

Abstract
Fine-scale population structure characterizes most continents and is especially pronounced in non-cosmopolitan populations. Roughly half of the world's population remains non-cosmopolitan and even populations within cities often assort along ethnic and linguistic categories. Barriers to random mating can be ecologically extreme, such as the Sahara Desert, or cultural, such as the Indian caste system. In either case, subpopulations accumulate genetic differences if the barrier is maintained over multiple generations. Genome-wide polymorphism data, initially with only a few hundred autosomal microsatellites, have clearly established differences in allele frequency not only among continental regions, but also within continents and within countries. We review recent evidence from the analysis of genome-wide polymorphism data for genetic boundaries delineating human population structure and the main demographic and genomic processes shaping variation, and discuss the implications of population structure for the distribution and discovery of disease-causing genetic variants, in the light of the imminent availability of sequencing data for a multitude of diverse human genomes.
INTRODUCTION
Although the majority of human genetic variation is shared across human populations (e.g. Fst among continental groups generally does not exceed 15%), nearly four decades of empirical human genetic research have documented the existence of thousands of genetic markers with marked allele frequency differences among populations. For example, seminal work on documenting global patterns of protein polymorphisms by Cavalli-Sforza et al. (1) culminated in ‘The History and Geography of Human Genes', which describes spatial genetic gradients across continents and highlights the concordance between gene trees and language groupings. Rapidly evolving mtDNA and Y-chromosome molecular polymorphisms were later used (and continue to be used) to trace hundreds of sex-specific migrations among groups. More recently, autosomal microsatellites and single nucleotide polymorphism (SNP) arrays have been used to reveal population structure via analysis of allele frequency differences among populations. A variety of statistical approaches have been used, ranging from classical descriptors, such as Wright's F statistics [which are akin to factors in hierarchical analysis of variance (2)] to non-parametric methods [such as principal component analysis of individual genotype data (3)] to fully parametric models of the joint site-frequency spectrum (4) or shared haplotype patterns from multiple populations (5). Genome-wide polymorphism data, initially with only a few hundred autosomal microsatellites, have clearly established differences in allele frequency among continental regions (6–8). More recently, data derived from single nucleotide polymorphism arrays found evidence of appreciable fine-scale structure within continents, and even within countries. For example, genome-wide autosomal SNP data have revealed strong statistical evidence for genetic substructure within Europe (9–12), the Near East (7), India (13,14), China (7), the Americas (15) and Africa (B.M.H., in preparation), to highlight only a few.
Often, patterns of genetic substructure suggest that geographic barriers to gene flow play a key role. For example, using nearly 200 000 common SNPs, Novembre et al. (9) recreated a ‘geographic map' of Europe by projecting the first two principal axes of genetic variation onto geographic coordinates (10–12). This gradient of variation across Europe indicates how population structure need not have sharp boundaries; if individuals in a uniform extended territory tend to choose mates from neighboring groups, the genetic composition of the population will vary continuously with geography. This phenomenon can clearly extend to variation within countries, and genetic evidence exists for fine-scale substructure within Finland (16), Japan (17), Mexico (18), Qatar (19) and South Africa (20). However, not all regions appear to have clearly differentiated populations; western Africa is striking for having very little fine-scale structure, at least at the level of resolution captured by common SNP data (21,22), even though these data included populations of Bantu and Non-Bantu Niger-Kordofanian, Afro-Asiatic and Nilo-Saharan speakers spread out over broad geographic regions. This suggests extensive gene flow in the region, a large effective population size and, by consequence, high levels of genomic diversity.
Broad patterns of population structure among humans are largely the result of ancient demographic events. According to the standard model for human evolution (23,24), ∼50 000–60 000 years ago, a small group of eastern Africans, perhaps as few as 1000 individuals, drifted out of Africa and into similar terrain in the Near East. In individuals living outside of Africa, this 50 000-year-old bottleneck during the Out of Africa exit remains the most visible demographic imprint reflected in their genomes, contributing substantially to reduced heterozygosity in non-Africans. After this bottleneck, the population rapidly diverged; one population traced the coast of the Indian Ocean, reaching Australia in only a few thousand years (25). Another, separate population expanded more slowly from the Near East northward into the European continent (24). Serial founder migrations, where populations sequentially grow and bud off to find the next population, continued until the majority of the Eurasian and Oceanic landmasses were populated by 30 000 years ago; the North and South American continents were populated by a similar process beginning about 15 000 years ago. Clustering algorithms and principal component analysis of autosomal markers clearly reconstruct these continental boundaries among human populations (Fig. 1). Subsequent migrations associated with multiple inventions of agriculture have served to homogenize genetic variation within continents and, to a lesser extent, between major geographic regions when, for example, Neolithic agriculturalists expanded from the Near East into Europe about 8000 years ago. The genetic proportion of European ancestry that traces back to these recent Neolithic farmers remains under considerable debate (1). In contrast, fine-scale population structure probably stems from demographic processes that have occurred mostly in the past few thousand years, reflecting recent endogamous mating within populations that either have a similar language or culture, or live in a geographically circumscribed location such as an island.
