

Abstract
High-risk human papillomavirus (HR HPV) requires differentiating epithelial cells to continue to divide in order to replicate the viral DNA. To achieve this, HPV perturbs several regulatory pathways, including cellular apoptosis and senescence signals. HPV E6 has been identified as a regulator of the NFκB signaling pathway, a pathway important in many cellular processes, as well as regulation of virus-host cell interactions. We report here that NFX1-91, an endogenously expressed transcriptional regulator of human telomerase reverse transcriptase (hTERT) that is targeted by HPV type 16 (HPV16) E6/E6-associated protein (E6AP) for degradation, is also critical for regulation of the NFκB pathway by HPV16 E6. Microarray analysis revealed induction of NFκB-responsive genes and reduction of NFκB inhibitors with knockdown of NFX1-91. Knockdown of NFX1-91 induced downregulation of p105, an NFκB inhibitor in both primary human foreskin keratinocytes (HFKs) and HCT116 cells. Chromatin immunoprecipitation assays further confirmed that NFX1-91 bound to the p105 promoter and upregulated its expression. Similarly, in HPV16 E6-positive cells, reduction of p105 expression was observed, paralleling knockdown of NFX1-91 expression. Overall, our data suggest a mechanism for HPV16 E6 activation of the NFκB pathway through NFX1-91. Also, it provides evidence that NFX1-91 can function as a dual regulator, not only a transcriptional repressor, but also a transcriptional activator, when bound to DNA.
Human papillomaviruses (HPVs) are small DNA viruses that infect epithelial tissues. High-risk HPV (HR HPV) types, such as HPV types 16 (HPV16) and 18, encode two main oncoproteins, E6 and E7, which play central roles in viral replication and oncogenic progression (48, 68). The best-characterized function of E6 is to interact with a cellular E3 ubiquitin ligase, E6-associated protein (E6AP). Together, this heterodimer targets the tumor suppressor p53 for degradation, blocking apoptotic and cellular senescence signals due to DNA damage (34, 59). In addition to targeting p53 for degradation, E6 also mediates the E6AP-dependent degradation of several cellular PDZ domain proteins that have roles in signal transduction, transcriptional regulation, receptor assembly, and cellular polarity (24, 27, 34, 40, 44, 53, 64). Furthermore, we and others have previously demonstrated that HPV16 E6 induces human telomerase reverse transcriptase (hTERT) expression in an E6AP-dependent manner (26, 47, 71). In addition to the role of HR HPV E6 in cellular transformation, the E6 protein also has multiple functions important in viral life cycle regulation, including viral DNA replication, episomal maintenance, and cellular antiviral defenses (43, 67).
NFX1 (nuclear factor, X box binding) was originally identified in a screen for proteins that bound to the X-box region of class II major histocompability genes and was implicated in a feedback loop to limit the immune response following infection (62). In a yeast two-hybrid screen, NFX1 was found to be associated with HPV16 E6/E6AP (26). In epithelial cells, NFX1 is expressed as two splice variants, encoding isoforms with identical N termini but unique C termini. Interestingly, both isoforms of NFX1 have been shown to be involved in hTERT regulation, but in opposing ways. The shorter isoform, NFX1-91, represses hTERT expression at the promoter through direct interaction with mSin3A and histone deacetylases (HDACs) to maintain a transcriptionally inactive state at the hTERT promoter (26, 71). The longer isoform, NFX1-123, posttranscriptionally upregulates hTERT expression in concert with cytoplasmic poly(A) binding proteins (PABPCs) by increasing hTERT mRNA stability (37, 38). In the presence of HPV16 E6, E6AP can target only the shorter isoform, NFX1-91, for proteasome-mediated degradation through polyubiquitination of the unique C terminus of NFX1-91, which derepresses hTERT transcriptional expression (26). The differences between the roles of the NFX1 proteins can be explained by their different C termini and their different localizations in cells (37, 38, 71).
Induction of telomerase is a critical step for cell immortalization, transformation, and human cancer development (13). However, it is unclear how the induction of telomerase benefits the HPV viral life cycle. HPV infects basal cells in the cutaneous or mucosal epithelium, and it induces cellular division of the differentiating cells to replicate its own viral DNA and to express its viral capsid proteins for packaging. However, HPV completes its life cycle entirely within the epithelium, and the number of population doublings that an HPV-infected epithelial cell requires to produce virions is unlikely to create critically shortened telomeres. Therefore, the question remains as to how induction of telomerase benefits HPV. Three possible explanations have been considered. First, HPV likely remains latent or persistent in epithelial cells, and the induction of telomerase might confer long-lasting replicative potential. Second, telomerase has activities other than elongation of telomeres that may be beneficial to HPVs. Third, NFX1-91 likely represses many genes in addition to the hTERT gene, and derepression of some of these other genes by E6/E6AP, through degradation of NFX1-91, may be important for the virus life cycle. NFX1-91 is ubiquitously expressed (data not shown) and is known to function as a transcriptional regulator. However, other functions of NFX1-91 have not been well studied. Therefore, we focused on identifying other targets of NFX1-91 that may be beneficial to the HPV life cycle. Using microarray analyses to identify genes that are upregulated when NFX1-91 is knocked down, we found that loss of NFX1-91 expression by stable RNA interference (RNAi) upregulated NFκB signaling pathways. This included upregulation of NFκB-responsive genes and downregulation of NFκB inhibitors.
NFκB/Rel proteins comprise a family of structurally related eukaryotic transcription factors, including RelA (p65), RelB, c-Rel, p50, and p52. These proteins play a central role in immune-mediated inflammatory- and acute-phase responses (31, 36). They have also been implicated in the control of a variety of other physical processes, such as proliferation, cell survival, and cellular differentiation (49, 57). In the epidermis, NFκB signaling has been shown to play important roles in maintaining epithelium homeostasis and cellular differentiation (9, 22, 58, 60, 61). NFκB activation by HPV oncoproteins has been previously reported, suggesting that E6 can modulate NFκB activity and apoptosis via alteration of gene expression (35). Furthermore, expression profile analysis has revealed that NFκB-responsive genes, such as nerve growth factor, nerve growth factor receptor, and inflammatory cytokines, can be induced by E6 expression (28, 54). E6 has also been found to protect cells from tumor necrosis factor (TNF)-induced apoptosis (18, 20, 21).
p105, a precursor of p50, functions as an NFκB inhibitor. Both p105 and p50 play key roles in regulating the NFκB signaling pathway. p105 is widely expressed in cells, and its promoter region contains binding sites for NFκB and other transcription factors, like E2F, Sp1, and Ets (7, 11, 12, 42, 46). The majority of studies on regulation of p105 expression have been limited to its autoregulation by an activated NFκB pathway (65), ensuring a negative-feedback loop. In this study, we show that NFX1-91 regulates p105 expression by binding to its promoter and upregulating its transcriptional activity. This in turn modulates NFκB activity and NFκB-dependent gene expression. This study provides a novel mechanism for HPV16 E6 regulation of the NFκB pathway. Moreover, it provides evidence that NFX1-91, through direct DNA binding, functions not only as a transcriptional repressor, but also as a transcriptional activator.
