

Abstract
The DNA glycosylase MutY homologue (MYH or MUTYH) removes adenines misincorporated opposite 8-oxoguanine as part of the base excision repair pathway. Importantly, defects in human MYH (hMYH) activity cause the inherited colorectal cancer syndrome, MYH-associated polyposis (MAP). A key feature of MYH activity is its coordination with the cell cycle checkpoint via interaction with the Rad9-Rad1-Hus1 (9-1-1) complex. The 9-1-1 complex facilitates cell cycle checkpoint activity and coordinates this activity with ongoing DNA repair. The interdomain connector between the catalytic and 8-oxoG recognition-domains of hMYH (IDC, residues 295-350) is a critical element that maintains interactions with the 9-1-1 complex. We report the first crystal structure of a eukaryotic MutY protein, a fragment of hMYH (residues 65-350) that consists of the catalytic domain and the IDC. Our structure reveals that the IDC adopts a stabilized conformation projecting away from the catalytic domain to form a docking scaffold for 9-1-1. We further examined the role of the IDC using Schizosaccharomyces pombe MYH (SpMyh1) as a model system. In vitro studies of SpMyh1 identified residues I261 and E262 of the IDC (equivalent to V315 and E316 of the hMYH IDC) as critical for maintaining the MYH/9-1-1 interaction. We determined that the eukaryotic IDC is also required for DNA damage selection and robust enzymatic activity. Our studies also provide the first evidence that disruption of the MYH/9-1-1 interaction diminishes repair of oxidative DNA damage in vivo. Thus, preserving the MYH/9-1-1 interaction contributes significantly to minimizing the mutagenic potential of oxidative DNA damage.
Keywords: DNA glycosylase, DNA repair, cell cycle checkpoint, Rad9-Rad1-Hus1, crystal structure
INTRODUCTION
The genome is vulnerable to DNA-damaging agents of both endogenous and environmental origin. Guanine is very susceptible to oxidation and can be converted to 7,8-dihydro-8-oxoguanine (8-oxoG), which is one of the most stable and deleterious products of oxidative DNA damage1. There are approximately 103 and 105 8-oxoG lesions in normal and cancerous tissues respectively per cell per day2. Importantly, if 8-oxoG is not repaired, adenine is misincorporated opposite 8-oxoG during DNA replication3, ultimately leading to G:C to T:A mutations within the genome4-6.
Eukaryotic MutY homologues such as human MYH (hMYH or hMUTYH) and Schizosaccharomyces pombe MYH (SpMyh1) are vital DNA glycosylases that carry out the first step in the base excision repair (BER) pathway to excise adenines or 2-hydroxyadenines mispaired with 8-oxoGs or guanines. MYH cleaves the N-glycosidic bond between the target base and its deoxyribose sugar leaving an apurinic/apyrimidinic (AP) site7. The phosphodiester bond 5′ to the AP site is then cleaved by AP endonuclease (APE1) and downstream BER enzymes complete the repair process. The functional importance of MYH is observed both experimentally and clinically: (1) deletion of the Spmyh1+8 or mouse MYH9 genes results in a substantial increase in the mutation rate in vivo and (2) biallelic hMYH mutations permit downstream mutations in tumor suppressors (i.e. APC) and proto-oncogenes (i.e. K-ras) causing colorectal adenomas and carcinomas (as in the syndrome MYH-associated polyposis, or MAP)10-13. To date, 85 MAP-associated mutations have been identified, spread throughout the entire length of the gene14. However, only 11 MAP-associated hMYH variants have been characterized via functional studies14-18 and of these variants, only 2 (V232F and Q324R/H) have mutations within putative protein-interaction domains. Additional studies must be performed to investigate the potential involvement of impaired protein interactions with hMYH variants in the development of colorectal cancer in some MAP patients.
In eukaryotes, detection and correction of DNA damage is coordinated, through protein-protein interactions, with signaling pathways that regulate DNA replication, cell cycle progression, and apoptosis19,20. We have shown that MYH directly associates with proliferating cell nuclear antigen (PCNA) in both S. pombe and human cells21,22. We also provided direct evidence that the association between SpMyh1 and PCNA is biologically important for SpMyh1 function in mutation avoidance21. By coupling the base excision repair pathway with DNA replication, the association between MYH and PCNA is believed to direct repair towards daughter DNA strands21-25. The connection between DNA repair and cell cycle checkpoints provides an additional mechanism to preserve genomic integrity20. In response to DNA damage, checkpoint proteins initiate cell cycle arrest to allow more time for enhanced DNA repair. In cases of extreme DNA damage, apoptosis can be triggered.
The checkpoint controls are highly conserved from yeast to humans. In fission yeast S. pombe, six checkpoint sensor proteins (Rad9, Rad1, Hus1, Rad17, Rad3, and Rad26) are proposed to initiate the proper DNA damage response under DNA replication block or stress26,27. Rad9, Rad1, and Hus1 form a heterotrimeric complex (the 9-1-1 complex). The structure of the 9-1-1 complex was recently determined28-30 and exhibited striking structural similarity with the PCNA sliding clamp31-33. Besides serving as a damage sensor34-36, the 9-1-1 complex has been shown to interact with and stimulate many enzymes in the BER repair pathway37. During normal DNA replication, MYH coordinates with PCNA; however, in the event of DNA damage, MYH is proposed to recruit the 9-1-1 complex which then enhances MYH glycosylase activity38,39. Importantly, mammalian cell cycle checkpoint proteins are recognized as key tumor-suppressors40 and their direct role in DNA repair, such as with the 9-1-1/MYH interaction, can prevent the accumulation of mutations.
Although bacterial MutY structures have been published41,42, MYH contains extra sequence information that encodes for structural domains mediating its interactions with enzymes involved with DNA replication, mismatch repair, and cell cycle checkpoints37. As the checkpoint response is unique to eukaryotes, the region of MYH critical for interaction with the 9-1-1 complex is absent in the prokaryotic enzyme. We have shown that the major 9-1-1 binding site is located within residues 295-350 of hMYH and residues 245-293 of SpMyh139. In this study, we use structural and biochemical approaches to further examine the interaction between MYH and the 9-1-1 complex, and its significance to DNA repair. Here we report the first eukaryotic structure of MYH containing the human MYH catalytic domain and 9-1-1 binding region (within the interdomain connector, or IDC). The structure of hMYH (residues 65-350) has been solved to 2.3 Å resolution and shows that the IDC differs in size and orientation from its bacterial counterparts. We further examined the significance of the interaction between the SpMyh1-IDC and the 9-1-1 complex. We previously determined that mutation of V315 of hMYH and I261 of SpMyh1 attenuates the interaction with 9-1-139 to a modest extent. In an attempt to elicit a biological effect in vivo, we mutated the adjacent highly-conserved glutamate to glutamine (E262Q). In this report, we demonstrate that residues I261 and E262 of SpMyh1 are key mediators of its interaction with the 9-1-1 complex and that disruption of the interaction itself hinders DNA repair in vivo. In particular, the SpMyh1(I261A/E262Q) mutant cannot complement the mutator phenotype of myh1Δ cells and interruption of the interaction between SpMyh1 and 9-1-1 increases cell sensitivity to hydrogen peroxide. To further examine the role of the IDC in MYH function, we created a SpMyh1-Chimera construct that included the N- and C-terminal domains of SpMyh1 but replaced the IDC with the Escherichia coli MutY linker region. We determined that, although the IDC of SpMyh1 is not needed for DNA-binding, it is required for DNA substrate selection and robust enzymatic activity of the eukaryotic protein. Our results demonstrate that the interaction between MYH and the 9-1-1 complex is an important step in DNA repair. Furthermore, these results strengthen the possibility that impaired hMYH-protein interactions contribute to the development of colorectal cancer in some MAP patients.
RESULTS
hMYH(65-350) contains the six-helix barrel and [4Fe-4S] cluster domains
Structures of bacterial MutY proteins show a catalytic domain and a C-terminal domain connected by a linker region41,42. The catalytic domain consists of the six-helix barrel and [4Fe-4S] cluster domains while the C-terminal domain has structural similarity to MutT43,44 and plays an important role in the recognition of 8-oxoG lesions43,45,46. Overall, hMYH shares a moderate amount of sequence identity with bacterial MutY proteins: 37% with E. coli MutY (EcMutY) and 33.6% with Bacillus stearothermophilus MutY (BstMutY). Upon closer examination, sequence alignments indicate that the catalytic and C-terminal domains of hMYH and SpMyh1 share significant homology with the equivalent domains of EcMutY and BstMutY. In contrast, the interdomain connector (IDC) between the two domains of eukaryotic MYHs diverges significantly in sequence and length from the bacterial MutY linkers. Such a marked change in an otherwise well-conserved homologue suggests a distinct role for the eukaryotic IDC in MYH function. In an attempt to visualize structural differences between the bacterial MutY linkers and the hMYH IDC, we crystallized a construct containing the catalytic domain (residues 65-292) and the IDC (residues 293-350) of hMYH (the full-length protein is 535 residues). The 31.7 kDa hMYH(65-350) protein contains the binding domains for 9-1-1 (residues 295-350 of hMYH)39, APE1 (residues 293-318 of hMYH)22, and hMSH6 (residues 232-254 of hMYH)24. As the catalytic core of E. coli MutY (Ec-cMutY) is sufficient on its own to preserve glycosylase activity42, we confirmed that hMYH(65-350) maintains adenine glycosylase activity with DNA containing an A/8-oxoG mispair (Fig. SD1).
The crystal structure of hMYH(65-350) was determined at 2.3 Å resolution using a combination of single-wavelength anomalous diffraction (SAD) and molecular replacement for phasing (Fig. 1 and Table 1). The final model of hMYH(65-350) contains 271 residues for the first monomer in the asymmetric unit and 272 residues for the second monomer. Residues 65-67, 310-314, and 344-350 (monomer 1; 65-67, 311-314, and 344-350 for monomer 2) are not visible in the structure and therefore are not included in the final model. As expected, the structure of the catalytic domain of hMYH(65-350) is similar to that of Ec-cMutY42 (RMSD = 1.5 Å2, 207 Cα residues; Fig. 1). Two α-helical domains comprise the catalytic domain: (1) a six-helix barrel domain composed of alpha helices α2-α7 and (2) a [4Fe-4S] cluster domain composed of alpha helices α1 and α8-α11 surrounding an [4Fe-4S] cluster. The six-helix barrel domain contains the helix-hairpin-helix (HhH) motif (α6-α7) including the hairpin residues L198, P199, G200, V201, G202 which are also conserved in bacterial MutY enzymes42. Similar to the MutY enzymes, the HhH motif in hMYH is followed by a glycine-rich domain and a catalytically essential aspartate (D222). The [4Fe-4S] cluster domain contains four cysteine residues (C276, C283, C286, C292) which ligand the [4Fe-4S] cluster. Corresponding cysteine residues in EcMutY and BstMutY exist and the [4Fe-4S] cluster of hMYH is superimposable with the [4Fe-4S] clusters of these bacterial MutY enzymes. Also, based on structure-based sequence alignments with Ec-cMutY and BstMutY, the hMYH residues 266-QAAME-270 of α10 and 120-EVMLQATA-127 of α3 are predicted to interact with adenine as part of the adenine specificity pocket at the interface between the six-helix barrel and [4Fe-4S] cluster domains. Of the thirteen BstMutY residues that contact the adenine nucleobase47, eleven are invariant; the exceptions are E188 (Q266 in hMYH) and I191 (M269 in hMYH).