Figure 1.

Population structure of worldwide human populations. Individual ancestry proportions are shown for 1112 individuals from 42 different populations globally distributed (from left to right: 114 Africans, 100 African-Americans, 255 Europeans, 108 South Asians, 100 East Asians, 36 Oceanians, 186 Native Americans and 212 Hispanic/Latinos). Each individual is represented by a vertical line on each barplot and sorted in the same order across panels. A clustering algorithm (implemented in ADMIXTURE) was used to infer global genome estimates from K = 5 up to K = 9 ancestral populations per individual. At K = 5 clusters roughly correspond to continental regions and at K = 9 fine-scale subcontinental structure can be detected (note the South–North gradient in Europeans and the light blue component decreasing from North to South in Native Americans). Admixed populations such as African-Americans and Hispanic/Latinos show dramatic variation in admixture proportions between individuals within populations and among populations. The plot was generated using 73 901 autosomal SNP markers by intersecting genome-wide genotype data from four publicly available data sets [i.e. HGDP (7), POPRES (26), data from Mao et al. (55) and data from Bryc et al. (21)].
FINE-SCALE POPULATION STRUCTURE
If we think of fine-scale population structure as emerging from very recent patterns of endogamous mating, we may visualize populations as extended, multi-generational pedigrees. Even though such a perspective has been traditionally reserved for a small number of isolated populations, a survey of 70 populations across the world found that individuals within populations often share large identical fragments of their genome by common descent from shared ancestors (B.M.H., in preparation). Individuals can even contain long runs of homozygosity (i.e. identical tracts on both chromosomes) that indicate a recent shared ancestor; runs of homozygosity are elevated, for example, in Mexicans and Scottish island populations (26,27). The amount of genomic sharing can be easily quantified and the degree of relationships between a random pair of individuals in a population can be estimated. For example, a random pair of individuals in the Ashkenazi Jewish population are as genetically similar, on average, as fourth cousins (28), indicating that recent genealogy may be of importance. The impact of inbreeding on the frequency of rare diseases has been demonstrated in historically endogamous populations, such as the Ashkenazim, Hutterites and some island groups (29,30). However, if many geographic regions throughout the world are finely structured as suggested by identical-by-descent (IBD) analysis, then populations in those regions may also have elevated rates of rare alleles and correspondingly of unique rare diseases. Indeed, in terms of the average amount of genomic segments' IBD, the Ashkenazim are not outliers in the global sample mentioned above. Many populations simply have not been systematically surveyed for unique variants or frequencies of rare diseases, which may differ from a random cosmopolitan sample.
SOURCES OF ADDITIONAL DIFFERENTIATION: DEMOGRAPHY AND NATURAL SELECTION
The population differentiation caused by endogamous mating can be amplified by additional evolutionary forces. Demographic history may vary wildly between groups within substructured populations and contribute to genetic differentiation. Although populations outside of Africa have experienced an ancient, severe bottleneck, regional populations may have been differentially affected by more recent founder events [e.g. Hutterites (31), Ashkenazim (28), Québécois (32)] or rapid population growth due to technological innovations such as agriculture. The amount of genetic diversity as measured, for example, by the distribution of allele frequencies in a subpopulation (i.e. the site-frequency spectrum) can be used to distinguish between different possible demographic histories of the subpopulations (33,34). Demography, therefore, also influences the amount of standing variation on which natural selection can act. Populations that have undergone recent growth have a higher proportion of rare (<5% in frequency), young variants. Recent variants have had less time to be affected by natural selection, and therefore are proportionally more likely to be deleterious (35): demography can therefore impact the number of deleterious alleles and heterozygous sites a given individual is expected to carry.