MATERIALS AND METHODS
Cell culture.
HCT116 and HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco-BRL, Carlsbad, CA) plus 10% fetal bovine serum (FBS) plus penicillin-streptomycin. Primary human foreskin keratinocytes (HFKs) were derived from neonatal foreskin and grown in Epilife medium supplemented with 60 μM calcium chloride and human keratinocyte growth supplement (Cascade Biologics, Portland, OR).
Plasmids and RNAi.
The luciferase reporter construct, driven by the artificial promoter containing four κB sites, and the β-galactosidase (β-Gal) reporter construct were gifts from Zhengui Xia (14). The p105-luciferase reporter construct driven by the p105 promoter was a gift from A. S. Baldwin (11). The HPV early or late promoter region was cloned into the pGL3-luciferase construct by PCR from a plasmid containing the HPV16 genome. The primers were as follows: forward primer, GCGAAGCTTTCTGCAGACCTAGA; reverse primer for the HPV16 early promoter, GATCCATGGCAGTTCTCTTTTGGT; reverse primer for the HPV16 late promoter, GGCAGCCCATGGTAGATTATGG. HPV16 E6 cDNA was cloned into the LXSN vector as previously described (25). NFX1-91 number 1 or number 2 short hairpin RNA (shRNA) (shN91 number 1 or number 2), β-Gal shRNA (shβ-Gal), and the scrambled shRNA (shScr) were cloned into the pLenti-viral vector FUGW, as previously described (71). NFX1-91 small interfering RNA (siRNA) is a mixture of two NFX1-91 siRNAs: number 1 (UCCCACUACUGGGCGUCUA) and number 2 (CAUCGUAUGUUCUCAAUU). The scrambled siRNA is a mixture of two siRNAs: number 1 (UGGUUUACAUGUCGACUAA) and number 2 (UGGUUUACAUGUUUUCUGA).
Transfection and infection.
DNA was transiently transfected into HFK and HCT116 cells using Fugene 6 (Roche, Alameda, CA). siRNA was transiently transfected into HFK and HCT116 cells using RNAiMax (Invitrogen, Carlsbad, CA). Cells stably expressing HPV16 E6 or a control vector, LXSN, and cells stably expressing short hairpin RNA by viral transduction were previously described (25).
Western blot analysis.
Western blotting was performed as previously described (26) with the following antibodies: rabbit anti-p105/p50 (sc-1190), mouse anti-p65 (sc-8008), and mouse anti-nucleolin (sc-8031) (Santa Cruz Biotechnology, Santa Cruz, CA); mouse anti-p53 (Calbiochem, San Diego, CA); and mouse anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Abcam, Cambridge, MA). Rabbit anti-NFX1-91 and the preimmune serum were previously described (26).
Quantitative RT-PCR.
Total RNA was extracted from tissue culture plates using an RNeasy minikit (Qiagen, Valencia, CA). One microgram of total RNA was used to synthesize cDNA using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative PCR using power SYBR green dye (ABI Prism, London, United Kingdom) was conducted on an ABI Prism 9700 and analyzed with SDS 2.2.2 software. The primers used were as follows: p105 primers, F (AGAAGTCTTACCCTCAGGTCAAA) and R (TCCAGCAGTTACAGTGCAGAT); cIAP2 primers, F (TCCTGGATAGTCTACTAACTGCC) and R (GCTTCTTGCAGAGAGTTTCTGAA); IκBa primers, F (TCGCAGTGGACCTGCAAAAT) and R (TGAGCTGGTAGGGAGAATAGC); TNF primers, F (ATGAGCACTGAAAGCATGATCC) and R (GAGGGCTGATTAGAGAGAGGTC). Primers for NFX1-91, hTERT, and 36B4 (as an internal control) used in quantitative RT-PCR were previously described (71).
mRNA stability assay.
HCT116 cells stably expressing shN91 number 1 or shScr were treated with actinomycin D (5 μg/ml; Sigma, St. Louis, MO), and total RNA was collected at each time point indicated following the procedure described above for quantitative RT-PCR. The total amount of p105 mRNA was normalized to the internal-control 36B4 mRNA at each time point, and the ratio at the zero time point was set to 100%. Nonlinear regression was performed with Excel software.
Luciferase reporter assay.
HFKs and HeLa and HCT116 cells stably expressing shN91 number 1 or shScr or cells stably expressing E6 or LXSN were grown to 50 to 60% confluence in 24-well plates and cotransfected with 1.2 μg luciferase reporter and 0.15 μg β-Gal reporter in each well, using a 1:3 DNA/Fugene 6 (Roche, Alameda, CA) ratio. The cells were incubated for 48 h after transfection, rinsed in phosphate-buffered saline (PBS), and lysed in the well by freeze-thawing in 100 μl of reporter lysis buffer (Promega, Madison, WI). Cellular debris was removed by centrifugation. The luminescence was quantified using 25 μl per lysate after mixing it with luciferase assay buffer (Promega, Madison, WI), using a Fluroskan Ascent FL luminometer. Luciferase activity was normalized to either the β-Gal activity or the protein concentration. Each experiment was done multiple times in triplicate.
Chromatin immunoprecipitation assay.
A chromatin immunoprecipitation (ChIP) assay for anti-NFX1-91 antibody was conducted in HCT116 cells using a protocol from Santa Cruz Biotechnology (Santa Cruz, CA). The average chromatin size of the fragments obtained was approximately 400 bp. NFX1-91 was immunoprecipitated by anti-NFX1-91 antibody (with preimmune serum as a control). The primers targeted to the p105 promoter for the ChIP assay included the following: site 1 (nucleotides [nt] −839 to −705), F (AAGCAGCTCAGATGCCAGTG) and R (AGCAATGGTTTTGGTTCACAGC); site 2 (nt −123 to −15), F (CCCTAGAAGTGCGGGCTTCC) and R (TTCCCACTGACGTCGAGAGAGC). The primers targeted to the control β-globin gene promoter for ChIP assay were previously described (71).
Microarray analysis.
Total RNA was purified from TRIzol using an RNeasy kit (Qiagen, Valencia, CA), and 5 μg was used for preparation of the target for array hybridization. An Affymetrix GeneChip human genome HGU133 (A2.0) (Affymetrix, Santa Clara, CA) was used. Target preparation and hybridization were conducted as described in the Affymetrix technical manual. Three independent pools of RNA from sample cells (shN91 number 1) and control cells (shβ-Gal) were included in the analysis. Data were normalized, expression values were generated using dCHIP (http://biosun1.harvard.edu/complab/dchip/) (45), and expression profiles between the two groups were analyzed using CyberT (http://cybert.microarray.ics.uci.edu/). Default CyberT settings were applied to all data sets. Genes were found to be differentially expressed between the two groups by ranking the Bayesian P values (i.e., “Bayes.p”) and using a false-discovery rate (FDR) methodology to account for multiple testing (41). An FDR of 5% was used for all analyses.
GEO Series accession number.