Fig. 1.

Domain architecture of the catalytic domain (composed of the six-helix barrel and [4Fe-4S] cluster domains) and interdomain connector (IDC) of hMYH. Stereo diagram of hMYH(65-350) including the six-helix barrel domain (magenta and red) with the signature helix-hairpin-helix element (red) found in HhH-GPD superfamily members. Cysteine residues of the [4Fe-4S] cluster domain (blue) coordinate the iron (orange) and sulfur (yellow) atoms of the [4Fe-4S] cluster. The IDC (cyan) connects the N-terminal catalytic domain to the C-terminal 8-oxoG recognition domain (not included in this structure). Residues V315 and E316 (brown) of hMYH are indicated (*) and correspond with residues I261 and E262 of SpMyh1. The schematic above the stereo diagram depicts the full-length hMYH protein and is color-coded as described above to show the elements that comprise the hMYH(65-350) crystal structure. The line in the schematic (brown, *) also represents residues V315 and E316 of hMYH. The residues of hMYH that interact with MutSα (232-254), APE1 (293-318), and 9-1-1 (295-350) are indicated above the schematic. Please note that other experiments in this paper were conducted with SpMyh1 and that the 9-1-1 binding site consists of residues 245-293 of SpMyh1.
Table 1.
Data collection and refinement statistics
| hMYH(65-350) | |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 60.31, 82.17, 63.46 |
| α, β, γ (°) | 90, 100.9, 90 |
| Resolution (Å) | 2.3 |
| R sym | 0.097 (0.437)a |
| I / σI | 18.7 (4.6) |
| Completeness (%) | 95.8 (93.4) |
| Redundancy | 7.6 (7.8) |
| Refinement | |
| Resolution (Å) | 2.3 |
| No. reflections | 24,674 |
| Rwork / Rfree | 20.6/25.1 |
| No. atoms | 4,225 |
| Protein | 4,150 |
| Ligand/ ion | 19 |
| Water | 56 |
| B-factors | |
| Protein | 66.165 |
| Ligand/ ion | 63.634 |
| Water | 65.734 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.017 |
| Bond angles (°) | 1.991 |
One crystal was used for the structure of hMYH(65-350).
Values in parentheses are for the highest-resolution shell.
Despite the many structural similarities existing between hMYH and the bacterial MutY enzymes, some minor and major differences were evident. In the Ec-cMutY and the DNA-bound BstMutY structures, helix α1 of the [4Fe-4s] cluster domains begin at residues 3 and 9, respectively while the corresponding helix α1 of hMYH(65-350) begins at residue 76. Residues 65-75 of hMYH exist in an extended conformation and the structure and function of residues 1-64 of hMYH remain unknown. Nonetheless, residues 1-75 of hMYH account for additional structural domains that are not present in the bacterial MutY enzymes. Additionally, α1 of EccMutY is 3 residues shorter and is angled slightly further away from the globular center of the enzyme than α1 of hMYH(65-350). Another minor structural difference in hMYH(65-350) is observed at helices α2-α3. α2 of hMYH is longer than the corresponding helices in EcMutY and BstMutY by 5 and 3 residues, respectively, while α3 of hMYH is longer than the corresponding helices by 4 and 3 residues. The loop between α2-α3 of hMYH is 3 residues long while the corresponding loops in EcMutY and BstMutY are 4 and 2 residues long, respectively. Most of the structural differences in the catalytic domain of the hMYH and MutY enzymes are modest. The most significant differences exist in the hMYH IDC versus the bacterial MutY linkers, described in detail below.
hMYH contains a unique interdomain connector (IDC)
The hMYH IDC (residues 292-353 of hMYH) is 41 residues longer than and possesses little sequence homology with the linker regions found in EcMutY (residues 208-228) and BstMutY (residues 214-234) (Fig. 2a). In the bacterial structures41,42, the short linker region extends only 5 Å away from the globular N-terminal catalytic domain before traversing a relatively direct path to the C-terminal 8-oxoG recognition domain. The MutY linker follows this path in both the apo-Ec-cMutY and BstMutY-DNA structures suggesting that there is no major conformational shift in the MutY linker upon binding to substrate DNA. Strikingly, the hMYH IDC consists of a short helical structure, α12 (Figs. 2b and 2c, colored cyan; Fig. SD2), projecting 18.5 Å away from the catalytic domain (residues 293 to 305) before transitioning into an extended conformation. There are no crystal contacts that stabilize the helix itself, suggesting that the helical extension persists in the full-length protein. The orientation of α12 is stabilized by a covalent bond between residue C292 and the [4Fe-4S] cluster, plus nine hydrogen bonds among nearby residues (Fig. 2d), suggesting that the orientation observed in the hMYH(65-350) structure reflects that seen in the full-length protein. No visible density for residues 310-314 or 344-350 can be identified; however residues 315-343 continue on a path away from the globular catalytic domain. Of functional significance, the structure of hMYH(65-350) shows that the proposed 9-1-1-binding region of hMYH (residues 295-350) is within the IDC which projects away from the catalytic domain. In this position and conformation, the IDC would provide an ideal scaffold without structural obstacles to promote the interaction between hMYH and 9-1-1.
Fig. 2.

The hMYH interdomain connector (IDC) projects away from the catalytic domain. (a) Structure-based sequence alignment of the linker regions of prokaryotic MutY proteins and IDCs of eukaryotic species. Sequences are: H. sapiens MYH (hMYH, accession no. U63329), M. musculus MYH (mMYH, accession no. AY007717), R. norvegicus MYH (rMYH, accession no. Q8R5G2), S. pombe MYH (SpMYH, accession no. Z69240), B. stearothermophilus MutY (BstMutY, accession no. 46015544 ), and E. coli MutY (EcMutY, accession no. P17802). Identical amino acid residues present in at least four sequences are boxed in black and conserved residues are boxed in gray. The hMYH IDC includes the residues required for interaction with APE1 (residues 293-318 of hMYH) and Hus1 (residues 295-350 of hMYH). The stars indicate SpMyh1 residues, I261 and E262, which are important for the 9-1-1 interaction. The gray cylinder above the sequence alignment depicts helix α12 of hMYH(65-350) at the beginning of the IDC. The following solid line indicates the additional residues of the IDC that are in an extended conformation. The portions of the line that are dotted indicate residues 310-314 and 344-350 for which no electron density was identified from the hMYH(65-350) crystal structure. (b, c) The hMYH structure is overlaid with the apo-Ec-cMutY (b, orange) and DNA-bound-BstMutY (c, red) structures to highlight differences between the hMYH IDC and the bacterial MutY linkers. The hMYH IDC (b, c; cyan) projects 18.5 Å away from the catalytic domain differing from the more direct paths of the bacterial MutY linkers to the C-terminal domain. In both the apo-EccMutY and DNA-bound-BsMYH structures, the linkers only extend 5 Å away from the catalytic domain. (d) The orientation of the hMYH IDC is stabilized by a covalent bond between residue C292 and the [4Fe-4S] cluster. (e) The orientation of the hMYH IDC is further stabilized by nine hydrogen bonds (black dashed lines). The oxygen (red) and nitrogen (blue) atoms involved are shown. Importantly, some of these hydrogen bonds involve R231, V232, and R295 which are each associated with MAP mutations.
I261 and E262 of the SpMyh1 IDC are important mediators of the Sp9-1-1 interaction
In SpMyh1, the IDC comprises residues 245-293. Using a glutatione S-transferase (GST)-pulldown assay, we previously showed that mutation of I261 of the SpMyh1 IDC to alanine [SpMyh1(I261A)] attenuates its interaction with SpHus139. Of the three 9-1-1 subunits, SpHus1 is the preferred binding partner of SpMyh1, and is therefore used in pulldown assays to estimate relative 9-1-1 binding proficiency. Because the I261A mutation resulted in partial disruption of the 9-1-1 interaction, we sought to further disrupt the interaction between SpHus1 and SpMyh1 by creating a mutation at E262, another highly conserved residue within the proposed Hus1-binding region of SpMyh139. In addition, both I261 and E262 reside in the extended region that lies just beyond the helical structure of the IDC (equivalent to V315 and E316 of hMYH, Figs. 1 and 2a). Thus, we performed site-directed mutagenesis of SpMyh1(I261A) to replace E262 with glutamine (E262 → Q262). As predicted, the interaction between the SpMyh1(I261A/E262Q) mutant and SpHus1 is significantly weakened compared to the SpMyh1(I261A) and SpHus1 interaction (Fig. 3a, lanes 3 and 4).
Fig. 3.

I261 and E262 of SpMyh1 are important for 9-1-1 binding. (a) Physical interactions between SpHus1 and SpMyh1 mutants examined via a GST-pulldown assay. Lane 1 contains 10% input of E. coli cell extracts containing His-SpHus1. Lanes 2-5 are pellets containing His-SpHus1 from E. coli cell extracts pulled down by GST-SpMyh1 (wild-type), GSTSpMyh1(I261A), GST-SpMyh1(I261A/E262Q), or GST alone, respectively. The pellets were fractionated on a 10% SDS-PAGE gel and Western blot analysis was performed with anti-His6 antibody. Equal amounts of GST and GST-fusion proteins were immobilized onto glutathione-Sepharose 4B beads (data not shown). (b-d) S. pombe 9-1-1 complex stimulates glycosylase activity of SpMyh1 mutants. Lane 1 of each panel is DNA substrates containing A/8-oxoG. The DNA substrate (0.18 nM) was incubated with recombinant SpMyh1 (0.2 nM) (lanes 2 of each panel). Lanes 3-7 are similar to lane 2 but with added 0.313, 0.625, 1.25, 2.5, and 5 nM S. pombe 9-1-1 complex purified from E. coli, respectively. Reactions were carried out at 30°C for 30 min and the products were separated on a 14% DNA sequencing gel. The gel images were viewed on a PhosphorImager and quantified using the ImageQuant software (GE Healthcare). Arrows mark the intact DNA substrate (I) and the nicked product (N). (e) Quantitative analyses of fold stimulation of S. pombe 9-1-1 complex on wild-type- (open circles), I261A- (closed diamonds), and I261A/E262Q- (closed triangles) SpMyh1. The area at the product position in the control lane (no protein; lanes 1 of b-d) was subtracted as background signal. The SpMyh1 cleavage activity was calculated by the percentage of nicked product over total DNA (product plus substrate bands). SpMyh1 glycosylase activities from three experiments are shown. The error bars reported are the standard deviations of the averages.