Historic differences in selection pressure among populations are another archetypal cause of population allele frequency differentiation (36,37). A survey of loci under selection in eight different regions of the world found that the top candidates for selective sweeps were generally non-overlapping (38). The exception was Europe, the Middle East and Central Asian populations that generally shared similar signals for selection; this may be due to recent shared ancestry and dietary environments following the diffusion of agriculture through this region. However, closely related or geographically proximate populations need not necessarily have experienced identical selective histories. For example, Simonson et al. (39) recently described decreased hemoglobin count (i.e. affected by variation in the EGLN1 and PPARA genes) as an adaptation to the high-altitude environment of the Tibetan Plateau. They confirm that this high-altitude adaption was not present in lowland Chinese and posit that the adaptive process occurred over a very brief evolutionary time span, as they infer the time of divergence between Tibetans and Han Chinese to be only about 3000 years.
Conversely, even if populations have experienced similar selection pressures for similar phenotypes, different genetic variants in separate populations can sweep to high frequency. Both the evolution of light skin pigmentation in Europeans and East Asians and the evolution of lactase persistence exemplify phenotypic convergence in distinct human populations, but the causal variants behind these phenotypes are independent. Scans for positive selection between continentally defined populations revealed that numerous genomic loci related to skin pigmentation are in the upper tail of distributions of allele frequency differences (30). Light skin pigmentation is highly correlated with UV exposure, primarily on latitudinal gradients (38,40). Both European and East Asian populations occupy higher latitudes, but at least four genes (TRYP1, SLC45A2, SLC24A5 and MC1R) affecting skin pigmentation were identified in markers with the highest 5% of Fst between these two groups, suggesting differential fixation of selected alleles may underlie some degree of skin color adaptations. In another example, at least four different lactase persistence alleles, an adaptation for prolonged, adult consumption of sugar from milk, have arisen in pastoralist groups around the world, including northern Europe, the Near East (41) and eastern Africa (42), although these alleles are all upstream of a single gene, LCT. In sum, natural selection can differentiate populations at a variety of genotypes and phenotypes, such as hemoglobin count, skin pigmentation, lactase persistence as well as medically relevant phenotypes such as type II diabetes (38) and kidney disease (43).
LARGE-SCALE RESEQUENCING AND POPULATION STRUCTURE
Large-scale patterns of human population genetic structure are evident from allele frequency differences among a host of classical (i.e. protein polymorphism, mtDNA, Y-chromosome) genetic markers. The past decade has seen characterization of fine-scale population structure through genetic analysis and compounding of small allele frequency differences across hundreds of thousands of common genetic markers. What remains unknown is the degree to which these slight differences in allele frequency, while allowing us to characterize recent genetic ancestry for individuals, are relevant for the design and interpretation of medical genomic studies of populations. In particular, it is critical to assess the extent to which relatively rare (0.1–5%) genetic variation is shared among populations. Our understanding of the ‘rare allele frequency spectrum', and its implications for medical genetics, history and human evolution, therefore has the most to gain from genomic studies sequencing a large number of samples within a population or across a set of closely related populations. Multiple existing and projected large-scale data sets and experimental programs fulfill this criterion, including the Human Genome Diversity Panel (HGDP) (44), the Personal Genome Project (45), the UK10K project (www.uk10k.org), the HapMap project (www.hapmap.org), the NIEHS environmental genome project (http://www.niehs.nih.gov/research/supported/programs/egp/), ENCODE (http://www.genome.gov/10005107) and the 1000 Genomes Project (1000G) (1000 Genomes Consortium, in preparation).
Many of these projects have multiple goals, combining medical and fundamental research. The choice of a sequencing or genotyping strategy reflected these goals and took into account population structure. A common design, used by the 1000G, as well as HGDP, NIEHS and HapMap, consists of choosing samples comprising numerous individuals from a limited number of populations around the world, in the hope of capturing both large-scale diversity and local variation.
The 1000G sets out to dramatically improve both the number of diverse populations with sequenced individuals and the number of individuals sequenced per population. One stated goal of the project is to identify 95% of the genome-wide variants above 1% in frequency, down to 0.1% frequency in coding sequence. The project has already released whole-genome data for 180 individuals in the YRI, CEU, CHB and JPT populations, with a mean sequencing coverage of 2–4×, and target capture data for exons of 1000 genes for additional populations and samples (Table 1). The main project will include both whole-genome (low coverage) and whole-exome (high coverage) data for 2500 samples drawn from 27 populations distributed in five groups (America, East Asia, Europe, South Asia, West Africa, see http://www.1000genomes.org). Interestingly, the role of population structure in the design of the TGP was not limited to sample choice, but also impacted the sequencing methodology: to compensate for the low coverage necessary to affordably sequence a very large sample size, data from similar populations are pooled and population structure is explicitly used to improve the quality of the variant calls (1000 Genomes Consortium, in preparation).