The data discussed in this article have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE23674 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE23674).
RESULTS
Knockdown of NFX1-91 in epithelial cells upregulated the NFκB signaling pathway.
To begin investigating other roles of NFX1-91 in epithelial cells, we carried out gene expression analysis of NFX1-91 downstream targets. NFX1-91 was knocked down by lentiviral stable shRNA expression. NFX1-91 expression was reduced by 90% with shN91 number 1 compared to a control shRNA (shβ-Gal) in HCT116 cells. As the same vector reduced NFX1-91 in HFKs by only 50%, we chose to carry out gene expression analysis in HCT116 cells.
Analysis of deregulated genes following NFX1-91 knockdown indicated activation of NFκB signaling pathways. Knockdown of NFX1-91 induced expression of NFκB-responsive inflammatory cytokines (interleukins [ILs]), inflammatory cytokine receptors (ILRs), tumor necrosis factor receptors (TNFRs), CXCR4, and the apoptotic regulator BCL2 (Table 1). Furthermore, reduction of NFX1-91 also induced downregulation of the NFκB inhibitors p105 (NFκB1), IκBα (NFκBIA), and IκBɛ (NFκBIE) (Table 1), all of which function to sequester active NFκB members in the cytoplasm.
TABLE 1.
Deregulated genes involved in the NFκB signaling pathway when NFX1-91 was knocked downa
| Affymetrix identifier | UniGene identifier | Symbol | Bayes.lnp | Fold change |
|---|---|---|---|---|
| 205403_at | Hs. 25333 | IL-1R2 | 0.00297 | 2.96 |
| 205227_at | Hs. 143527 | IL-1RAP | 0.00017 | 2.51 |
| 206467_x_at | Hs. 348183 | TNFRSF6B | 1.22E−10 | 2.06 |
| 205992_s_at | Hs. 528402 | IL-15 | 3.83E−06 | 2.05 |
| 204773_at | Hs. 204891 | IL-11RA | 8.92E−05 | 2.02 |
| 217028_at | Hs. 421986 | CXCR4 | 0.00017 | 1.98 |
| 204781_s_at | Hs. 82359 | TNFRSF6 | 9.72E−09 | 1.88 |
| 204780_s_at | Hs. 82359 | TNFRSF6 | 7.18E−10 | 1.83 |
| 216252_x_at | Hs. 82359 | TNFRSF6 | 3.13E−07 | 1.82 |
| 207160_at | Hs. 673 | IL-12A | 0.00087 | 1.8 |
| 215719_x_at | Hs. 82359 | TNFRSF6 | 7.54E−08 | 1.78 |
| 202948_at | Hs. 82112 | IL-1R1 | 2.72E−06 | 1.77 |
| 210654_at | Hs. 129844 | TNFRSF10D | 3.96E−05 | 1.7 |
| 205945_at | Hs. 193400 | IL-6R | 0.00092 | 1.69 |
| 217371_s_at | Hs. 528402 | IL-15 | 0.000222 | 1.64 |
| 210916_s_at | Hs. 306278 | CD44 | 5.02E−05 | 1.57 |
| 221463_at | Hs. 247838 | CCL24 | 0.00968 | 1.56 |
| 203684_s_at | Hs. 79241 | BCL2 | 0.008922 | 1.55 |
| 201502_s_at | Hs. 81328 | NFκBIA | 5.01E−06 | −1.5 |
| 203927_at | Hs. 458276 | NFκBIE | 0.00011 | −1.53 |
| 209239_at | Hs. 160557 | NFκB1 | 2.17E−12 | −2.45 |
Three independent Affymetrix microarray analyses were performed in HCT116 cells stably expressing shN91 number 1 or shβ-Gal. Bayes.lnp, P value associated with the t test for log-transformed data.
As it appeared that NFX1-91 directly affected multiple regulators of the NFκB pathway, we wanted to determine whether this effect was achieved through an increase in NFκB activity. We transfected a luciferase reporter driven by an artificial promoter containing four consecutive κB binding sites in HFKs and HCT116 and HeLa cells. Knockdown of NFX1-91 increased luciferase activity by 2-fold in HCT116 cells and HFKs and 3-fold in HeLa cells compared to the control cells (Fig. 1A).
FIG. 1.

Knockdown of NFX1-91 induced NFκB-responsive genes by activating NFκB signaling. (A) Knockdown of NFX1-91 induced NFκB activity. HCT116 cells, HFKs, and HeLa cells with knockdown of NFX1-91 by RNA interference were transiently transfected with a luciferase reporter driven by four κB sites, and luciferase activity was significantly increased in all three cell types compared to the control cells (*, P < 0.05). The data are representative of two independent experiments, each of which was conducted in triplicate. (B and C) Confirmation of gene expression in HCT116 cells and HFKs. Total RNA was extracted from HCT116 cells and HFKs stably expressing shN91 number 1 or shScr and subjected to qPCR analysis with 36B4 as an internal control. The error bars indicate standard errors.
We next confirmed changes in gene expression first seen in our microarray analysis through quantitative PCR (qPCR). As shown in Fig. 1B, NFX1-91 mRNA was reduced to less than 10% by shN91 number 1 in HCT116 cells. Upregulation of TNF by 2-fold and downregulation of p105 and IκBα by 50% and 20%, respectively, were detected in shN91 number 1 cells (Fig. 1B). cIAP2, an NFκB-responsive gene often used as an indicator of upregulation of NFκB activity, was also increased by 2-fold (Fig. 1B).
The expression levels of these genes were also examined by qPCR in HFKs when NFX1-91 expression was reduced to 40% in shN91 number 1 HFKs. As shown in Fig. 1C, the cIAP2 mRNA level increased by more than 2-fold in shN91 number 1 HFKs. Furthermore, the mRNA levels of p105 and IκBα were reduced to 50% and 70%, respectively (Fig. 1C). hTERT mRNA levels increased by 3-fold in HFKs (Fig. 1C), which is consistent with what we previously reported (71). It should be noted that we did not detect significant change in the expression of hTERT in shN91 number 1 HCT116 cells (Fig. 1B); we attribute this to the high basal level of hTERT expression in this cell type.
We wanted to confirm that the knockdown of NFX1-91 was needed to decrease p105. Therefore, we designed a second shRNA target, the unique C terminus of NFX1-91 (shN91 number 2). Like shN91 number 1, shN91 number 2 efficiently reduced the mRNA level of NFX1-91 by 90% in HCT116 cells (Fig. 2A), and shN91 number 2 decreased the mRNA level of p105 similarly (Fig. 2A).
FIG. 2.

Knockdown of NFX1-91 decreased p105 expression. (A) qPCR of the mRNA levels of NFX1-91 and p105 normalized to 36B4 in HCT116 cells stably expressing two different NFX1-91 shRNAs (shN91 number 1 and number 2). The error bars indicate standard errors. (B) Western blot analysis in both HeLa and HCT116 cells (with nucleolin as a loading control) showed that protein levels of p105 and its cleavage product, p50, were reduced in NFX1-91 knockdown cells. (C) Western blot analysis in HeLa and C33A cells (with GAPDH as a loading control) showed that protein levels of both NFX1-91 and p53 were reduced in HeLa cells compared to C33A cells.
