. Four yeast spliceosomal proteins (PRP5, PRP9, PRP11, and PRP21) interact to promote U2 snRNP binding to pre-mRNA. Genes Dev. 7:1909-1925. [DOI] [PubMed] [Google Scholar]
- 57.Russell, I. D., and D. Tollervey. 1992. NOP3 is an essential yeast protein which is required for pre-rRNA processing. J. Cell Biol. 119:737-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen, E. C., M. F. Henry, V. H. Weiss, S. R. Valentini, P. A. Silver, and M. S. Lee. 1998. Arginine methylation facilitates the nuclear export of hnRNP proteins. Genes Dev. 12:679-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Silverman, E. J., A. Maeda, J. Wei, P. Smith, J. D. Beggs, and R. J. Lin. 2004. Interaction between a G-patch protein and a spliceosomal DEXD/H-box ATPase that is critical for splicing. Mol. Cell. Biol. 24:10101-10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith, V., and B. G. Barrell. 1991. Cloning of a yeast U1 snRNP 70K protein homologue: functional conservation of an RNA-binding domain between humans and yeast. EMBO J. 10:2627-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spingola, M., and M. Ares, Jr. 2000. A yeast intronic splicing enhancer and Nam8p are required for Mer1p-activated splicing. Mol. Cell 6:329-338. [DOI] [PubMed] [Google Scholar]
- 62.Staley, J. P., and J. L. Woolford, Jr. 2009. Assembly of ribosomes and spliceosomes: complex ribonucleoprotein machines. Curr. Opin. Cell Biol. 21:109-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tadesse, H., J. Deschenes-Furry, S. Boisvenue, and J. Cote. 2008. KH-type splicing regulatory protein interacts with survival motor neuron protein and is misregulated in spinal muscular atrophy. Hum. Mol. Genet. 17:506-524. [DOI] [PubMed] [Google Scholar]
- 64.Tardiff, D. F., S. A. Lacadie, and M. Rosbash. 2006. A genome-wide analysis indicates that yeast pre-mRNA splicing is predominantly posttranscriptional. Mol. Cell 24:917-929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tardiff, D. F., and M. Rosbash. 2006. Arrested yeast splicing complexes indicate stepwise snRNP recruitment during in vivo spliceosome assembly. RNA 12:968-979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trinkle-Mulcahy, L., S. Boulon, Y. W. Lam, R. Urcia, F. M. Boisvert, F. Vandermoere, N. A. Morrice, S. Swift, U. Rothbauer, H. Leonhardt, and A. Lamond. 2008. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 183:223-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wachtel, C., and J. L. Manley. 2009. Splicing of mRNA precursors: the role of RNAs and proteins in catalysis. Mol. Biosyst. 5:311-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wahl, M. C., C. L. Will, and R. Luhrmann. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136:701-718. [DOI] [PubMed] [Google Scholar]
- 69.Wu, J. Y., and T. Maniatis. 1993. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell 75:1061-1070. [DOI] [PubMed] [Google Scholar]
- 70.Yamagata, K., H. Daitoku, Y. Takahashi, K. Namiki, K. Hisatake, K. Kako, H. Mukai, Y. Kasuya, and A. Fukamizu. 2008. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell 32:221-231. [DOI] [PubMed] [Google Scholar]
- 71.Yu, M. C., F. Bachand, A. E. McBride, S. Komili, J. M. Casolari, and P. A. Silver. 2004. Arginine methyltransferase affects interactions and recruitment of mRNA processing and export factors. Genes Dev. 18:2024-2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu, M. C., D. W. Lamming, J. A. Eskin, D. A. Sinclair, and P. A. Silver. 2006. The role of protein arginine methylation in the formation of silent chromatin. Genes Dev. 20:3249-3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.