Table 1.
Currently available HapMap and 1000G populations, with numbers of individuals genotyped in HapMap or sequenced in the 1000 Genomes Project pilot phase
| Population and location (abbreviation) | HapMap: sample size | 1000 genomes: sample size |
|
|---|---|---|---|
| Whole genome | Capture | ||
| African-American in southwest USA (ASW) | 90 | ||
| European ancestry in Utah, USA (CEU) | 180 | 60 | |
| Chinese from Beijing, China (CHB) | 90 | 30 | 109 |
| Chinese in Denver, USA (CHD) | 100 | 107 | |
| Gujurati from Houston, USA (GIH) | 100 | ||
| Japanese from Tokyo, Japan (JPT) | 91 | 30 | 105 |
| Luhya from Webuye, Kenya (LWK) | 100 | 108 | |
| Maasai from Kinyawa, Kenya (MKK) | 180 | ||
| Mexican from Los Angeles, USA (MXL) | 90 | ||
| Tuscans from Italy (TSI) | 100 | 66 | |
| Yoruba from Ibadan (YRI) | 180 | 59 | 112 |
The capture data targeted the exons of 1000 genes. Additional data for the 2500 samples currently or soon to be to be sequenced by the 1000G project can be found at www.1000genomes.org.
ADMIXTURE
Patterns of endogamy that give rise to population structure are not static: as migration to urban centers has increased and migration between continents accelerated over the past 500 years, individuals from many different, divergent populations have come into contact, and many models predict that only a limited amount of migration is required to largely eliminate differences in population frequencies.
However, the time to reach equilibrium is dependent on many factors (most notably degree of non-random mating) and, in this respect, population structure is still relevant in cosmopolitan communities or populations that have absorbed large numbers of migrants. Admixed individuals may contain haplotypes with deeply divergent ancestries (Fig. 2), such as African-Americans who average roughly 75% western African, 20% European and 5% Native American ancestry (21,46). Furthermore, individuals can vary widely in their proportions of different ancestries (Fig. 1). Differences in genome-wide ancestry proportions may reflect different population histories between proximal populations. Such is the case of the Caribbean populations which exhibit much higher proportions of West African ancestry as a result of their proximity to classical slave trade routes, compared with other Latin American populations from the continental landmass (Fig. 1).
Figure 2.
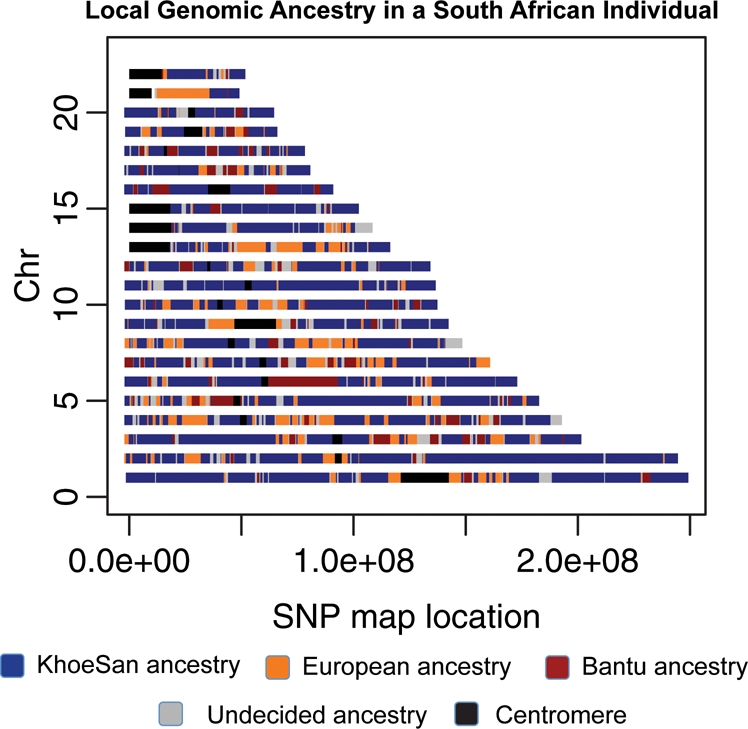
Local ancestry assigned along the genome for a South African individual. Using SNPs from the Illumina 550K array platform, we use a principal component-based method to assign different ancestries to segments of a genome (21). We assumed three possible ancestral source populations for an admixed South African individual: indigenous southern KhoeSan ancestry, Bantu ancestry represented by the Kenyan Luhya and recent European (Italian) ancestry (data from HapMap3 and Henn et al., in preparation). Data were phased and the distance from each admixed haplotype to the nearest ancestral source population was measured in 40 SNP windows. Only one phase for each chromosome is plotted (for ease of visualization), and chromosome X is not shown. Ancestry with a low posterior probability is plotted in gray.