The purified SpMyh1(I261A/E262Q) mutant protein exhibits glycosylase activity with the A/8-oxoG substrate similar to that of the wild-type (WT) and SpMyh1(I261A) enzymes (compare lanes 2 in Figs. 3b-d). However, consistent with its weakened association with SpHus1, SpMyh1(I261A/E262Q) requires greater amounts of the S. pombe 9-1-1 complex (Fig. 3d and Fig. 3e) to increase its glycosylase activity to the same extent as that seen for SpMyh1-WT (Fig. 3b and Fig. 3e). Two-fold stimulation of SpMyh1-WT (0.2 nM) requires a slight molar excess of S. pombe 9-1-1 (0.3 nM) but a 25-fold molar excess of S. pombe 9-1-1 (5 nM) is needed for two-fold stimulation of SpMyh1(I261A/E262Q) (Fig. 3e). At a concentration of 5 nM, the S. pombe 9-1-1 complex stimulates the glycosylase activities of SpMyh1-WT, SpMyh1(I261A), and SpMyh1(I261A/E262Q) by approximately 5.5, 3.3, and 2.1-fold, respectively (Fig. 3e).
Expression of the I261A/E262Q IDC mutant of SpMyh1 in myh1Δ cells confers a mutator phenotype
We have shown that the S. pombe myh1Δ strain displays a mutator phenotype8 (Table 2, line 2) and expression of wild-type SpMyh1 in these cells reduces the mutation frequency to the same level as wild-type cells (Table 2, line 3). To test whether interaction with the 9-1-1 complex is important for the in vivo SpMyh1 function, we examined the mutation frequency of JSP303-Y4 (myh1Δ) yeast cells expressing the SpMyh1(I261A/E262Q) mutant. The expression level of SpMyh1(I261A/E262Q) protein in yeast cells is comparable to the level of wild-type SpMyh1 under the same conditions (data not shown). The mutation frequency of myh1Δ yeast cells expressing the SpMyh1(I261A/E262Q) mutant is 28-fold higher than that of the wild-type strain (Table 2, compare line 4 to line 1) (P = 0.003) and is 2-fold lower than that of the parental myh1Δ strain (Table 2, compare line 4 to line 2) (P = 0.05). Thus, the SpMyh1(I261A/E262Q) mutant cannot complement the chromosomal myh1 deletion. These results provide direct evidence that the interaction between SpMyh1 and the S. pombe 9-1-1 complex is important to maintain the SpMyh1 biological function of mutation avoidance.
Table 2.
Mutation frequencies of S. pombe strains
| Strain | Mutation Frequency (FOAR/108 cells) |
Fold |
|---|---|---|
| 1. JSP303 (WT) | 3.3 ± 2.4a | 1 |
| 2. myh1Δ | 167 ± 40a | 50 |
| 3. myh1Δ + WT SpMyh1 | 7.4 ± 1.9a | 2 |
| 4. myh1Δ + I261A/E262Q SpMyh1 | 95 ± 12 | 28 |
These values (means with standard deviations) in this study are comparable to those derived from Chang et al. (2001) Mol. Genet. Genomics. 266:336-342.
A peptide consisting of the SpMyh1 IDC (residues 245-293) interacts with the 9-1-1 complex
We have previously shown that residues 245-293 of SpMyh1 are required for 9-1-1 binding using deletion constructs39. To further demonstrate that these residues associate with 9-1-1, we expressed His-tagged and green florescence protein (GFP)-tagged SpMyh1(245-293) using the plasmids pRep41X and p4XG, respectively. Expression of the SpMyh1(245-293) peptide is demonstrated by Western blot analysis (Fig. 4a, lane 2; Fig. 4c, lane 1). Because the nmt1 promoter controls transcription of cDNA in pREP41X and p4XG, SpMyh1(245-293) expression is regulated by varying the concentrations of thiamine (Vitamin B1) used in the minimal media during growth of the yeast cells. At 5 μg/ml of thiamine, expression of the His-tagged and GFP-tagged peptides is almost completely suppressed (Fig. 4a, lane 3; Fig. 4c, lane 2).
Fig. 4.

(a) Expression of His-tagged SpHIP peptide derived from residues 245-293 of SpMyh1 in S. pombe. Equal protein amounts were loaded on a 20% SDS-PAGE gel and Western blot analysis was performed with antibody against SpMyh1. Lane 1, extract from myh1Δ yeast cells; lane 2, extract from myh1Δ cells expressing the His-tagged SpHIP peptide in the absence of thiamine (B1); lane 3, similar to lane 2 except the His-tagged SpHIP peptide is not expressed in the presence of 5 μg/ml thiamine. (b) Interaction of SpHus1, SpRad1, and SpRad9 with His-tagged SpHIP. S. pombe cells were transfected with plasmid containing His-tagged SpHIP peptide derived from residues 245-293 of SpMyh1. Extracts derived from these yeast cells were incubated with GST-SpHus1, GST-SpRad1, or GST-SpRad9 immobilized on beads to observe the binding interactions between SpHIP and the 9-1-1 complex components. Equal amounts of GST and GST-fusion proteins were loaded (data not shown). His-tagged SpHIP in the pellets were detected by Western blot analysis using anti-SpMyh1 antibody. (c) Expression of GFP-tagged SpHIP peptide derived from residues 245-293 of SpMyh1 in Hus1-MYC S. pombe cells. Lane 1, extract from Hus1-MYC cells expressing the GFP-SpHIP in the absence of thiamine (B1); lane 2, similar to lane 1 except GFP-SpHIP is not expressed in the presence of 5 μg/ml thiamine. Lane 3, extract from Hus1-MYC cells expressing the GFP in the absence of thiamine; lane 4, similar to lane 3 except GFP is not expressed in the presence of 5 μg/ml thiamine. (d) Co-immunoprecipitation of SpHus1 with GFP-SpHIP by anti-GFP antibody. S. pombe cells containing Myc-tagged SpHus1 were transfected with plasmid containing GFP-SpHIP or GFP alone. Immunoprecipitation was performed with anti-GFP antibody and Western blotting was detected by anti-Myc antibody. S and P represent supernatant and pellet, respectively. (e) SpHIP inhibits the SpMYH1-SpHus1 interaction. Lane 1, purified SpMyh1 (0.1 μg) was incubated with GST-SpHus1 immobilized on beads. Lanes 2-4, increasing amounts of extracts containing SpHIP (as indicated) were added to reactions similar to lane 1 with immobilized GST-SpHus1 and purified SpMyh1. Both SpMyh1 and SpHIP were detected in the pellets by Western blot analysis using anti-SpMyh1 antibody.
The association between the SpMyh1(245-293) peptide and the 9-1-1 proteins was examined with GST pull-down assays. GST-tagged hHus1, hRad1, and hRad9 proteins were immobilized on three separate bead preparations and used to pull down His-tagged SpMyh1(245-293) peptide from yeast extracts. As shown in Fig. 4b, the SpMyh1(245-293) peptide binds to GST-hHus1 (lane 2) and GST-hRad1 (lane 3). However, the same peptide cannot bind to GST-Rad9 (lane 4) which displays a binding level similar to the negative control of GST alone (lane 5). Thus, the SpMyh1(245-293) peptide binds to the 9-1-1 complex asymmetrically. The weak binding of SpRad9 to the peptide is consistent with our published data that Rad9 is the weakest binding partner of the 9-1-1 complex subunits for both intact SpMyh1 and hMYH38,39. As a result, we named the SpMyh1(245-293) peptide “SpHus1 interacting peptide” (SpHIP).
The interaction of SpHIP with the 9-1-1 complex was also demonstrated by coimmunoprecipitation. GFP-tagged SpHIP was expressed in the Hus1-MYC strain of S. pombe cells which expresses Myc-tagged SpHus1 (Table SD1, line 3). The GFP antibody was used to precipitate the GFP-tagged SpHIP from the cell extracts. The SpHus1 protein is co-precipitated with GFP-tagged SpHIP (Fig. 4d, lane 3) but not with GFP alone (Fig. 4d, lane 6), indicating that SpHIP can interact with 9-1-1 in vivo. Finally, we tested whether SpHIP interferes with the SpMyh1-SpHus1 interaction. Increasing amounts of yeast extracts containing SpHIP were added to the GST pull-down reactions with immobilized GST-SpHus1 and purified SpMyh1. As shown in Fig. 4e, His-tagged SpHIP inhibits the interaction between SpHus1 and SpMyh1.
Expression of SpHIP renders S. pombe cells more sensitive to hydrogen peroxide
To further study the interaction between SpMyh1 and 9-1-1 in vivo, we expressed SpHIP in S. pombe cells and analyzed its influence on hydrogen peroxide sensitivity. S. pombe cells were transfected with plasmid containing GFP-SpHIP and grown in minimal media with or without 5 μg/ml thiamine. At H2O2 concentrations higher than 1.5 mM, expression of SpHIP markedly increases H2O2 sensitivity (Fig. 5, gray bars) compared with cells not expressing SpHIP (Fig. 5, white bars). The expression of GFP-SpHIP alone does not affect the growth rate of the S. pombe cells (data not shown).
Fig. 5.
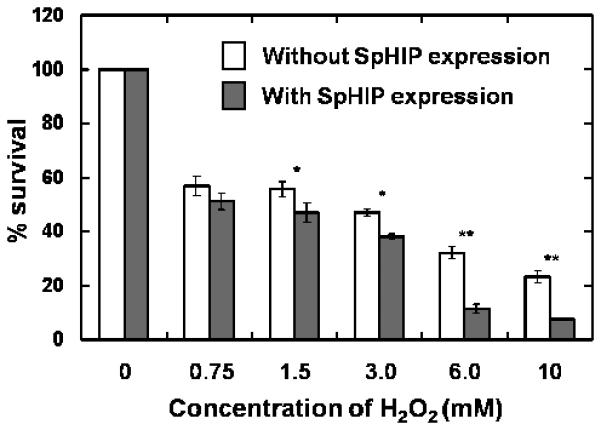
H2O2-sensitivity of S. pombe cells expressing GFP-SpHIP. S. pombe BM2681 cells were transfected with plasmid containing GFP-SpHIP and grown in minimal medium with or without 5 μg/ml thiamine. The expression of GFP-SpHIP is inhibited with thiamine. Cells were treated with H2O2 for 30 minutes and recovered in fresh media without H2O2 for an additional two hours. The percentages of surviving cells after H2O2 treatment were measured. At H2O2 concentrations higher than 1.5 mM, expression of SpHIP increased H2O2 sensitivity compared with cells not expressing SpHIP. For the cells exposed to H2O2 concentrations of 1.5, and 3.0 mM, the measured increased sensitivities were statistically significant with P < 0.02 (*). For the cells exposed to H2O2 concentrations of 6.0 and 10.0 mM, the measured increased sensitivities were statistically significant with P < 0.001 (**).