We confirmed a decrease in p105 protein expression by Western blot analysis. As shown in Fig. 2B, NFX1-91 protein levels were decreased in both HeLa and HCT116 cells expressing shN91 number 1. As expected, p105 and its cleavage product, p50, were reduced in both cell types with NFX1-91 knockdown (Fig. 2B). We did not detect any change in the protein levels of other NFκB members, such as p65, in NFX1-91 knockdown cells (Fig. 2B).
We have previously shown that high-risk HPV16 E6 can induce NFX1-91 degradation (26, 71). Although HeLa cells are an HPV18-positive cervical cancer cell line, the NFX1-91 protein was detected at similar levels in HeLa cells and HCT116 cells, as shown in Fig. 2B. To further test the protein level of NFX1-91 in HPV18-positive cells, we compared NFX1-91 protein levels in HeLa cells and a corresponding HPV-negative cervical cancer cell line, C33A. As shown in Fig. 2C, we detected lower expression of NFX1-91 and p53 in HeLa cells than in C33A cells.
NFX1-91 regulated p105 gene promoter activity.
An mRNA half-life assay was performed to determine whether the decrease in p105 mRNA seen when NFX1-91 was knocked down was caused by its destabilization. HCT116 cells stably expressing shN91 number 1 or shScr were treated with actinomycin D to inhibit new transcription. Total RNA was collected at the time points indicated in Fig. 3A, and the relative p105 mRNA level was normalized to an internal control, 36B4. No significant difference in RNA stability was detected (P = 0.503) (Fig. 3A), excluding the possibility that NFX1-91 regulated p105 expression by affecting its mRNA stability.
FIG. 3.

Knockdown of NFX1-91 decreased p105 transcription. (A) p105 mRNA stability assay. HCT116 cells stably expressing shN91 number 1 or shScr cells were treated with actinomycin D, and total RNA was extracted at the indicated time points and detected by qPCR. The RNA ratio was normalized to an internal control, 36B4. (B) Knockdown of NFX1-91 decreased p105 promoter activity. HCT116 cells with knockdown of NFX1-91 were transiently cotransfected with a luciferase reporter driven by the p105 promoter and a β-Gal reporter. Luciferase activity was significantly reduced in NFX1-91 knockdown cells compared to the control cells (*, P < 0.02). These data are representative of two independent experiments, each of which was conducted in triplicate. The error bars indicate standard errors.
To test the possibility that NFX1-91 regulated p105 transcriptionally, a reporter construct driven by the natural core promoter of the p105 gene was tested in NFX1-91 knockdown HCT116 cells. Luciferase expression was reduced by half when NFX1-91 was knocked down (Fig. 3B), indicating that NFX1-91 regulated p105 expression transcriptionally and not posttranscriptionally.
NFX1-91 bound the p105 promoter.
We have previously shown that NFX1-91 directly binds to the hTERT promoter and functions as a transcriptional repressor of hTERT (26, 71). To test whether NFX1-91 could bind the p105 promoter, we performed ChIP assays in HCT116 cells using an antibody to NFX1-91 and probed with two sets of primers that targeted the p105 core promoter (Fig. 4A). As shown in Fig. 4B, a 2-fold enrichment of NFX1-91 was repeatedly detected at both sites compared to a negative-control site on the β-globin gene. To further test the specificity of NFX1-91 binding to the p105 promoter, ChIP assays were performed in cells with NFX1-91 knockdown. With the knockdown of NFX1-91, the binding of NFX1-91 to the p105 promoter was abolished (Fig. 4C). These results together indicated that NFX1-91 likely regulated p105 activity by directly binding to its promoter.
FIG. 4.

NFX1-91 bound the p105 promoter. (A) Locations of the two pairs of primers designed for ChIP assay at the p105 promoter (TSS, transcription start site). (B) ChIP assay in HCT116 cells using NFX1-91-specific antibody (with β-globin as a negative control) indicated that NFX1-91 bound to the p105 promoter, with similar levels at both sites. (C) ChIP assay in HCT116 cells stably expressing shN91 (number 1 and number 2) or shScr indicated that knockdown of NFX1-91 abolished the binding of NFX1-91 to the p105 promoter at both sites. The error bars represent standard errors of multiple PCRs from two independent experiments.
HPV16 E6 expression mimicked knockdown of NFX1-91 on p105 expression.
We previously showed that HPV16 E6 expression can target NFX1-91 for proteasome-mediated degradation (26, 71). The effect of E6 expression therefore would be similar to that of knockdown of NFX1-91 expression by shRNA. To test this, we stably infected both HFKs and HCT116 cells with HPV16 E6 or a vector control, LXSN. E6 expression and function were assayed indirectly by examining the levels of p53 protein and hTERT mRNA. As shown in Fig. 5A, E6 expression led to degradation of p53 protein in both HFKs and HCT116 cells. As expected, E6 induced hTERT expression in HFKs 5-fold and in HCT116 cells 2- to 3-fold versus control cells (Fig. 5B). Moreover, we also found that E6 upregulated cIAP2 mRNA levels by 2- to 3-fold in both cell types (Fig. 5B), consistent with previous reports (35). HCT116 cells stably expressing E6 were used for a p105 luciferase reporter assay. E6 expression reduced luciferase expression to less than 20% (Fig. 5C). Furthermore, protein levels of both p105 and p50 were reduced in E6-expressing HFKs and HCT116 cells (Fig. 5D). These results were consistent with our data obtained by knockdown of NFX1-91.
FIG. 5.

E6 expression decreased the expression level of p105, which mimicked the effect of NFX1-91 knockdown in HFKs. (A) Western blot analysis (with GAPDH as a loading control) showed that p53 levels were reduced in both HFKs and HCT116 cells expressing E6 compared to the LXSN control cells. (B) qPCR showed that the mRNA levels of hTERT and cIAP2 were induced in E6-expressing cells. (C) E6 expression decreased p105 promoter activity. Cells stably expressing E6 or LXSN were transiently transfected with a luciferase reporter driven by the core p105 promoter. (D) Western blot analysis (with nucleolin as a loading control) showed that the levels of p105 and p50 were reduced in E6-expressing cells. The numbers under the rows indicate the quantification of each signal, performed using Image J software. (E) E6 expression decreased both HPV early and late promoter activities. Cells stably expressing E6 or LXSN were transiently transfected with a luciferase reporter driven by either the early or late HPV promoter. (F) Knockdown of NFX1-91 decreased both HPV early and late promoter activities. NFX1-91 knockdown cells were transiently transfected with a luciferase reporter driven by either the early or late HPV promoter. The data in panels C, E, and F are representative of two independent experiments, each of which was conducted in triplicate. The error bars indicate standard errors.