In many cases, it is possible to identify the ancestry of genomic segments: the most accurate method of assigning ancestry in admixed individuals is an area of active research (21,47,48). Such local ancestry assignment, or ‘admixture deconvolution', can be exploited in admixture mapping, and in inference of past demographic events (49). Local ancestry assignment will be empowered by the increased description of rare variants (at frequencies of 0.1–5%), such as those discovered by the 1000 Genomes Project. Rare variants are likely to have recently arisen and segregate between populations and are informative markers of ancestry.
CONCLUSION
New sequencing technology enables genetic studies with larger and larger sample sizes, increasing our power to detect associations between genetic variants and medically relevant traits, especially in the case of rare variants. Understanding patterns of admixture and population structure is an important part of maximizing this detection power and reducing confounding factors in genome sequencing-to-phenotype association studies (50–54). As we have seen in the analysis of dense array genotype data, the same sequencing technology that enables sequencing/phenotype mapping will also enable us to improve our knowledge of population structure at a fine scale. This improved knowledge should be of assistance not only in identifying structure in association studies, but also in the description of human history and genetic adaptation, and in the development of personalized medicine tools.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by NIH grants 1R01GM083606 (C.D.B., A.M.-E.), 2R01HG003229 (C.D.B., B.M.H.) and 1RC2HL102926 (C.D.B., S.G.).
REFERENCES
- 1.Cavalli-Sforza L.L., Menozzi P., Piazza A. The History and Geography of Human Genes. Princeton University Press; 1994. [Google Scholar]
- 2.Nievergelt C.M., Libiger O., Schork N.J. Generalized analysis of molecular variance. PLoS Genet. 2007;3:e51. doi: 10.1371/journal.pgen.0030051. doi:10.1371/journal.pgen.0030051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price A.L., Butler J., Patterson N., Capelli C., Pascali V.L., Scarnicci F., Ruiz-Linares A., Groop L., Saetta A.A., Korkolopoulou P., et al. Discerning the ancestry of European Americans in genetic association studies. PLoS Genet. 2008;4:e236. doi: 10.1371/journal.pgen.0030236. doi:10.1371/journal.pgen.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gutenkunst R.N., Hernandez R.D., Williamson S.H., Bustamante C.D. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009;5:e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beaumont M.A., Zhang W., Balding D.J. Approximate Bayesian computation in population genetics. Genetics. 2002;162:2025–2035. doi: 10.1093/genetics/162.4.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.-C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R., et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. doi:10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 7.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. doi:10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg N.A., Pritchard J.K., Weber J.L., Cann H.M., Kidd K.K., Zhivotovsky L.A., Feldman M.W. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. doi:10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 9.Novembre J., Johnson T., Bryc K., Kutalik Z., Boyko A.R., Auton A., Indap A., King K.S., Bergmann S., Nelson M.R., et al. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. doi:10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heath S.C., Gut I.G., Brennan P., McKay J.D., Bencko V., Fabianova E., Foretova L., Georges M., Janout V., Kabesch M., et al. Investigation of the fine structure of European populations with applications to disease association studies. Eur. J. Hum. Genet. 2008;16:1413–1429. doi: 10.1038/ejhg.2008.210. doi:10.1038/ejhg.2008.210. [DOI] [PubMed] [Google Scholar]