The SpMyh1 IDC is required to promote DNA damage selection and robust glycosylase activity of the eukaryotic enzyme
In order to further examine the functional impact of differences between the linker regions of prokaryotic MutY proteins and the IDCs of eukaryotic MYH proteins, we constructed a SpMyh1-EcMutY Chimera (SpMyh1-Chimera) which contains the N-terminal domain (residues 1-244) and C-terminal domain (residues 289-461) of SpMyh1 connected by the shorter linker region (residues 214-227) of EcMutY (Fig. 6a). The SpMyh1-Chimera was designed to maintain the N-terminal catalytic and C-terminal 8-oxoG recognition domains of the glycosylase, while eliminating the 9-1-1 interaction domain found only in eukaryotic MYH proteins. We designed this construct in such a manner as to ensure that the chimeric linker is long enough to traverse the DNA and allow SpMyh1-Chimera to encircle it, as is required for high-affinity binding41,48,49. As a preliminary check on our design, we used SWISS-MODEL50 to create a homology model of the SpMyh1-Chimera (Fig. SD3); in the model the linker appears to be of sufficient length to allow the C-terminal domain to access the 8-oxoG lesion. In addition, the observed affinity of SpMyh1-Chimera for abasic product DNA (see below) indicates that the E. coli linker in the context of the SpMyh1-Chimera is long enough to position the SpMyh1 C-terminal domain on the lesion side of the DNA.
Fig. 6.

The SpMyh1 linker domain is important for DNA damage specificity and glycosylase activity. (a) Schematic depicting the SpMyh1-Chimera protein. The SpMyh1-Chimera is composed of the N- and C-terminal domains of S. pombe Myh1 (residues 1-244 and 289-461 of SpMyh1) connected by the E. coli MutY linker (residues 214-227 of E. coli MutY). (b) Glycosylase activity of SpMyh1-Chimera. Lane 1 is DNA substrate (0.18 nM) containing A/8-oxoG. The DNA substrate was incubated with SpMyh1-WT (26 nM) in lane 2. In lanes 3-5, the DNA substrate was incubated with increasing concentrations of SpMyh1-Chimera (26 nM, 260 nM, 2600 nM respectively). Reactions were carried out at 30 °C and 25 °C for 60 min for SpMyh1-WT and SpMyh1-Chimera, respectively. The products were separated on a 14% DNA sequencing gel and the gel image was viewed on a PhosphorImager. Arrows mark the intact DNA substrate (I) and the nicked product (N). Although WT-SpMyh1 has robust enzymatic activity at a concentration of 26 nM, no glycosylase activity was observed for SpMyh1-Chimera at the same concentration. At a 10-fold and 100-fold increase in protein concentration (260 nm and 2600 nM, respectively), SpMyh1-Chimera has some glycosylase activity but not at a level equal to that of WT-SpMyh1. (c) Abasic DNA product affinities of WT-SpMyh1 and SpMyh1-Chimera. Fluorescein-labeled 20-base pair duplex DNA with a centrally located 8-oxoG base opposite an abasic site was incubated with either WT-SpMyh1 or SpMyh1-Chimera over a range of protein concentrations. Binding isotherms were fit for each protein and the relative affinities for the substrate DNA were approximated based on the calculated midpoint concentrations. (d) DNA-substrate specificity of SpMyh1-Chimera. Reactions were pre-incubated at room temperature for 30 minutes with 150 nM SpMyh1-Chimera or WT-SpMyh1, 1 nM fluorescein-labeled 20-base pair duplex DNA with a centrally located A/8-oxoG mispair, and unlabeled competitor substrates (with a centrally located A/8-oxoG mispair or C:G pair) over a range of concentrations (0-1000 nM). The unlabeled A/8-oxoG substrate (black circles) can displace the fluorescein-labeled A/8-oxoG substrate bound to SpMyh1-Chimera or WT-SpMyh1. The unlabeled C:G substrate can displace the fluorescein-labeled A/8-oxoG substrate bound to SpMyh1-Chimera (right panel, red diamonds) but not WT-SpMyh1 (left panel, red diamonds).
We compared the glycosylase activity of SpMyh1-Chimera to that of the wild type enzyme to assess the SpMyh1-Chimera as a functional glycosylase. For accurate comparison, both SpMyh1-Chimera and SpMyh1-WT were expressed with the same maltose-binding protein (MBP)-affinity tag and purified using similar protocols. As shown in Fig. 6b, while SpMyh1-WT has robust glycosylase activity at a concentration of 26 nM with A/8-oxoG-containing DNA, no enzymatic activity is observed for SpMyh1-Chimera at the same concentration. At a 10-fold increase in protein concentration (260 nM), SpMyh1-Chimera displays only minimal enzymatic activity. At 2600 nM, SpMyh1-Chimera shows increased enzymatic activity but still not at a level equal to that of SpMyh1-WT at 26 nM. Although the enzymatic activity of SpMyh1-Chimera is not completely abolished, it is markedly reduced compared to that of SpMyh1-WT.
To investigate the potential cause of the reduced enzymatic activity of SpMyh1-Chimera, we compared the DNA-binding affinities of SpMyh1-Chimera versus SpMyh1-WT utilizing fluorescence anisotropy experiments. We incubated fluorescein-labeled 20-base pair duplex DNA containing the product of the SpMyh1 glycosylase reaction (an AP/8-oxoG mispair) with either SpMyh1-Chimera or SpMyh1-WT over a range of protein concentrations (Fig. 6c). Binding isotherms were fit for each protein using a transformed Hill equation (see Materials and Methods) which yields a parameter ([P]1/2) that approximates the protein concentrations at which half-maximal binding is achieved. Unexpectedly, the SpMyh1-Chimera and SpMyh1-WT proteins have very similar affinities for the DNA substrate containing an AP/8-oxoG mispair, with half-maximal binding at 12 ± 3 nM and 10 ± 2 nM, respectively. Both proteins display apparent binding cooperativity with Hill coefficients of 2.6 ± 1.1 for SpMyh1-Chimera and 3.7 ± 1.1 for SpMyh1-WT.
To clarify the DNA substrate preference of SpMyh1-Chimera, we performed a competition assay. We compared the abilities of unlabeled substrates containing either a C:G pair or an A/8-oxoG mispair to displace a fluorescein-labeled A/8-oxoG substrate bound to SpMyh1-Chimera or SpMyh1-WT. As expected, the unlabeled A/8-oxoG substrate (Fig. 6d, black circles) can displace the fluorescein-labeled A/8-oxoG substrate bound to SpMyh1-Chimera or SpMyh1-WT with measured apparent inhibition constants (Ki,app) of 42 ± 9 nM and 14 ± 9 nM, respectively. Thus, the competition assay indicates that the SpMyh1-Chimera binds an A/8-oxoG DNA with less affinity than the wild-type protein. However, the C:G substrate is an ineffective competitor for SpMyh1-WT and is unable to displace the fluorescein-labeled A/8-oxoG substrate to any measurable extent (Fig. 6d, left panel, red diamonds). Strikingly, the C:G substrate can displace the fluorescein-labeled A/8-oxoG substrate bound to SpMyh1-Chimera (Ki,app = 17 ± 4 nM) (Fig. 6d, right panel, red diamonds) and is therefore an effective competitor. In addition, when measuring direct binding to fluorescein-labeled 20 base-pair C:G substrate, SpMyh1-Chimera binds the substrate with high affinity whereas SpMyh1-WT does not bind the substrate to any measurable extent (Fig. SD4). These results indicate that SpMyh1-Chimera exhibits only a modest preference for binding A/8-oxoG-containing DNA relative to undamaged DNA.
DISCUSSION
In this study, we solved the first eukaryotic MYH structure and examined the significance of the interaction between MYH and the 9-1-1 complex for promoting DNA repair. Our studies provide a structural rationale for the additional residues found in eukaryotic MYH IDCs. We have demonstrated that two residues of SpMyh1 (I261 and E262), which reside on the extended region of the IDC, are key mediators of the interaction between SpMyh1 and 9-1-1. Importantly, disruption of the interaction between SpMyh1 and the 9-1-1 complex via mutation [SpMyh1(I261A/E262Q)] has a deleterious impact on oxidative DNA repair in vivo. When the IDC of SpMyh1 is replaced by the EcMutY linker, the protein binds abasic product DNA with normal affinity, but also binds undamaged DNA with abnormally high affinity, resulting in substantially abrogated glycosylase activity. Thus, the IDC of eukaryotic MYH serves as a structural scaffold to mediate important protein interactions and simultaneously serves as a structural hinge to properly position the N- and C- terminal domains for A/8-oxoG recognition and catalysis.
Transient interactions between hMYH and other proteins coordinate MYH BER with DNA replication, other DNA repair pathways, and DNA damage response6. On a basic level, these interactions may promote the efficient transfer of the product of one step of repair to the next enzyme in the repair pathway. At first glance, the effects of these interactions appear minor, as the catalytic activity of MYH increases only five-fold, at best, in the presence of a high excess of a given stimulatory protein. However, upon closer inspection, the observed effects seem to primarily foster cycling through the BER pathway and, if necessary, transitioning to other processes. This “BER relay” system appears to operate at the expense of maximizing the catalytic turnover of any particular enzyme. Such a regulatory network of malleable protein interactions affords the BER pathway sufficient flexibility to repair multiple types of damage. In that regard, high affinity interactions between MYH and interacting proteins are likely not optimal for the seamless incorporation of MYH-BER into other pathways of DNA metabolism.
Thus far, a partial sketch of this protein interaction regulatory network has been assembled through systematic, pairwise investigation of the effects of interacting partners on MYH activity. APE1, a downstream BER enzyme, interacts with MYH22 and enhances its glycosylase activity51. This interaction likely promotes MYH turnover and prevents release of potentially cytotoxic AP sites. MYH activity can be also stimulated by the mismatch recognition protein MSH2/MSH6 (MutSα)24. In particular, the repair of A/8-oxoG mispairs requires communication between the BER and mismatch repair pathways and coupling to DNA replication21-25 to ensure that the mis-incorporated adenine on the daughter strand DNA is repaired rather than the 8-oxoG on the parental strand. Finally, MYH interacts with 9-1-1, resulting in an increase in MYH glycosylase activity38,39. The interaction with 9-1-1 is enhanced by stresses such as H2O2 and ionizing radiation exposure38,39, which is consistent with the suggestion that 9-1-1 might replace PCNA under stress52 to arrest the cell cycle and simultaneously enhance BER.