It has been shown that a functional NFκB site on the HPV long control region (LCR) negatively regulates HPV promoter activity (23). To further test the possible effect of NFκB activity induced by E6 expression or NFX1-91 degradation on HPV gene regulation, we transiently transfected HPV16 early and late promoter-driven luciferase reporter constructs in E6-expressing or NFX1-91 knockdown cells, respectively. As shown in Fig. 5E and F, expression of E6 or knockdown of NFX1-91 was found to be able to similarly reduce luciferase expression driven by either the HPV early or late promoter, suggesting E6 expression or knockdown of NFX1-91 downregulates HPV16 early and late promoter activities. Altogether, these results indicated that NFX1-91 plays a specific role in E6-regulated NFκB activity in general, which is important for the HPV viral life cycle.
DISCUSSION
Infection with HR HPVs is a major risk factor for the development of cervical cancer (5, 73, 74). Two main oncoproteins, E6 and E7, contribute to the immortalization and transformation of HR HPV-infected cells (48, 51). E7 binds and targets retinoblastoma protein (Rb) for degradation, allowing the activation of E2F target genes involved in cell cycle progression (19, 52). E6 targets p53 for degradation and activates hTERT expression (34, 71). The NFκB pathway is an important signaling pathway implicated in many cellular physiological processes, such as antiapoptosis, cell proliferation, cell migration, and angiogenesis (30, 32, 56). Moreover, the NFκB signaling pathway plays important roles in regulating the immune response to viral infection (56). Many studies have suggested that HPV infection regulates NFκB activity, and E6 and E7 specifically have been shown to be involved in regulating the NFκB signaling pathway (2, 17, 35, 63, 72). However, the mechanisms of HPV-induced NFκB activity have remained unclear.
NFX1-91 is a novel target of the E6/E6AP E3 ubiquitin ligase complex (26). Degradation of NFX1-91 induced by E6 expression activates hTERT expression (26, 71). Here, we found that, in addition to the activation of hTERT expression, knockdown of NFX1-91 by shRNA also induced NFκB-responsive genes and reduced the expression of NFκB inhibitors by microarray analysis. Specifically, we demonstrated that NFX1-91 bound the p105 promoter in vivo and activated its transcription in epithelial cells (Fig. 6). Reduction of NFX1-91, either by knockdown of NFX1-91 by RNA interference or by E6 expression-induced degradation, led to a reduction in p105 expression and activation of the NFκB signaling pathway (Fig. 6). p105 is a member of the IκB family that sequesters NFκB dimers in the cytoplasm and plays important roles in regulating both the NFκB pathway and the immune response (4, 55). The decrease in p105 expression also led to the reduction of its cleavage product, p50 (Fig. 2B), which functions as an active NFκB member in the canonical NFκB pathway. Therefore, reduction of the p50 protein in NFX1-91 knockdown cells indicates that knockdown of NFX1-91-induced NFκB activation may be through an NFκB complex that is independent of p50. Previous studies have shown that the low basal level of p105 expression in T-All cells results in the formation and activation of the atypical p65/c-Rel complex in the nucleus and its increased transcriptional activity (8, 70). One study has indicated the formation of nuclear p52-containing NFκB complexes induced by E6 expression to an NFκB binding element induces NFκB-responsive promoter activity and expression of the NFκB-responsive cIAP2-encoding gene (35). Induced NFκB activity has been shown to play important roles in multiple physiological processes. Specifically, for the HPV life cycle, induced NFκB activity has been shown to regulate antiapoptosis, epithelial homeostasis, and HPV gene regulation (35, 58, 60, 61). Here, we have demonstrated that either E6 expression or NFX1-91 knockdown downregulates HPV16 promoter activity to affect the HPV viral life cycle (Fig. 6).
FIG. 6.
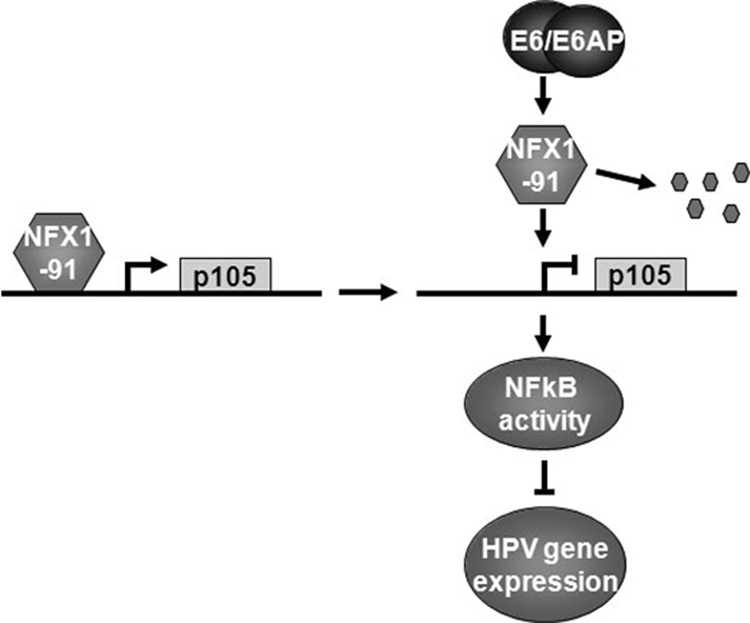
Model of NFX1-91 involved in E6-activated NFκB activity. NFX1-91 binds to the p105 promoter and activates p105 expression. E6/E6AP induces degradation of NFX1-91, which reduces expression of p105 and its cleavage product, p50, and possibly induces the formation of NFκB heterodimers lacking p50, such as p65/c-Rel. These NFκB complexes inhibit HPV gene expression.
TNF is a well-studied activator to induce NFκB activity through the canonical p50-dependent NFκB pathway. TNF treatment (10 ng/ml) in both HCT116 and HeLa cells can induce about a 3-fold increase in NFκB activity (data not shown). Knockdown of NFX1-91 in TNF-treated cells did not significantly change NFκB activity induced by TNF (data not shown), which is reasonable, because knockdown of NFX1-91 increased NFκB activity by reducing the expression of an NFκB inhibitor, p105, and possibly inducing the formation of an NFκB complex, which is independent of p50. However, reduction of p105 and p50 expression impaired TNF-induced NFκB activity to some degree, so the overall NFκB activity is regulated by the balance of TNF and knockdown of NFX1-91.
p53 is another E6 target for degradation, and cross talk between p53 and the NFκB signaling pathway has been proposed (66). By comparing the HCT116 wild-type and p53-null cells, we detected about a two-fold increase in NFκB activity in p53-null HCT116 cells (data not shown). However, knockdown of NFX1-91 in p53-null HCT116 cells further increased NFκB activity, suggesting that E6 utilizes many mechanisms to regulate NFκB activity and that both p53 and NFX1-91 play roles in E6-induced NFκB activity. E6 has also been shown to be able to induce NFκB activity under hypoxia conditions (35). Under hypoxia conditions, E6 can specifically induce degradation of CYLD, an inhibitor of the NFκB pathway, to activate the NFκB pathway (2). This was not seen under normoxia conditions, suggesting that E6 utilizes different mechanisms to regulate NFκB activity under different conditions.