- 11.Lao O., Lu T.T., Nothnagel M., Junge O., Freitag-Wolf S., Caliebe A., Balascakova M., Bertranpetit J., Bindoff L.A., Comas D., et al. Correlation between genetic and geographic structure in Europe. Curr. Biol. 2008;18:1241–1248. doi: 10.1016/j.cub.2008.07.049. doi:10.1016/j.cub.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 12.McEvoy B.P., Montgomery G.W., McRae A.F., Ripatti S., Perola M., Spector T.D., Cherkas L., Ahmadi K.R., Boomsma D., Willemsen G., et al. Geographical structure and differential natural selection among North European populations. Genome Res. 2009;19:804–814. doi: 10.1101/gr.083394.108. doi:10.1101/gr.083394.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reich D., Thangaraj K., Patterson N., Price A.L., Singh L. Reconstructing Indian population history. Nature. 2009;461:489–494. doi: 10.1038/nature08365. doi:10.1038/nature08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majumder P.P. The human genetic history of South Asia. Curr. Biol. 2010;20:R184–R187. doi: 10.1016/j.cub.2009.11.053. doi:10.1016/j.cub.2009.11.053. [DOI] [PubMed] [Google Scholar]
- 15.Risch N., Choudhry S., Via M., Basu A., Sebro R., Eng C., Beckman K., Thyne S., Chapela R., Rodriguez-Santana J.R., et al. Ancestry-related assortative mating in Latino populations. Genome Biol. 2009;10:R132. doi: 10.1186/gb-2009-10-11-r132. doi:10.1186/gb-2009-10-11-r132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jakkula E., Rehnström K., Varilo T., Pietiläinen O.P.H., Paunio T., Pedersen N.L., deFaire U., Järvelin M.-R., Saharinen J., Freimer N., et al. The genome-wide patterns of variation expose significant substructure in a founder population. Am. J. Hum. Genet. 2008;83:787–794. doi: 10.1016/j.ajhg.2008.11.005. doi:10.1016/j.ajhg.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamaguchi-Kabata Y., Nakazono K., Takahashi A., Saito S., Hosono N., Kubo M., Nakamura Y., Kamatani N. Japanese population structure, based on SNP genotypes from 7003 individuals compared to other ethnic groups: effects on population-based association studies. Am. J. Hum. Genet. 2008;83:445–456. doi: 10.1016/j.ajhg.2008.08.019. doi:10.1016/j.ajhg.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silva-Zolezzi I., Hidalgo-Miranda A., Estrada-Gil J., Fernandez-Lopez J.C., Uribe-Figueroa L., Contreras A., Balam-Ortiz E., del Bosque-Plata L., Velazquez-Fernandez D., Lara C., et al. Analysis of genomic diversity in Mexican Mestizo populations to develop genomic medicine in Mexico. Proc. Natl Acad. Sci. USA. 2009;106:8611–8616. doi: 10.1073/pnas.0903045106. doi:10.1073/pnas.0903045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunter-Zinck H., Musharoff S., Salit J., Al-Ali K.A., Chouchane L., Gohar A., Matthews R., Butler M.W., Fuller J., Hackett N.R., et al. Population genetic structure of the people of Qatar. Am. J. Hum. Genet. 2010;87:17–25. doi: 10.1016/j.ajhg.2010.05.018. doi:10.1016/j.ajhg.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Wit E., Delport W., Rugamika C.E., Meintjes A., Möller M., van Helden P.D., Seoighe C., Hoal E.G. Genome-wide analysis of the structure of the South African coloured population in the Western Cape. Hum. Genet. 2010;128:145–153. doi: 10.1007/s00439-010-0836-1. doi:10.1007/s00439-010-0836-1. [DOI] [PubMed] [Google Scholar]
- 21.Bryc K., Auton A., Nelson M.R., Oksenberg J.R., Hauser S.L., Williams S., Froment A., Bodo J.-M., Wambebe C., Tishkoff S.A., et al. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc. Natl Acad. Sci. USA. 2010;107:786–791. doi: 10.1073/pnas.0909559107. doi:10.1073/pnas.0909559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zakharia F., Basu A., Absher D., Assimes T.L., Go A.S., Hlatky M.A., Iribarren C., Knowles J.W., Li J., Narasimhan B., et al. Characterizing the admixed African ancestry of African Americans. Genome Biol. 2009;10:R141. doi: 10.1186/gb-2009-10-12-r141. doi:10.1186/gb-2009-10-12-r141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavalli-Sforza L.L., Feldman M.W. The application of molecular genetic approaches to the study of human evolution. Nat. Genet. 2003;33:266–275. doi: 10.1038/ng1113. doi:10.1038/ng1113. [DOI] [PubMed] [Google Scholar]
- 24.Klein R.G. Out of Africa and the evolution of human behavior. Evol. Anthropol. Issues News Rev. 2008;17:267–281. doi:10.1002/evan.20181. [Google Scholar]