Our crystal structure of hMYH(65-350) further supports the idea that transient protein interactions regulate the activity of hMYH. While the hMYH IDC is required to maintain a physical link to the 9-1-1 complex and to APE1, the structure of hMYH(65-350) reveals that the hMYH IDC possesses no regular secondary or tertiary structure beyond the helical extension (residues 293-305; Fig. 1 and Fig. 2b-d). Interestingly, the 9-1-1 interacting regions of other DNA glycosylases, including hNEIL1 (residues 290-350)53 and hTDG (residues 67-110)54, may also be flexible. No identifiable density beyond residue 290 can be detected in the crystal structure of hNEIL1 containing residues 2-34353. Similarly, NMR data indicate that residues 67-110 of hTDG are unstructured. Thus, a common feature of the 9-1-1 binding motif appears to be that it adopts a flexible structure, possibly to enable transient interactions with multiple protein partners. Still, it is possible that this region becomes more structured in the presence of the 9-1-1 complex, resulting in a conformational change that promotes the catalytic activities of DNA glycosylases. Of note, it has been observed that many unstructured protein segments do not fold until they bind to their biological targets, thus permitting protein promiscuity55.
Structure-based sequence alignment shows that the IDCs of hMYH and SpMyh1 are 41 and 34 residues longer, respectively, than the linker of the bacterial MutY proteins (Fig. 2a). Eukaryotic MYH family members possess few conserved stretches within their IDCs, with only ~ 25% sequence identity between the hMYH and SpMyh1 IDCs. Our hMYH(65-350) structure provides a potential rationale for the added length of the IDC. The additional length of the hMYH IDC appears to serve in part to project the 9-1-1-interacting region away from the surface of bound DNA (Fig. SD5). Without projection of the IDC away from the catalytic domain and DNA-binding site, the modest features of the IDC might be obscured by the negative charge of DNA and thus prevent the interaction between MYH and the 9-1-1 complex. The orientation of the hMYH IDC is stabilized by the covalent bond between residue C292 and the [4Fe-4S] cluster, plus nine additional hydrogen bonds (Fig. 2d). Any significant reorientation of the IDC would require accommodation of the hydrogen bonding groups without exposure of the 4Fe-4S cluster to solvent, a further indication that the orientation we observe is likely fixed. Significantly, some of these hydrogen bonds involve residues R231, V232, and R295, each of which has an associated MAP mutation14. Additionally, residues R295, Q324, F344, and P345 of hMYH are all within the IDC and are associated with MAP mutations14 (Fig. 2d). It will be interesting to see whether any newly-discovered MAP mutations will include mutations that disrupt the interaction between hMYH and 9-1-1.
Although the interaction between MYH and 9-1-1 produces a modest effect on catalytic activity in vitro, the interaction is still of great physiological significance. In previous studies39, we showed that mutation of I261 to alanine alone could attenuate the interaction between SpMyh1 and 9-1-1 without perturbing catalytic activity. However, the effect was modest. We demonstrated here that the interaction with 9-1-1 is more severely compromised for the SpMyh1(I261A/E262Q) mutant than for the SpMyh1(I261A) mutant (Fig. 3). This allowed us to assess the impact of disrupting the interaction between SpMyh1 and 9-1-1 on oxidative DNA damage repair in vivo. Unlike SpMyh1-WT, SpMyh1(I261A/E262Q) does not reduce the mutation frequency of myh1Δ cells (Table 2). In a separate approach, we showed that disruption of the interaction between MYH and 9-1-1 in S. pombe cells through expression of SpHIP (Fig. 4) makes cells more sensitive to H2O2 (Fig. 5), reducing the DNA repair capacity of the cells. Since the IDC of hMYH also contains the hAPE1 binding site (residues 295-318)22, it is possible that SpHIP may also interfere with the interaction between SpMyh1 and APE1.
By making more radical changes to the IDC, we demonstrate that its impact extends beyond mediating protein interactions. We created a chimeric protein that replaced the region of the SpMyh1 IDC implicated in 9-1-1 interactions with the E. coli MutY linker in an attempt to retain catalytic activity while abolishing 9-1-1 interactions. However, characterization of SpMyh1-Chimera reveals that the eukaryotic IDC is designed not only to promote protein-protein interactions, but also to foster substrate selection and catalytic activity (Fig. 6). Despite preservation of the catalytic and C-terminal domains (Fig. 6a), the SpMyh1-Chimera has significantly reduced glycosylase activity (Fig. 6b). In contrast, SpMyh1-Chimera maintains normal affinity for the abasic DNA product (Fig. 6c). Such high affinity binding requires extensive interactions between both the N-terminal and C-terminal domains and the bound DNA. In particular, isolated N-terminal domains of both E. coli MutY48 and hMYH (Toth, unpublished) exhibit a marked reduction in affinity for abasic product. Furthermore, the isolated E. coli MutY C-terminal domain has no intrinsic affinity for DNA56. Thus, high affinity binding requires successful positioning of both domains simultaneously on DNA. If the creation of the SpMyh1-Chimera retains wild type abasic product affinity, some deficit in substrate recognition must explain the severe catalytic defect. In fact, our data show that SpMyh-Chimera binds undamaged DNA with abnormally high affinity (Fig. 6d and Fig. SD4). This gain of function (i.e. non-specific DNA binding) relative to the wild type enzyme was unexpected and suggests an active role for the IDC in promoting catalysis. It appears that the extra length of the IDC might be required to properly orient the catalytic and C-terminal domains on substrate DNA to optimize the contacts required for preferential binding to an A/8-oxoG mispair. The fact that SpMyh1-Chimera catalyzes the glycosylase reaction poorly with the radical change in DNA-binding behavior suggests that the process of encountering the lesion might be impaired. In effect, the SpMyh1-Chimera might spend far more time engaged with undamaged DNA than the wild type enzyme, and perhaps as a result only infrequently recognizes A/8-oxoG mispairs. These data provide an additional potential explanation for the presence of large insertions (41 residues in hMYH, 34 residues in SpMyh1) in eukaryotic IDCs. The IDCs provide an accessible platform for protein-protein interactions while at the same time retaining the ability to help orient the N- and C-terminal domains for catalysis. Satisfying these simultaneous constraints likely necessitated the large insertions observed in eukaryotic IDCs rather than the more modest changes observed in the N- and C-terminal domains.
Here we demonstrate for the first time that the eukaryotic MYH IDC is not merely an inert tether that connects the catalytic and C-terminal 8-oxoG recognition domains, but rather it is essential for the in vitro and in vivo functions of MYH. Our work provides insight into how protein interactions of modest affinity, such as that between MYH and 9-1-1, can modulate BER and play an important role in mutation avoidance. Even slight changes to the MYH IDC can diminish the ability of the enzyme to mitigate the mutagenic potential of oxidative DNA damage in vivo. Despite relatively modest structural differences between the eukaryotic IDCs and prokaryotic linker regions, the hMYH IDC provides an ideal “docking station” for 9-1-1 (and APE1), a feature which distinguishes eukaryotic MYH from prokaryotic MutY. Of clinical value, our work provides the first structural and biochemical data to implicate impaired cell-signaling as another possible mechanism underlying the mutagenic potential of some hMYH mutants in MAP patients.
MATERIALS AND METHODS
Creation of expression constructs
The sequences of all constructs were verified before undertaking subsequent experiments.
hMYH(65-350)
Primers C4F-B and R-EC5 (all of the oligonucleotides used are listed in Table SD2) were used to amplify the hMYH(65-350) region of the hMYH gene from template pET11a-hMYH22. The hMYH(65-350) polymerase chain reaction (PCR) product was cleaved by NdeI and XhoI and ligated into a modified pET-19b vector (Novagen) with an N-terminal decahistidine tag and a PreScission Protease cleavage site.
SpMyh1-I261A/E262Q double mutant
The I261A/E262Q double mutant of the Spmyh1+ gene was constructed by the PCR splicing overlap extension method57. Primers CHANG219/Sp-IA-E262Q-R and Sp-IA-E262QF/CHANG220 were used to amplify the N-terminal and C-terminal regions of the Spmyh1+ gene from template pET11a-SpMyh1-IA39. Next, both purified PCR products were used as templates for another PCR reaction with the CHANG219 and CHANG220 primers. The final PCR products were cleaved by NdeI and BamHI, and ligated into the NdeI-BamHI-digested pET11a vector (EMD Biosciences). This parent construct served as the starting point for the subcloning of the SpMyh1-IA/EQ double mutant into a bacterial expression vector for production of the glutathione S-transferase (GST)-tagged mutant and subcloning into the yeast expression vector pREP41X (American Type Culture Collection). The primers used for creation of these constructs are listed in Table SD2.
GST-SpRad9, SpRad1, and SpHus1
The cDNA fragments containing SpRad9, SpRad1, and SpHus1 fused to the GST gene were obtained by PCR using the primers listed in Table SD2 and templates pET21a-SpRad9, pET21aSpRad1, and pET21a-SpHus138, respectively. The PCR products were digested with BamHI and ligated into the BamHI-digested pGEX-4T-2 vector (Amersham Biosciences, Inc.).
SpMyh1 peptide corresponding to residues 245 to 293 (SpHIP)
The SpHus1 binding region in SpMyh1 has been mapped between residues 245 to 29339. To express this SpHus1 interacting peptide (SpHIP), the Spmyh1+ cDNA fragment coding residues 245 to 293 was amplified by PCR from full-length cDNA template, pSPMYH1958 with primers SpMYH245-Xho and SpMYH245-Bam, and ligated into p4X-G which contains a coding sequence of green fluorescence protein (GFP)59. Spmyh1+ cDNA coding residues 245-293 was also synthesized by PCR with primers SpMYH245-Xho-ATG and SpMYH245-His-Xma and ligated into pREP41X.
MBP-SpMyh1-WT and MBP-SpMyh1-Chimera
Primers TOTH382/SpMyh-F and TOTH371/SpMyh-R were used to amplify the Spmyh1+ gene from template pET11a-SpMyh1. The PCR product was digested with KpnI and BamHI and ligated into a KpnI-BamHI-digested dual N-terminal hexahistidine (His6) -maltose-binding-protein (MBP) pLM303 fusion vector.