Although upregulation of p105 and p100 has been reported in some HPV-positive oral cancer and cervical cancer tissues (6, 50), the upregulation of both p105 and p100 has been shown to be highly associated with the increased severity of these lesions (6, 50). It is likely that expression of p105 and p100 is induced with the progression of disease. In our study, we consistently detected the reduction of p105 expression in NFX1-91 knockdown cells and E6-expressing cells. No significant changes in p100 expression were detected (data not shown). Another report has shown increased p105 expression, with a differential subcellular localization, in E6-, E7-, or E6/E7-overexpressing HFKs (29). These data were partially explained by the expression of different splice variants of E6. It is possible that the different splice variants of E6 have different effects on p105 expression. In our study, we examined the expression of E6 functionally by testing knockdown of p53 and induction of hTERT expression.
In addition to regulating NFκB activity, p105 has also been shown to play important roles in regulating the mitogen-activated protein kinase (MAPK) pathway by interacting with Tpl2, a MAPK kinase, to maintain Tpl2 protein stability and inhibit its kinase activity. Loss of p105 releases Tpl2 and induces rapid activation of MAPK signaling pathways (3, 10, 69). Both NFκB and MAPK signaling are involved in regulating multiple physiological processes and play important roles in regulating epidermis homeostasis and cell differentiation in the epidermis (39, 60, 61). Current studies on HPV-induced NFκB activity have focused on its proliferative capacity and cell protection from apoptosis in tissue culture. It will be interesting to determine the cross talk between the NFκB and MAPK pathways and the roles they play in epithelial cells and/or epidermis formation that is important for HPV.
We previously showed that NFX1-91 functions as a transcriptional repressor of hTERT by interacting with the mSin3A-HDAC complex to keep chromatin inactive in primary HFKs (26, 71). Here, we demonstrated that NFX1-91 bound the p105 promoter in vivo and activated its transcription in epithelial cells. Our findings are the first to indicate that NFX1-91 may function as a dual regulator, both a repressor and an activator. Previous studies have revealed that transcription regulators can switch between activator and repressor depending upon both protein-protein interactions and the promoter sequence context (15, 16, 33). There are two possible reasons that NFX1-91 could be a dual regulator. NFX1-91 has a PHD motif in the N terminus, which has been shown to be involved in regulating protein-protein interactions and chromatin-mediated transcriptional regulation (1). Therefore, it is likely that NFX1-91 interacts with different proteins that affect the NFX1-91 regulatory function on different genes in trans. Second, two X-box-like elements in opposite orientations were found at the p105 promoter, which are located at nt −810 to −797 and −113 to −100, respectively. It is possible that the different DNA contexts and structures affect the NFX1-91 cis regulatory function at genes. Further studies to determine other proteins that function with NFX1-91 and the chromatin structure surrounding the X boxes to which NFX1-91 bind will be considered.
Acknowledgments
We thank Kristin L. Robinson for assisting with tissue culture; Portia Vliet-Gregg for critical reading of the manuscript; Jeffrey J. Delrow, Ryan S. Basom, and Andy Marty in the genomic facility at FHCRC for helping with array analysis; and Zhengui Xia and Albert S. Baldwin for providing multiple luciferase reporter constructs used in this study.
Footnotes
Published ahead of print on 25 August 2010.
REFERENCES
- 1.Aasland, R., T. J. Gibson, and A. F. Stewart. 1995. The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem. Sci. 20:56-59. [DOI] [PubMed] [Google Scholar]
- 2.An, J., D. Mo, H. Liu, M. S. Veena, E. S. Srivatsan, R. Massoumi, and M. B. Rettig. 2008. Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell 14:394-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babu, G. R., W. Jin, L. Norman, M. Waterfield, M. Chang, X. Wu, M. Zhang, and S. C. Sun. 2006. Phosphorylation of NF-kappaB1/p105 by oncoprotein kinase Tpl2: implications for a novel mechanism of Tpl2 regulation. Biochim. Biophys. Acta 1763:174-181. [DOI] [PubMed] [Google Scholar]
- 4.Beinke, S., and S. C. Ley. 2004. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 382:393-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosch, F. X., M. M. Manos, N. Munoz, M. Sherman, A. M. Jansen, J. Peto, M. H. Schiffman, V. Moreno, R. Kurman, and K. V. Shah. 1995. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J. Natl. Cancer Inst. 87:796-802. [DOI] [PubMed] [Google Scholar]
- 6.Branca, M., C. Giorgi, M. Ciotti, D. Santini, L. Di Bonito, S. Costa, A. Benedetto, D. Bonifacio, P. Di Bonito, P. Paba, L. Accardi, L. Mariani, M. Ruutu, S. Syrjanen, C. Favalli, and K. Syrjanen. 2006. Upregulation of nuclear factor-kappaB (NF-kappaB) is related to the grade of cervical intraepithelial neoplasia, but is not an independent predictor of high-risk human papillomavirus or disease outcome in cervical cancer. Diagn. Cytopathol. 34:555-563. [DOI] [PubMed] [Google Scholar]
- 7.Chang, P. Y., K. Draheim, M. A. Kelliher, and S. Miyamoto. 2006. NFKB1 is a direct target of the TAL1 oncoprotein in human T leukemia cells. Cancer Res. 66:6008-6013. [DOI] [PubMed] [Google Scholar]
- 8.Chang, P. Y., and S. Miyamoto. 2006. Nuclear factor-kappaB dimer exchange promotes a p21(waf1/cip1) superinduction response in human T leukemic cells. Mol. Cancer Res. 4:101-112. [DOI] [PubMed] [Google Scholar]
- 9.Chaturvedi, V., J. Z. Qin, M. F. Denning, D. Choubey, M. O. Diaz, and B. J. Nickoloff. 1999. Apoptosis in proliferating, senescent, and immortalized keratinocytes. J. Biol. Chem. 274:23358-23367. [DOI] [PubMed] [Google Scholar]
- 10.Cho, J., and P. N. Tsichlis. 2005. Phosphorylation at Thr-290 regulates Tpl2 binding to NF-kappaB1/p105 and Tpl2 activation and degradation by lipopolysaccharide. Proc. Natl. Acad. Sci. U. S. A. 102:2350-2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cogswell, P. C., M. W. Mayo, and A. S. Baldwin, Jr. 1997. Involvement of Egr-1/RelA synergy in distinguishing T cell activation from tumor necrosis factor-alpha-induced NF-kappa B1 transcription. J. Exp. Med. 185:491-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cogswell, P. C., R. I. Scheinman, and A. S. Baldwin, Jr. 1993. Promoter of the human NF-kappa B p50/p105 gene. Regulation by NF-kappa B subunits and by c-REL. J. Immunol. 150:2794-2804. [PubMed] [Google Scholar]