- 25.Macaulay V., Hill C., Achilli A., Rengo C., Clarke D., Meehan W., Blackburn J., Semino O., Scozzari R., Cruciani F., et al. Single, rapid coastal settlement of Asia revealed by analysis of complete mitochondrial genomes. Science. 2005;308:1034–1036. doi: 10.1126/science.1109792. doi:10.1126/science.1109792. [DOI] [PubMed] [Google Scholar]
- 26.Auton A., Bryc K., Boyko A., Lohmueller K., Novembre J., Reynolds A., Indap A., Wright M.H., Degenhardt J., Gutenkunst R., et al. Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome Res. 2009;19:795–803. doi: 10.1101/gr.088898.108. doi:10.1101/gr.088898.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McQuillan R., Leutenegger A., Abdel-Rahman R., Franklin C., Pericic M., Barac-Lauc L., Smolej-Narancic N., Janicijevic B., Polasek O., Tenesa A. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. doi:10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atzmon G., Hao L., Pe'er I., Velez C., Pearlman A., Palamara P.F., Morrow B., Friedman E., Oddoux C., Burns E., et al. Abraham's children in the genome era: major Jewish diaspora populations comprise distinct genetic clusters with shared Middle Eastern Ancestry. Am. J. Hum. Genet. 2010;86:850–859. doi: 10.1016/j.ajhg.2010.04.015. doi:10.1016/j.ajhg.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reich D.E., Lander E.S. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/s0168-9525(01)02410-6. doi:10.1016/S0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- 30.Rudan I., Rudan D., Campbell H., Carothers A., Wright A., Smolej-Narancic N., Janicijevic B., Jin L., Chakraborty R., Deka R. Inbreeding and risk of late onset complex disease. J. Med. Genet. 2003;40:925–932. doi: 10.1136/jmg.40.12.925. doi:10.1136/jmg.40.12.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pichler I., Fuchsberger C., Platzer C., Caliskan M., Marroni F., Pramstaller P.P., Ober C. Drawing the history of the Hutterite population on a genetic landscape: inference from Y-chromosome and mtDNA genotypes. Eur. J. Hum. Genet. 2010;18:463–470. doi: 10.1038/ejhg.2009.172. doi:10.1038/ejhg.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tremblay M., Vézina H. Genealogical analysis of maternal and paternal lineages in the Quebec population. Hum. Biol. 2010;82:179–198. doi: 10.3378/027.082.0204. doi:10.3378/027.082.0204. [DOI] [PubMed] [Google Scholar]
- 33.Keinan A., Mullikin J.C., Patterson N., Reich D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat. Genet. 2007;39:1251–1255. doi: 10.1038/ng2116. doi:10.1038/ng2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myers S., Fefferman C., Patterson N. Can one learn history from the allelic spectrum? Theor. Popul. Biol. 2008;73:342–348. doi: 10.1016/j.tpb.2008.01.001. doi:10.1016/j.tpb.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Lohmueller K.E., Indap A.R., Schmidt S., Boyko A.R., Hernandez R.D., Hubisz M.J., Sninsky J.J., White T.J., Sunyaev S.R., Nielsen R., et al. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. doi:10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barreiro L.B., Laval G., Quach H., Patin E., Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nat. Genet. 2008;40:340–345. doi: 10.1038/ng.78. doi:10.1038/ng.78. [DOI] [PubMed] [Google Scholar]
- 37.Xue Y., Zhang X., Huang N., Daly A., Gillson C.J., Macarthur D.G., Yngvadottir B., Nica A.C., Woodwark C., Chen Y., et al. Population differentiation as an indicator of recent positive selection in humans: an empirical evaluation. Genetics. 2009;183:1065–1077. doi: 10.1534/genetics.109.107722. doi:10.1534/genetics.109.107722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pickrell J., Coop G., Novembre J., Kudaravalli S., Li J., Absher D., Srinivasan B., Barsh G., Myers R., Feldman M., et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:711–722. doi: 10.1101/gr.087577.108. doi:10.1101/gr.086652.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simonson T.S., Yang Y., Huff C.D., Yun H., Qin G., Witherspoon D.J., Bai Z., Lorenzo F.R., Xing J., Jorde L.B., et al. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329:72–75. doi: 10.1126/science.1189406. doi:10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- 40.Jablonski N.G., Chaplin G. Human skin pigmentation as an adaptation to UV radiation. Proc. Natl Acad. Sci. USA. 2010;107:8962. doi: 10.1073/pnas.0914628107. doi:10.1073/pnas.0914628107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Enattah N.S., Jensen T.G.K., Nielsen M., Lewinski R., Kuokkanen M., Rasinpera H., El-Shanti H., Seo J.K., Alifrangis M., Khalil I.F. Independent introduction of two lactase-persistence alleles into human populations reflects different history of adaptation to milk culture. Am. J. Hum. Genet. 2008;82:57–72. doi: 10.1016/j.ajhg.2007.09.012. doi:10.1016/j.ajhg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tishkoff S.A., Reed F.A., Ranciaro A., Voight B.F., Babbitt C.C., Silverman J.S., Powell K., Mortensen H.M., Hirbo J.B., Osman M., et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 2007;39:31–40. doi: 10.1038/ng1946. doi:10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oleksyk T.K., Nelson G.W., An P., Kopp J.B., Winkler C.A. Worldwide distribution of the MYH9 kidney disease susceptibility alleles and haplotypes: evidence of historical selection in Africa. PLoS ONE. 2010;5:e11474. doi: 10.1371/journal.pone.0011474. doi:10.1371/journal.pone.0011474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cann H.M., de Toma C., Cazes L., Legrand M.-F., Morel V., Piouffre L., Bodmer J., Bodmer W.F., Bonne-Tamir B., Cambon-Thomsen A., et al. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. doi:10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 45.Church G.M. The personal genome project. Mol. Syst. Biol. 2005;1:0030. doi: 10.1038/msb4100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tishkoff S., Reed F., Friedlaender F., Ehret C., Ranciaro A., Froment A., Hirbo J., Awomoyi A., Bodo J., Doumbo O., et al. The genetic structure and history of Africans and African Americans. Science. 2009;324:1035–1044. doi: 10.1126/science.1172257. doi:10.1126/science.1172257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price A.L., Tandon A., Patterson N., Barnes K.C., Rafaels N., Ruczinski I., Beaty T.H., Mathias R., Reich D., Myers S. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5:e1000519. doi: 10.1371/journal.pgen.1000519. doi:10.1371/journal.pgen.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sankararaman S., Sridhar S., Kimmel G., Halperin E. Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 2008;82:290–303. doi: 10.1016/j.ajhg.2007.09.022. doi:10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pool J.E., Nielsen R. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics. 2009;181:711–719. doi: 10.1534/genetics.108.098095. doi:10.1534/genetics.108.098095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li M., Reilly M.P., Rader D.J., Wang L.-S. Correcting population stratification in genetic association studies using a phylogenetic approach. Bioinformatics. 2010;26:798–806. doi: 10.1093/bioinformatics/btq025. doi:10.1093/bioinformatics/btq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Price A.L., Zaitlen N.A., Reich D., Patterson N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010;11:459–463. doi: 10.1038/nrg2813. doi:10.1038/nrg2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenberg N.A., Huang L., Jewett E.M., Szpiech Z.A., Jankovic I., Boehnke M. Genome-wide association studies in diverse populations. Nat. Rev. Genet. 2010;11:356–366. doi: 10.1038/nrg2760. doi:10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teo Y.Y., Small K.S., Kwiatkowski D.P. Methodological challenges of genome-wide association analysis in Africa. Nat. Rev. Genet. 2010;11:149–160. doi: 10.1038/nrg2731. doi:10.1038/nrg2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian C., Gregersen P.K., Seldin M.F. Accounting for ancestry: population substructure and genome-wide association studies. Hum. Mol. Genet. 2008;17:143–150. doi: 10.1093/hmg/ddn268. doi:10.1093/hmg/ddn268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mao X., Bigham A.W., Mei R., Gutierrez G., Weiss K.M., Brutsaert T.D., Leon-Velarde F., Moore L.G., Vargas E., McKeigue P.M. A genomewide admixture mapping panel for Hispanic/Latino populations. Am. J. Hum. Genet. 2007;80:1171–1178. doi: 10.1086/518564. doi:10.1086/518564. [DOI] [PMC free article] [PubMed] [Google Scholar]