The SpMyh1-Chimera construct was derived from pET11a-SpMyh1. First, the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) was used to create a SalI restriction enzyme cut site within the Spmyh1+ gene using primers, SpMyh-Sal-F and SpMyh-Sal-R. The SalI site was created immediately 3′ to the segment of DNA that encodes for the SpMyh1 IDC region and immediately 5′ to the segment of DNA that encodes for the SpMyh1 C-terminal domain. The pET11a-SpMyh1-SalI mutagenesis product was digested with NdeI and SalI and the digested DNA fragment containing the pET11a vector and the DNA encoding for the C-terminal domain of SpMyh1 (pET11a-CTDSpMyh1) was gel purified. Simultaneously, PCR was completed to amplify DNA containing a 5′-NdeI cut site and the 5′-end of the Spmyh1+ gene up to the beginning of the section of DNA that encodes for the SpMyh1 linker region with primers SpMyh-NdeI and SpMyh-SalI. The SpMyh-SalI primer used in the PCR reaction included DNA to synthesize the specified section of Spmyh1+, the E. coli MutY linker region, and a SalI cut site. This PCR product was digested with NdeI and SalI and ligated into the NdeI-SalI digested pET11a-CTDSpMyh1. Using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene) and primers, SpCHIM-Sal-to-Nat-F and SpCHIM-Sal-to-Nat-R, mutagenesis was completed again to remove the SalI site. The pET11a-SpMyh1-Chimera construct was used as a template for subcloning into the pLM303 vector.
Protein purification
hMYH(65-350)
hMYH(65-350) was overexpressed in E. coli Rosetta™2(DE3) (Novagen) cells. Following cell lysis, the supernatant was loaded onto a nickel-sepharose (GE Healthcare) affinity column in buffer containing 50 mM Na2HPO4 pH 8.0, 300 mM NaCl, and 10 mM imidazole. After washing, the protein was eluted from the column with 250 mM imidazole and then dialyzed at 4 °C overnight in buffer containing 20 mM KH2PO4 pH 7.4, 300 mM KCl, and 1 mM dithiothreitol (DTT). Subsequently the protein solution was dialyzed for 2 hours in a low-salt buffer (50 mM KCl). hMYH(65-350) was further purified with a Q-sepharose anion-exchange column (GE Healthcare) using a salt gradient of 0.05 - 1 M KCl. Most of the protein was retrieved from the flow through and wash. To lower its conductivity, the collected hMYH(65-350) was diluted in a 1:1 ratio with 20 mM KH2PO4 pH 7.4 and 1 mM DTT. Heparin-sepharose affinity chromatography (GE Healthcare) was used for the final purification step and the column was developed with a salt gradient of 0.05 – 1 M KCl. Peak fractions were pooled together and incubated with PreScission Protease (GE Healthcare) according to the manufacturer's instructions resulting in complete removal of the decahistidine tag. The protein was dialyzed at 4 °C overnight in buffer containing 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 1 mM EDTA, and 1 mM DTT. Purified hMYH(65-350) was concentrated to ~8 mg ml−1 and stored at − 80 °C.
SpMyh1-Wild Type, -I261A, and -I261A/E262Q
The non-tagged wild type and mutant SpMyh1 proteins were purified according to the described procedures58.
MBP-SpMyh1-WT and MBP-SpMyh1-Chimera
The MBP-SpMyh1-WT fusion protein was overexpressed in E. coli Rosetta-2(DE3) (Novagen) cells. Following cell lysis in the presence of Benzonase (Novagen) nuclease, polyethyleneimine (PEI) was added to the supernatant to a final concentration of 1% (v/v) to precipitate contaminating nucleic acids. A partial protein purification step was completed with the addition of ammonium sulfate to a final concentration of 30% (w/v) to precipitate a subset of the contaminants. Next, ammonium sulfate was added to the remaining solution to a final concentration of 50% (w/v) to precipitate MBP-SpMyh1-WT. The precipitated protein was resuspended in Buffer T (20 mM Tris-HCl pH 7.5, 200 mM NaCl, 10 mM β-mercaptoethanol (BME), 0.1 mM EDTA, and 0.1% Triton X-100). After a two-hour dialysis step in Buffer T, the protein was loaded onto an amylose-sepharose (New England Biolabs) affinity column. After washing, the protein was eluted from the column with Buffer T containing 10 mM maltose. To reduce the ionic strength of the eluted protein sample, it was diluted with Buffer H (20 mM KH2PO4 pH 7.5, 1 mM DTT, 0.2 mM phenylmethylsulfonyl fluoride (PMSF), 10% glycerol, and 0.1% Triton X-100). A heparin-sepharose (GE Healthcare) affinity column was used for the final purification step using a salt gradient of 0.05 – 0.6 M KCl. The peak fractions were pooled, filtered, and stored at − 80 °C.
The purification protocol for the MBP-SpMyh1-Chimera fusion protein began the same way as the protocol used for the MBP-SpMyh1-WT fusion protein. However, after the two-hour dialysis in Buffer T, MBP-SpMyh1-Chimera was loaded onto a diethylaminoethyl (DEAE) -cellulose (Whatman) anion exchanger column in-tandem with the amylose-sepharose (New England Biolabs) affinity column. The DEAE column was used here to bind any remaining contaminating nucleic acids. After a thorough wash step, the DEAE column was removed and the protein was eluted from the amylose column with Buffer T containing 10 mM maltose. To reduce the ionic strength of the eluted protein sample, it was diluted with Buffer S (25 nM HEPES pH 7.5, 1 mM DTT, 0.25 mM EDTA, 1% glycerol, and 0.1% Triton X-100). At this point, the protein was loaded onto an SP-sepharose (GE Healthcare) cation exchanger column. The column was developed with a salt gradient of 0.05 − 1.0 M NaCl. The peak protein fractions were dialyzed for two hours in Buffer Q (20 mM KH2PO4 pH 7.5, 50 mM KCl, 1 mM DTT, and 0.1% Triton X-100). The final purification step employed anion exchange using a Q-sepharose (GE Healthcare) anion exchanger column. The Q-sepharose column was developed with a salt gradient of 0.05 – 0.6 M KCl. The purified protein was concentrated to about 1.5 mg/ml, filtered, and stored at − 80 °C.
Yeast expression
S. pombe strains and growth
Yeast strains used in this study are listed in Table SD1. Standard procedures and media were used for culture growth, transformation, and genetic analysis60. Yeast cells were grown in yeast extract-peptone-dextrose (YPD) medium for regular maintenance. For specific selection and mutation frequency measurements, cells were grown in Edinburgh Minimal Medium (EMM) with supplements as indicated.
Expression of the SpMyh1-I261A/E262Q mutant in Spmyh1 knockout cells
A clone containing the Spmyh1 gene (pREP41X-SpI261A/E262Q) was confirmed by DNA sequencing and transformed into Spmyh1 knockout cells, JSP303-Y4 (myh1Δ)8 by electroporation. Transformed cells acquired a Leu+ phenotype and were selected on Leu− yeast nitrogen base (YNB) agar plates. The pREP41X expression vector contains the nmt1 promoter that can be regulated with varying concentrations of thiamine; transcription at the nmt1 promoter is almost completely suppressed in the presence of 5 μg/ml of thiamine.
Expression of GFP- and His-tagged SpHIP
DNA from a confirmed GFP-SpHIP clone was incorporated via electroporation into the cells of BM2681 or the Hus1-MYC strain while DNA from a confirmed His-SpHIP clone was incorporated into TMN3309. Transformed GFP-SpHIP cells acquired a Ura+ phenotype and were selected on Ura− YNB agar plates. Meanwhile, transformed His-SpHIP cells acquired a Leu+ phenotype and were selected on Leu− YNB agar plates.
The transcription of GFP-tagged and His-tagged SpHIP in the expression vectors p4XG and pREP41X, respectively, is controlled by the thiamine-regulated nmt1 promoter. Yeast cells were grown in EMM media to OD600 of ~0.6 in the absence or presence of 5 μg/ml of thiamine. Cells were harvested and lysed as described8. The GFP-SpHIP product encoded by the sequences in p4XG was detected by antibodies against either SpMyh1 or GFP. Expression of His-tagged SpHIP in yeast cells was confirmed by Western blot analysis with polyclonal antibodies against full-length SpMyh1 as previously described21.
hMYH(65-350) crystallization and structure determination
hMYH(65-350) crystals grew within 1 day using sitting drop trays in a buffer containing 0.2 M magnesium acetate, 20% (v/v) polyethylene glycol (PEG) 3350, 5 mM Tris[2-carboxyethyl] phosphine (TCEP), 5% glycerol, and 10 mM spermidine. The crystallization buffer was supplemented with glycerol to a final concentration of 20% (v/v) for cryoprotection. The crystals are primitive monoclinic (P21) with cell dimensions a = 60.31 Å, b = 82.17 Å, c = 63.46 Å, β = 100.9, and contain a dimer in the asymmetric unit. X-ray diffraction data were collected at beamline X6A in the National Synchrotron Light Source of the Brookhaven National Laboratory. The images were processed and scaled using the HKL2000 program suite61. The [4Fe-4S] cluster within hMYHΔC5 enabled collection of single-wavelength anomalous diffraction (SAD) data at the iron absorption edge, 1.65 Å (7.5 keV), to 2.3 Å resolution. Computational programs within the Collaborative Computational Program Number 4 (CCP4)62 were used for structure determination. The positions of the two 4Fe-4S clusters in the asymmetric unit were determined by inspection of an anomalous difference Patterson map. After phasing and density modification, the resulting electron density maps were not of sufficient quality to allow model building to proceed. Thus, a combined approach using both the experimental phases derived from the iron positions and molecular replacement was employed. The CCP4 program CHAINSAW was used to generate a search model from the E. coli cMutY structure. Two rounds of molecular replacement were needed to obtain a solution for both hMYH(65-350) monomers in the asymmetric unit. Using the experimental phases of hMYH(65-350) from MLPHARE and the E. coli cMutY search model, one monomer of hMYH(65-350) was found with MOLREP. The model phases from this molecular replacement solution were used to complete rigid body refinement in REFMAC. The resulting model phases were used to initiate a second round of molecular replacement in MOLREP. All model building was carried out with the program COOT. Additional non-crystallographic symmetry (NCS) averaging was performed, using NCS operators derived from the correctly placed search model, along with solvent flattening and histogram matching using the program DM. The density-modified phases were used as input for restraints for the REFMAC mlhl target function. Additionally, TLS refinement was performed on the catalytic domain and IDC as separate domains. After several rounds of model building and the addition of waters, the Rfree was 25.1% and the Rcryst was 20.6%. The data collection and refinement statistics are presented in Table 1. Analysis of the Ramachandran plot shows that 94.4% of residues are in the favored regions, and 5.6% of residues are in allowed regions. Figures were made using the program PyMol63 .
Glycosylase activity assays of S. pombe MYH proteins
The glycosylase assay for purified recombinant SpMyh1 and the SpMyh1(I261A/E262Q) double mutant with an A/8-oxoG-containing DNA substrate was described previously58. The DNA substrate was a 20 base-pair duplex DNA containing a central A/8-oxoG mismatch. The SpRad9-Rad1-Hus1 complex was purified as described39. The glycosylase assay for purified SpMyh1-Chimera followed the same protocol except that the glycosylase reaction was performed at 25 °C instead of 30 °C.