- 13.Counter, C. M., A. A. Avilion, C. E. LeFeuvre, N. G. Stewart, C. W. Greider, C. B. Harley, and S. Bacchetti. 1992. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 11:1921-1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cude, K., Y. Wang, H. J. Choi, S. L. Hsuan, H. Zhang, C. Y. Wang, and Z. Xia. 2007. Regulation of the G2-M cell cycle progression by the ERK5-NFkappaB signaling pathway. J. Cell Biol. 177:253-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai, X., and L. B. Rothman-Denes. 1999. DNA structure and transcription. Curr. Opin. Microbiol. 2:126-130. [DOI] [PubMed] [Google Scholar]
- 16.Dove, S. L., J. K. Joung, and A. Hochschild. 1997. Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature 386:627-630. [DOI] [PubMed] [Google Scholar]
- 17.Du, J., G. G. Chen, A. C. Vlantis, H. Xu, R. K. Tsang, and A. C. van Hasselt. 2003. The nuclear localization of NFkappaB and p53 is positively correlated with HPV16 E7 level in laryngeal squamous cell carcinoma. J. Histochem Cytochem. 51:533-539. [DOI] [PubMed] [Google Scholar]
- 18.Duerksen-Hughes, P. J., J. Yang, and S. B. Schwartz. 1999. HPV 16 E6 blocks TNF-mediated apoptosis in mouse fibroblast LM cells. Virology 264:55-65. [DOI] [PubMed] [Google Scholar]
- 19.Dyson, N., P. M. Howley, K. Munger, and E. Harlow. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934-937. [DOI] [PubMed] [Google Scholar]
- 20.Filippova, M., T. A. Brown-Bryan, C. A. Casiano, and P. J. Duerksen-Hughes. 2005. The human papillomavirus 16 E6 protein can either protect or further sensitize cells to TNF: effect of dose. Cell Death Differ. 12:1622-1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Filippova, M., H. Song, J. L. Connolly, T. S. Dermody, and P. J. Duerksen-Hughes. 2002. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J. Biol. Chem. 277:21730-21739. [DOI] [PubMed] [Google Scholar]
- 22.Fisher, G. J., S. C. Datta, H. S. Talwar, Z. Q. Wang, J. Varani, S. Kang, and J. J. Voorhees. 1996. Molecular basis of sun-induced premature skin ageing and retinoid antagonism. Nature 379:335-339. [DOI] [PubMed] [Google Scholar]
- 23.Fontaine, V., E. van der Meijden, J. de Graaf, J. ter Schegget, and L. Struyk. 2000. A functional NF-kappaB binding site in the human papillomavirus type 16 long control region. Virology 272:40-49. [DOI] [PubMed] [Google Scholar]
- 24.Gardiol, D., C. Kuhne, B. Glaunsinger, S. S. Lee, R. Javier, and L. Banks. 1999. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 18:5487-5496. [DOI] [PubMed] [Google Scholar]
- 25.Gewin, L., and D. A. Galloway. 2001. E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-myc. J. Virol. 75:7198-7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gewin, L., H. Myers, T. Kiyono, and D. A. Galloway. 2004. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 18:2269-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glaunsinger, B. A., S. S. Lee, M. Thomas, L. Banks, and R. Javier. 2000. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 19:5270-5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Havard, L., P. Delvenne, P. Frare, J. Boniver, and S. L. Giannini. 2002. Differential production of cytokines and activation of NF-kappaB in HPV-transformed keratinocytes. Virology 298:271-285. [DOI] [PubMed] [Google Scholar]
- 29.Havard, L., S. Rahmouni, J. Boniver, and P. Delvenne. 2005. High levels of p105 (NFKB1) and p100 (NFKB2) proteins in HPV16-transformed keratinocytes: role of E6 and E7 oncoproteins. Virology 331:357-366. [DOI] [PubMed] [Google Scholar]
- 30.Hayden, M. S., and S. Ghosh. 2004. Signaling to NF-kappaB. Genes Dev. 18:2195-2224. [DOI] [PubMed] [Google Scholar]
- 31.Hayden, M. S., A. P. West, and S. Ghosh. 2006. NF-kappaB and the immune response. Oncogene 25:6758-6780. [DOI] [PubMed] [Google Scholar]
- 32.Hayden, M. S., A. P. West, and S. Ghosh. 2006. SnapShot: NF-kappaB signaling pathways. Cell 127:1286-1287. [DOI] [PubMed] [Google Scholar]
- 33.Hochschild, A., and S. L. Dove. 1998. Protein-protein contacts that activate and repress prokaryotic transcription. Cell 92:597-600. [DOI] [PubMed] [Google Scholar]
- 34.Huibregtse, J. M., M. Scheffner, and P. M. Howley. 1994. E6-AP directs the HPV E6-dependent inactivation of p53 and is representative of a family of structurally and functionally related proteins. Cold Spring Harbor Symp. Quant. Biol. 59:237-245. [DOI] [PubMed] [Google Scholar]
- 35.James, M. A., J. H. Lee, and A. J. Klingelhutz. 2006. Human papillomavirus type 16 E6 activates NF-kappaB, induces cIAP-2 expression, and protects against apoptosis in a PDZ binding motif-dependent manner. J. Virol. 80:5301-5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karin, M., and F. R. Greten. 2005. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5:749-759. [DOI] [PubMed] [Google Scholar]
- 37.Katzenellenbogen, R. A., E. M. Egelkrout, P. Vliet-Gregg, L. C. Gewin, P. R. Gafken, and D. A. Galloway. 2007. NFX1-123 and poly(A) binding proteins synergistically augment activation of telomerase in human papillomavirus type 16 E6-expressing cells. J. Virol. 81:3786-3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katzenellenbogen, R. A., P. Vliet-Gregg, M. Xu, and D. A. Galloway. 2009. NFX1-123 increases hTERT expression and telomerase activity post-transcriptionally in HPV 16E6 keratinocytes. J. Virol. [DOI] [PMC free article] [PubMed]
- 39.Khavari, T. A., and J. Rinn. 2007. Ras/Erk MAPK signaling in epidermal homeostasis and neoplasia. Cell Cycle 6:2928-2931. [DOI] [PubMed] [Google Scholar]
- 40.Kiyono, T., A. Hiraiwa, M. Fujita, Y. Hayashi, T. Akiyama, and M. Ishibashi. 1997. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. U. S. A. 94:11612-11616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klipper-Aurbach, Y., M. Wasserman, N. Braunspiegel-Weintrob, D. Borstein, S. Peleg, S. Assa, M. Karp, Y. Benjamini, Y. Hochberg, and Z. Laron. 1995. Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulin-dependent diabetes mellitus. Med. Hypotheses 45:486-490. [DOI] [PubMed] [Google Scholar]
