

Abstract
Purpose
Lung squamous cell carcinoma (SCC) is clinically and genetically heterogeneous and current diagnostic practices do not adequately substratify this heterogeneity. A robust, biologically-based SCC subclassification may describe this variability and lead to more precise patient prognosis and management. We sought to determine if SCC mRNA expression subtypes exist, are reproducible across multiple patient cohorts, and are clinically relevant.
Experimental Design
Subtypes were detected by unsupervised consensus clustering in five published discovery cohorts of mRNA microarrays, totaling 382 SCC patients. An independent validation cohort of 56 SCC patients was collected and assayed by microarrays. A nearest-centroid subtype predictor was built using discovery cohorts. Validation cohort subtypes were predicted and evaluated for confirmation. Subtype survival outcome, clinical covariates, and biological processes were compared by statistical and bioinformatic methods.
Results
Four lung SCC mRNA expression subtypes, named primitive, classical, secretory, and basal, were detected and independently validated (P < 0.001). The primitive subtype had the worst survival outcome (P < 0.05) and is an independent predictor of survival (P < 0.05). Tumor differentiation and patient sex were associated with subtype. The subtypes’ expression profiles contained distinct biological processes (primitive – proliferation, classical – xeniobiotics metabolism, secretory – immune response, basal – cell adhesion) and suggested distinct pharmacologic interventions. Comparison to lung model systems revealed distinct subtype to cell type correspondence.
Conclusions
Lung SCC consists of four mRNA expression subtypes that have different survival outcomes, patient populations, and biological processes. The subtypes stratify patients for more precise prognosis and targeted research.
Keywords: lung cancer, squamous cell carcinoma, subtype, cell type, gene expression
STATEMENT OF TRANSLATIONAL RELEVANCE.
Lung squamous cell carcinoma (SCC) has broad clinical, genetic and morphologic heterogeneity. Currently, there is no subclassification that adequately describes this variability and SCC patients are basically treated as though they have the same disease. One explanation for SCC variability is that SCC is not a singular disease but a mixture of multiple discrete diseases or subtypes defined by innate biological differences. Using five discovery cohorts and an independent validation cohort totaling 438 patients, we demonstrate that SCC is composed of four robust mRNA expression subtypes (named primitive, classical, secretory and basal). The subtypes have significantly different survival outcomes, patient populations, and biological processes. Using these subtypes as a basis for a future clinical diagnostic assay, patients could receive a more precise prognosis. Additionally, we described model system partners for the subtypes which can be used for targeted basic research.
INTRODUCTION
Lung cancer is the leading cause of cancer-related death worldwide (1). Squamous cell carcinoma (SCC) is a major histological type and comprises approximately 30% of all pulmonary tumors (2, 3). SCC is defined by the presence of cytoplasmic keratinization and/or desmosomes (intracellular bridges) (4). Clinically, SCC tumors occur more often in smokers and males compared to the other histological types (2, 5). Patients affected with SCC tumors show a wide range of clinical outcomes. For instance, 83% of autopsied SCC patients had regional metastases (5) and 68% of SCC stage I patients survived beyond 5 years (6). Within SCC, there is noticeable morphologic variability, especially among poorly differentiated tumors (4, 7). The WHO SCC type includes a stratification of this variability with four variants (papillary, small cell, clear cell, and basaloid) (4) but their prevalence, clinical and biologic significance remains unclear. Because there is significant pathologic and clinical outcome variability within the SCC histological type, a robust, biologically derived subclassification may be valuable.
Recent years have seen progress in classification of a variety of malignancies using full genome molecular assays, primarily those directed at mRNA expression (e.g. leukemia (8), breast (9), lung adenocarcinoma (10)). A successful approach is unsupervised class discovery, which detects naturally-occurring tumor classes (“mRNA expression subtypes”) without pre-specified characteristics such as patient survival (8). Preliminary efforts have been made in SCC, suggesting the existence of SCC mRNA expression subtypes. In independent analyses, investigators (11-13) discovered two mRNA expression subtypes with intriguing biological profiles and a corresponding patient survival difference. These studies show that SCC might be subclassified using mRNA expression into groups with clinical relevance; however, the studies were not performed in a manner which validated either the number or the nature of these intriguing classes. A validated mRNA expression classification could substantially progress patient care and research in lung SCC. In this study, we describe four novel reproducible expression subtypes (primitive, classical, secretory, and basal) of lung SCC. The SCC subtypes have different survival outcomes, patient demographics, physical characteristics, biological processes, and correspondence to normal lung cell types.