Glycosylase assay of hMYH(65-350)
The glycosylase assay for purified recombinant hMYH(65-350) was similar to the assay described previously24, except a different DNA substrate and incubation times were used. The DNA substrate was a 20 base-pair duplex DNA containing a central A/8-oxoG mismatch. The DNA strand containing the adenine was 5′-labeled with fluorescein (IDT). The hMYH glycosylase reaction mixtures were incubated for 30 minutes at 37 °C. The reactions were stopped by heating the samples for 30 minutes at 90 °C with NaOH to a final concentration of 0.1 M.
GST pull-down assay
Expression, immobilization of the GST fusion constructs, and the GST-pull-down assay were similar to the procedures described previously38. E. coli (BL21Star/DE3) cells (Stratagene) harboring the expression plasmids were cultured in Luria-Bertani broth containing 100 μg/ml ampicillin at 25 °C. Protein expression was induced as described above. The cell paste from a 0.5-liter culture was lysed and extracts were immobilized onto glutathione-Sepharose 4B (GE Healthcare). A control was run concurrently with immobilized GST alone. After washing, the pellets were fractionated on a 10% (for His-tagged SpHus1) or 20% (for SpHIP) SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. Western blot analyses were performed with antibody against His-tag (sc-8036, Santa Cruz Biotechnology) or SpMyh18.
Co-immunoprecipitation of GFP-SpHIP with SpHus1 protein
GFP-SpHIP expressed in Hus1-MYC cells was precipitated by an anti-GFP antibody. Extracts (1 mg) derived from S. pombe cells expressing GFP alone or GFP-SpHIP were precleared by incubation with protein A Sepharose (50 μl) in PBS with protease inhibitors (Sigma/Aldrich) for 4 hours at 4 °C. After removal of the beads, the supernatant was mixed with 4 μl of monoclonal anti-GFP antibody (Abcam) for 16 hours at 4 °C. Then, protein A Sepharose (50 μl) was added to precipitate GFP-SpHIP. After centrifugation at 1,000 × g, the supernatant was collected and the pellet was washed. Both the supernatant (10% of the total volume) and pellet fractions were resolved on a 12% SDS-PAGE gel. The Myc-tagged SpHus1 that co-precipitated with GFP-SpHIP was verified with Western blot analysis using antibodies against c-Myc (Santa Cruz Biotechnology).
Measurement of mutation frequency
A clone containing the Spmyh1 gene (pREP41X-SpI261A/E262Q) was transformed into Spmyh1 knockout cells, JSP303-Y4 (myh1Δ)8 by electroporation. Five independent yeast colonies were grown to late log phase in EMM containing 0.1 mg/ml uracil. Additional amino acids were supplemented for the wild-type strain (0.1 mg/ml Leu and His), and the myh1Δ strain (0.1 mg/ml Leu). Each culture was plated onto EMM agar plates containing 1 mg/ml 5-fluoro-orotic acid (FOA) and 0.1 mg/ml uracil. FOA-resistant colonies were counted after 5 days of growth. The cell titer was determined by plating 0.1 ml of a 10−4 dilution onto plates without FOA. The mutation frequency was calculated as the ratio of FOA-resistant cells to the total cells. The measurement was repeated at least three times to ensure reproducibility.
H2O2 treatment
For hydrogen peroxide (H2O2) treatment, 1.0 ml of an overnight yeast culture grown in EMM containing 5 μg/ml of thiamine was added to 20 ml of EMM medium in the absence or presence of 5 μg/ml of thiamine. At an OD600 of ~0.6, 2 ml of the culture were aliquoted into 30-ml test tubes followed by addition of H2O2 to each aliquot at various concentrations. After a 30-minute incubation step, the cells were pelleted and resuspended in fresh, H2O2-free medium and shaken at 32 °C for 1 or 2 hours. Cells were diluted 10,000 fold and plated on YPD plates. The number of colonies was scored after 3 days of incubation at 32 °C.
Measurement of the DNA-binding affinity via fluorescence anisotropy
Fluorescence anisotropy experiments were performed to measure the affinity of SpMhy1 or SpMyh1-Chimera for a 20 base-pair duplex DNA substrate containing a centrally located abasic site opposite an 8-oxoG nucleotide on the complementary strand. The DNA substrate was prepared by 5′-labeling the strand containing the abasic site with fluorescein (Integrated DNA Technologies). The binding experiments were conducted as described64 using 1 nM of labeled DNA. For SpMyh1, total fluorescence emission decreased as a function of added protein concentration requiring that an appropriate correction factor64 be applied to the measured anisotropies. Relative affinities were calculated from the binding isotherms using the program GraphPad Prism version 3.03 and a variant of the Hill equation64,65:
| (1) |
where Atotal is the measured anisotropy, ADNA is the inherent anisotropy of the DNA substrate, Acomp is the anisotropy of the saturated protein-DNA complex, and h is the Hill coefficient. This equation also estimates the midpoint of the binding isotherm ([P]m), which, in the case of a single binding site (i.e. h is constrained to be 1.0), is equivalent to the Kd. To determine the affinity for undamaged DNA, the identical method was used, with the exception that the substrate was a 19 base-pair duplex DNA substrate, with a centrally located C:G base pair, with one base overhanging at the 5′ end of the DNA strand containing guanine.
Competition Assay
To determine the substrate specificity of SpMyh1-Chimera, we measured the ability of competitor DNA substrates to displace an A/8-oxoG substrate bound to the glycosylase. We used a fluorescein-labeled 20-base pair duplex DNA with a centrally located A/8-oxoG mispair. The strand containing the adenine was 5′-labeled with fluorescein (IDT). The experiments also required the use of unlabeled 20-base pair duplex DNA substrates with either a centrally located C:G pair or an A/8-oxoG mispair. Reaction samples included 150 nM of either SpMyh1-Chimera or MBP-SpMyh1-WT, 1 nM of the 5′-fluorescein-labeled 20-base pair duplex DNA, and either the C:G or A/8-oxoG unlabeled 20-base duplex DNA substrate over a range of concentrations (0-1000 nM) (Fig. 6d). The reaction samples were pre-incubated at 25 °C in low ionic strength buffer for 30 minutes to allow the samples to reach equilibrium before measuring A/8-oxoG-binding to SpMyh1-Chimera or MBP-SpMyh1-WT in the presence of the unlabeled duplex DNA competitor with either the C:G pair or A/8-oxoG mispair. The measured anisotropy values were analyzed as a function of competitor DNA concentration, similar to what was described64. Plots were made of anisotropy versus competitor concentration to measure apparent inhibition constants (Ki,app) for the competitor DNA substrates. The Ki,app measurements were calculated using the equation64:
| (2) |
Where Atotal is the measured anisotropy, ADNA is the anisotropy of the labeled DNA alone, Amax is the maximum observed anisotropy shift (i.e. in the absence of competitor DNA), and [I] is the concentration of the competitor DNA. The Ki,app measurements estimate what concentrations of competitor DNA are needed to achieve half-maximal binding to SpMyh1-Chimera or MBPSpMyh1-WT.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Antony Carr (MRC Cell Mutation Unit, United Kingdom), Charles Hoffman (Boston College) and Paul Russell (The Scripps Research Institute) for kindly providing the S. pombe strains. We are grateful to Dr. Emma Warbrick at University of Dundee, United Kingdom for providing the plasmid p4XG. We acknowledge Dr. Laura Mizoue of the Center for Structural Biology at Vanderbilt University for providing the expression plasmid used to produce recombinant SpMyh1-WT and SpMyh-Chimera proteins. We also thank the staffs of the University of Maryland Marlene and Stewart Greenebaum Cancer Center Structural Biology Shared Service and beamline X6A (National Synchrotron Light Source, Brookhaven National Laboratory) for assistance with X-ray data collection. Our initial efforts were supported by an Institutional Research Grant from the American Cancer Society (IRG-97-153-07, to Alan Tomkinson). This work was further supported by grants (GM35132 and CA78391) from the National Institute of Health to AL and by grant RSG-09-058-01-GMC from the American Cancer Society to EAT.
Abbreviations used
- IDC
interdomain connector
- Ec-cMutY
Escherichia coli MutY catalytic domain
- BstMutY
Bacillus stearothermophilus MutY
- MYH or MUTYH
MutY homologue
- MAP
MYH-associated polyposis
- 8-oxoG
7,8-dihydro-8-oxoguanine
- BER
base excision repair
- AP
apurinic/apyrimidinic
- APE1
AP-endonuclease 1
- S. pombe
Schizosaccharomyces pombe
- GST
glutathione S-transferase
- 9-1-1
Rad9-Rad1-Hus1
- GFP
green fluorescent protein
- MBP
maltose-binding protein
- FOA
5-fluoro-orotic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS: The coordinates for the structure of hMYH(65-350) have been deposited in the Protein Data Bank with accession code 3N5N.