- 42.Lambert, P. F., M. J. Ludford-Menting, N. J. Deacon, I. Kola, and R. R. Doherty. 1997. The nfkb1 promoter is controlled by proteins of the Ets family. Mol. Biol. Cell 8:313-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee, C., and L. A. Laimins. 2004. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 78:12366-12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee, S. S., B. Glaunsinger, F. Mantovani, L. Banks, and R. T. Javier. 2000. Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J. Virol. 74:9680-9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li, C., and W. H. Wong. 2001. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl. Acad. Sci. U. S. A. 98:31-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li, Z., X. Wang, R. Y. Yu, B. B. Ding, J. J. Yu, X. M. Dai, A. Naganuma, E. R. Stanley, and B. H. Ye. 2005. BCL-6 negatively regulates expression of the NF-kappaB1 p105/p50 subunit. J. Immunol. 174:205-214. [DOI] [PubMed] [Google Scholar]
- 47.Liu, X., H. Yuan, B. Fu, G. L. Disbrow, T. Apolinario, V. Tomaic, M. L. Kelley, C. C. Baker, J. Huibregtse, and R. Schlegel. 2005. The E6AP ubiquitin ligase is required for transactivation of the hTERT promoter by the human papillomavirus E6 oncoprotein. J. Biol. Chem. 280:10807-10816. [DOI] [PubMed] [Google Scholar]
- 48.Mansur, C. P., and E. J. Androphy. 1993. Cellular transformation by papillomavirus oncoproteins. Biochim. Biophys. Acta 1155:323-345. [DOI] [PubMed] [Google Scholar]
- 49.Mercurio, F., and A. M. Manning. 1999. Multiple signals converging on NF-kappaB. Curr. Opin. Cell Biol. 11:226-232. [DOI] [PubMed] [Google Scholar]
- 50.Mishra, A., A. C. Bharti, P. Varghese, D. Saluja, and B. C. Das. 2006. Differential expression and activation of NF-kappaB family proteins during oral carcinogenesis: role of high risk human papillomavirus infection. Int. J. Cancer 119:2840-2850. [DOI] [PubMed] [Google Scholar]
- 51.Münger, K., W. C. Phelps, V. Bubb, P. M. Howley, and R. Schlegel. 1989. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 63:4417-4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Münger, K., B. A. Werness, N. Dyson, W. C. Phelps, E. Harlow, and P. M. Howley. 1989. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 8:4099-4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakagawa, S., and J. M. Huibregtse. 2000. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 20:8244-8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nees, M., J. M. Geoghegan, T. Hyman, S. Frank, L. Miller, and C. D. Woodworth. 2001. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J. Virol. 75:4283-4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pereira, S. G., and F. Oakley. 2008. Nuclear factor-kappaB1: regulation and function. Int. J. Biochem. Cell Biol. 40:1425-1430. [DOI] [PubMed] [Google Scholar]
- 56.Perkins, N. D. 2007. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 8:49-62. [DOI] [PubMed] [Google Scholar]
- 57.Perkins, N. D., and T. D. Gilmore. 2006. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ. 13:759-772. [DOI] [PubMed] [Google Scholar]
- 58.Qin, J. Z., V. Chaturvedi, M. F. Denning, D. Choubey, M. O. Diaz, and B. J. Nickoloff. 1999. Role of NF-kappaB in the apoptotic-resistant phenotype of keratinocytes. J. Biol. Chem. 274:37957-37964. [DOI] [PubMed] [Google Scholar]
- 59.Scheffner, M., J. M. Huibregtse, R. D. Vierstra, and P. M. Howley. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495-505. [DOI] [PubMed] [Google Scholar]
- 60.Seitz, C. S., H. Deng, K. Hinata, Q. Lin, and P. A. Khavari. 2000. Nuclear factor kappaB subunits induce epithelial cell growth arrest. Cancer Res. 60:4085-4092. [PubMed] [Google Scholar]
- 61.Seitz, C. S., R. A. Freiberg, K. Hinata, and P. A. Khavari. 2000. NF-kappaB determines localization and features of cell death in epidermis. J. Clin. Invest. 105:253-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song, Z., S. Krishna, D. Thanos, J. L. Strominger, and S. J. Ono. 1994. A novel cysteine-rich sequence-specific DNA-binding protein interacts with the conserved X-box motif of the human major histocompatibility complex class II genes via a repeated Cys-His domain and functions as a transcriptional repressor. J. Exp. Med. 180:1763-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spitkovsky, D., S. P. Hehner, T. G. Hofmann, A. Moller, and M. L. Schmitz. 2002. The human papillomavirus oncoprotein E7 attenuates NF-kappa B activation by targeting the Ikappa B kinase complex. J. Biol. Chem. 277:25576-25582. [DOI] [PubMed] [Google Scholar]
- 64.Takizawa, S., K. Nagasaka, S. Nakagawa, T. Yano, K. Nakagawa, T. Yasugi, T. Takeuchi, T. Kanda, J. M. Huibregtse, T. Akiyama, and Y. Taketani. 2006. Human scribble, a novel tumor suppressor identified as a target of high-risk HPV E6 for ubiquitin-mediated degradation, interacts with adenomatous polyposis coli. Genes Cells 11:453-464. [DOI] [PubMed] [Google Scholar]
- 65.Ten, R. M., C. V. Paya, N. Israel, O. Le Bail, M. G. Mattei, J. L. Virelizier, P. Kourilsky, and A. Israel. 1992. The characterization of the promoter of the gene encoding the p50 subunit of NF-kappa B indicates that it participates in its own regulation. EMBO J. 11:195-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tergaonkar, V., and N. D. Perkins. 2007. p53 and NF-kappaB crosstalk: IKKalpha tips the balance. Mol. Cell 26:158-159. [DOI] [PubMed] [Google Scholar]
- 67.Thomas, J. T., W. G. Hubert, M. N. Ruesch, and L. A. Laimins. 1999. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. U. S. A. 96:8449-8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tommasino, M., and L. Crawford. 1995. Human papillomavirus E6 and E7: proteins which deregulate the cell cycle. Bioessays 17:509-518. [DOI] [PubMed] [Google Scholar]
- 69.Waterfield, M. R., M. Zhang, L. P. Norman, and S. C. Sun. 2003. NF-kappaB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Mol. Cell 11:685-694. [DOI] [PubMed] [Google Scholar]
- 70.Wissink, S., A. van de Stolpe, E. Caldenhoven, L. Koenderman, and P. T. van der Saag. 1997. NF-kappa B/Rel family members regulating the ICAM-1 promoter in monocytic THP-1 cells. Immunobiology 198:50-64. [DOI] [PubMed] [Google Scholar]
- 71.Xu, M., W. Luo, D. J. Elzi, C. Grandori, and D. A. Galloway. 2008. NFX1 interacts with mSin3A/histone deacetylase to repress hTERT transcription in keratinocytes. Mol. Cell. Biol. 28:4819-4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yuan, H., F. Fu, J. Zhuo, W. Wang, J. Nishitani, D. S. An, I. S. Chen, and X. Liu. 2005. Human papillomavirus type 16 E6 and E7 oncoproteins upregulate c-IAP2 gene expression and confer resistance to apoptosis. Oncogene 24:5069-5078. [DOI] [PubMed] [Google Scholar]
- 73.zur Hausen, H. 1991. Human papillomaviruses in the pathogenesis of anogenital cancer. Virology 184:9-13. [DOI] [PubMed] [Google Scholar]
- 74.zur Hausen, H. 1991. Viruses in human cancers. Science 254:1167-1173. [DOI] [PubMed] [Google Scholar]