References
- 1.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 2.Collins AR. Oxidative DNA damage, antioxidants, and cancer. Bioessays. 1999;21:238–46. doi: 10.1002/(SICI)1521-1878(199903)21:3<238::AID-BIES8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 3.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–4. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 4.Moriya M, Grollman AP. Mutations in the mutY gene of Escherichia coli enhance the frequency of targeted G:C-->T:a transversions induced by a single 8-oxoguanine residue in single-stranded DNA. Mol Gen Genet. 1993;239:72–6. doi: 10.1007/BF00281603. [DOI] [PubMed] [Google Scholar]
- 5.Wood ML, Dizdaroglu M, Gajewski E, Essigmann JM. Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry. 1990;29:7024–32. doi: 10.1021/bi00482a011. [DOI] [PubMed] [Google Scholar]
- 6.Lu AL, Li X, Gu Y, Wright PM, Chang DY. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem Biophys. 2001;35:141–70. doi: 10.1385/CBB:35:2:141. [DOI] [PubMed] [Google Scholar]
- 7.Mol CD, Parikh SS, Putnam CD, Lo TP, Tainer JA. DNA repair mechanisms for the recognition and removal of damaged DNA bases. Annu Rev Biophys Biomol Struct. 1999;28:101–28. doi: 10.1146/annurev.biophys.28.1.101. [DOI] [PubMed] [Google Scholar]
- 8.Chang DY, Gu Y, Lu AL. Fission yeast (Schizosaccharomyces pombe) cells defective in the MutY-homologous glycosylase activity have a mutator phenotype and are sensitive to hydrogen peroxide. Mol Genet Genomics. 2001;266:336–42. doi: 10.1007/s004380100567. [DOI] [PubMed] [Google Scholar]
- 9.Hirano S, et al. Mutator phenotype of MUTYH-null mouse embryonic stem cells. J Biol Chem. 2003;278:38121–4. doi: 10.1074/jbc.C300316200. [DOI] [PubMed] [Google Scholar]
- 10.Al-Tassan N, et al. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–32. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 11.Halford SE, et al. Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. Am J Pathol. 2003;162:1545–8. doi: 10.1016/S0002-9440(10)64288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones S, et al. Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C-->T:A mutations. Hum Mol Genet. 2002;11:2961–7. doi: 10.1093/hmg/11.23.2961. [DOI] [PubMed] [Google Scholar]
- 13.Sampson JR, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39–41. doi: 10.1016/S0140-6736(03)13805-6. [DOI] [PubMed] [Google Scholar]
- 14.Cheadle JP, Sampson JR. MUTYH-associated polyposis--from defect in base excision repair to clinical genetic testing. DNA Repair (Amst) 2007;6:274–9. doi: 10.1016/j.dnarep.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Kundu S, Brinkmeyer MK, Livingston AL, David SS. Adenine removal activity and bacterial complementation with the human MutY homologue (MUTYH) and Y165C, G382D, P391L and Q324R variants associated with colorectal cancer. DNA Repair (Amst) 2009;8:1400–10. doi: 10.1016/j.dnarep.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai H, et al. Functional characterization of human MutY homolog (hMYH) missense mutation (R231L) that is linked with hMYH-associated polyposis. Cancer Lett. 2006 doi: 10.1016/j.canlet.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molatore S, et al. MUTYH mutations associated with familial adenomatous polyposis: functional characterization by a mammalian cell-based assay. Hum Mutat. 31:159–66. doi: 10.1002/humu.21158. [DOI] [PubMed] [Google Scholar]
- 18.Bai H, et al. Functional characterization of two human MutY homolog (hMYH) missense mutations (R227W and V232F) that lie within the putative hMSH6 binding domain and are associated with hMYH polyposis. Nucleic Acids Res. 2005;33:597–604. doi: 10.1093/nar/gki209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellacosa A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ. 2001;8:1076–92. doi: 10.1038/sj.cdd.4400948. [DOI] [PubMed] [Google Scholar]
- 20.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 21.Chang DY, Lu AL. Functional interaction of MutY homolog with proliferating cell nuclear antigen in fission yeast, Schizosaccharomyces pombe. J Biol Chem. 2002;277:11853–8. doi: 10.1074/jbc.M111739200. [DOI] [PubMed] [Google Scholar]
- 22.Parker A, et al. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J Biol Chem. 2001;276:5547–55. doi: 10.1074/jbc.M008463200. [DOI] [PubMed] [Google Scholar]
- 23.Boldogh I, et al. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 2001;29:2802–9. doi: 10.1093/nar/29.13.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu Y, et al. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J Biol Chem. 2002;277:11135–42. doi: 10.1074/jbc.M108618200. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi H, et al. Replication-associated repair of adenine:8-oxoguanine mispairs by MYH. Curr Biol. 2002;12:335–9. doi: 10.1016/s0960-9822(02)00686-3. [DOI] [PubMed] [Google Scholar]
- 26.Murakami H, Nurse P. DNA replication and damage checkpoints and meiotic cell cycle controls in the fission and budding yeasts. Biochem J. 2000;349:1–12. doi: 10.1042/0264-6021:3490001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinert T. DNA damage checkpoints update: getting molecular. Curr Opin Genet Dev. 1998;8:185–93. doi: 10.1016/s0959-437x(98)80140-8. [DOI] [PubMed] [Google Scholar]
- 28.Dore AS, Kilkenny ML, Rzechorzek NJ, Pearl LH. Crystal structure of the rad9-rad1-hus1 DNA damage checkpoint complex--implications for clamp loading and regulation. Mol Cell. 2009;34:735–45. doi: 10.1016/j.molcel.2009.04.027. [DOI] [PubMed] [Google Scholar]
- 29.Sohn SY, Cho Y. Crystal structure of the human rad9-hus1-rad1 clamp. J Mol Biol. 2009;390:490–502. doi: 10.1016/j.jmb.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 30.Xu M, et al. Structure and functional implications of the human rad9-hus1-rad1 cell cycle checkpoint complex. J Biol Chem. 2009;284:20457–61. doi: 10.1074/jbc.C109.022384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burtelow MA, Roos-Mattjus PM, Rauen M, Babendure JR, Karnitz LM. Reconstitution and molecular analysis of the hRad9-hHus1-hRad1 (9-1-1) DNA damage responsive checkpoint complex. J Biol Chem. 2001;276:25903–9. doi: 10.1074/jbc.M102946200. [DOI] [PubMed] [Google Scholar]
- 32.Shiomi Y, et al. Clamp and clamp loader structures of the human checkpoint protein complexes, Rad9-1-1 and Rad17-RFC. Genes Cells. 2002;7:861–8. doi: 10.1046/j.1365-2443.2002.00566.x. [DOI] [PubMed] [Google Scholar]
- 33.Venclovas C, Thelen MP. Structure-based predictions of Rad1, Rad9, Hus1 and Rad17 participation in sliding clamp and clamp-loading complexes. Nucleic Acids Res. 2000;28:2481–93. doi: 10.1093/nar/28.13.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.al-Khodairy F, Carr AM. DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. Embo J. 1992;11:1343–50. doi: 10.1002/j.1460-2075.1992.tb05179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.al-Khodairy F, et al. Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol Biol Cell. 1994;5:147–60. doi: 10.1091/mbc.5.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zou L, Cortez D, Elledge SJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu AL, Bai H, Shi G, Chang DY. MutY and MutY homologs (MYH) in genome maintenance. Front Biosci. 2006;11:3062–80. doi: 10.2741/2033. [DOI] [PubMed] [Google Scholar]
- 38.Chang DY, Lu AL. Interaction of checkpoint proteins Hus1/Rad1/Rad9 with DNA base excision repair enzyme MutY homolog in fission yeast, Schizosaccharomyces pombe. J Biol Chem. 2005;280:408–17. doi: 10.1074/jbc.M406800200. [DOI] [PubMed] [Google Scholar]
- 39.Shi G, et al. Physical and functional interactions between MutY glycosylase homologue (MYH) and checkpoint proteins Rad9-Rad1-Hus1. Biochem J. 2006;400:53–62. doi: 10.1042/BJ20060774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–8. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 41.Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–6. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- 42.Guan Y, et al. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol. 1998;5:1058–64. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 43.Noll DM, Gogos A, Granek JA, Clarke ND. The C-terminal domain of the adenine-DNA glycosylase MutY confers specificity for 8-oxoguanine.adenine mispairs and may have evolved from MutT, an 8-oxo-dGTPase. Biochemistry. 1999;38:6374–9. doi: 10.1021/bi990335x. [DOI] [PubMed] [Google Scholar]
- 44.Volk DE, et al. Structural similarities between MutT and the C-terminal domain of MutY. Biochemistry. 2000;39:7331–6. doi: 10.1021/bi000416p. [DOI] [PubMed] [Google Scholar]
- 45.Gogos A, Cillo J, Clarke ND, Lu AL. Specific recognition of A/G and A/7,8-dihydro-8-oxoguanine (8-oxoG) mismatches by Escherichia coli MutY: removal of the C-terminal domain preferentially affects A/8-oxoG recognition. Biochemistry. 1996;35:16665–71. doi: 10.1021/bi960843w. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Wright PM, Lu AL. The C-terminal domain of MutY glycosylase determines the 7,8-dihydro-8-oxo-guanine specificity and is crucial for mutation avoidance. J Biol Chem. 2000;275:8448–55. doi: 10.1074/jbc.275.12.8448. [DOI] [PubMed] [Google Scholar]
- 47.Lee S, Verdine GL. Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc Natl Acad Sci U S A. 2009;106:18497–502. doi: 10.1073/pnas.0902908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chmiel NH, Golinelli MP, Francis AW, David SS. Efficient recognition of substrates and substrate analogs by the adenine glycosylase MutY requires the C-terminal domain. Nucleic Acids Res. 2001;29:553–64. doi: 10.1093/nar/29.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manuel RC, Lloyd RS. Cloning, overexpression, and biochemical characterization of the catalytic domain of MutY. Biochemistry. 1997;36:11140–52. doi: 10.1021/bi9709708. [DOI] [PubMed] [Google Scholar]
- 50.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 51.Yang H, et al. Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res. 2001;29:743–52. doi: 10.1093/nar/29.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Helt CE, Wang W, Keng PC, Bambara RA. Evidence that DNA damage detection machinery participates in DNA repair. Cell Cycle. 2005;4:529–32. doi: 10.4161/cc.4.4.1598. [DOI] [PubMed] [Google Scholar]
- 53.Doublie S, Bandaru V, Bond JP, Wallace SS. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc Natl Acad Sci U S A. 2004;101:10284–9. doi: 10.1073/pnas.0402051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smet-Nocca C, Wieruszeski JM, Chaar V, Leroy A, Benecke A. The thymine-DNA glycosylase regulatory domain: residual structure and DNA binding. Biochemistry. 2008;47:6519–30. doi: 10.1021/bi7022283. [DOI] [PubMed] [Google Scholar]
- 55.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 56.Manuel RC, Czerwinski EW, Lloyd RS. Identification of the structural and functional domains of MutY, an Escherichia coli DNA mismatch repair enzyme. J Biol Chem. 1996;271:16218–26. doi: 10.1074/jbc.271.27.16218. [DOI] [PubMed] [Google Scholar]
- 57.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–9. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 58.Lu AL, Fawcett WP. Characterization of the recombinant MutY homolog, an adenine DNA glycosylase, from yeast Schizosaccharomyces pombe. J Biol Chem. 1998;273:25098–105. doi: 10.1074/jbc.273.39.25098. [DOI] [PubMed] [Google Scholar]
- 59.Warbrick E. A functional analysis of PCNA-binding peptides derived from protein sequence, interaction screening and rational design. Oncogene. 2006;25:2850–9. doi: 10.1038/sj.onc.1209320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- 61.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 62.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 63.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; Palo Alto, CA, USA: 2008. [Google Scholar]
- 64.Bernstein J, Patterson DN, Wilson GM, Toth EA. Characterization of the essential activities of Saccharomyces cerevisiae Mtr4p, a 3′->5′ helicase partner of the nuclear exosome. J Biol Chem. 2008;283:4930–42. doi: 10.1074/jbc.M706677200. [DOI] [PubMed] [Google Scholar]
- 65.Heilman-Miller SL, Thirumalai D, Woodson SA. Role of counterion condensation in folding of the Tetrahymena ribozyme. I. Equilibrium stabilization by cations. J Mol Biol. 2001;306:1157–66. doi: 10.1006/jmbi.2001.4437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.