MATERIALS AND METHODS
Tumor collection
Frozen, surgically-extracted, macro-dissected, primary tumors from treatment-naïve patients at the University of North Carolina with a lung SCC diagnosis were collected under Institutional Review Board approved protocols #90-0573 and #07-0120. Morphologic quality control was based on a review of a representative Hematoxylin-and-Eosin stained section from paraffin-embedded tissue immediately adjacent to the frozen tissue for confirmation of squamous histology by four pathologists (Supplement Fig. 1) and for quantification of tumor content. Tumor RNA was extracted (14) and assayed for mRNA expression using Agilent 44,000 probe microarrays for a total of 56 microarrays. Microarrays were processed by normexp background correction and loess normalization (15). This dataset is referred to as the “validation cohort” and was deposited at NCBI1.
Published datasets
A structured search for publicly available SCC mRNA expression microarray datasets was conducted via Gene Expression Omnibus and PubMed and manually selecting datasets that have a large number of lung SCC samples to permit subtype analysis and that have significant cross-dataset gene reliability, as measured by integrative correlations (16). This search yielded five datasets (referred to as the “discovery cohorts”) from the following studies: Bild et al (17), Expression Project for Oncology (Expo)2, Lee et al (18), Raponi et al (13), and Roepman et al (19). Published cohorts contained surgical-resections from treatment-naïve patients if indicated. Clinical data and raw or processed microarray data were obtained. Only microarrays with SCC histology were retained. Raw microarrays or gene lists from lung model systems were obtained (20-23). Microarrays were subjected to standard quality assessments, mapped to a common transcript database and processed into gene-level expression values (Supplement Table 1).
Unsupervised subtype discovery
The subtype discovery and validation procedure is depicted in a flowchart (Supplement Fig. 2). Genes with high reliability and variability were selected similar to previously described methods (9, 10, 12, 13, 16). Gene reliability was measured by integrative correlations and genes exceeding an estimated false discovery rate of 0.1% were retained (16). To select variable genes, genes in each discovery cohort were ranked by median absolute deviation in decreasing order. These ranks were averaged and re-ranked to make a single variable gene list. The top 25% of this ranked list, totaling 2,307 genes, was used for clustering. Prior to clustering, each dataset was gene median centered (24, 25). Subtypes were determined in each discovery cohort by the Consensus Clustering algorithm via ConsensusClusterPlus (26, 27). This algorithm completed 1,000 microarray subsamples at a proportion of 80% and clustered these subsamples by an agglomerative average-linkage hierarchical algorithm using 1 – Pearson correlation coefficient distance. Consensus values, the proportion that two microarrays occupy the same cluster, were calculated and then clustered by an agglomerative average-linkage hierarchical algorithm using Euclidean distance.
Subtype summarization by centroids
Centroids are median expression profiles of a group of arrays and were prepared using methods previously described (25, 28). Centroids were determined by taking a group of microarrays from a gene median centered cohort and obtaining the median of each gene. Multi-cohort centroids are determined by taking a group of centroids and taking the median of each gene.
Differentially expressed genes
Differentially expressed genes were determined by a standardized mean difference procedure that considers between cohort and within cohort variation (29) using the the GeneMeta Bioconductor library3 and a random effects option. Gene set enrichment analysis was used to determine gene sets significantly enriched in ranked gene lists (30).
Validation cohort subtype prediction
Subtype status of the validation cohort was predicted by a nearest-centroid classification algorithm following previously published methods (28). In brief, the predictor was built, using only the discovery cohorts, by adding genes to a balanced centroid, assessing subtype prediction error rates by leave-one-out cross validation, adding genes differentially expressed from the most mis-predicted subtype to its centroid, and stopping once accuracy failed to improve. Subtype predictor centroids, unsupervised gene lists, and all gene multi-cohort centroids are available online4.
Survival analysis
The R library survival was used for survival statistical analyses. Patients dead within one month following surgery were considered to have procedure-related complications and not considered in survival analyses. Five patients met this condition all from the UNC cohort. Relapse-free survival time was defined as the time from surgery until first relapse or death.
Immunohistochemistry
1 mm cores were taken from available UNC cohort tissue blocks and randomly organized into tissue microarray (TMA) blocks. Consequal 4 μm array block sections were assembled on array slides and stained with Hematoxylin & Eosin, MAC387 (Dako, #M0747), p63 (Dako, #M7247), CK7 (Leica Microsystems, #PA0942 RN7), and MCM6 (Santa Cruz Biotechnology, #SC-22781).
Computational procedures were executed using R version 2.7.15 and Bioconductor libraries6 unless otherwise specified.
RESULTS
Unsupervised discovery of lung SCC expression subtypes in five cohorts
Lung squamous cell carcinomas (SCC) are a heterogeneous group of tumors, and therefore, we performed a common set of mRNA expression analyses using 5 previously published lung SCC datasets in order to determine how many distinct subtypes/groups of disease might exist. These five “discovery cohorts” were analyzed for the presence of mRNA expression subtypes using the Consensus Clustering methodology (26) as previously described for lung cancer (10). Consensus Clustering is a semi-quantitative method for determining an optimal number of mRNA expression clusters/groups. Results show that all five cohorts contain four clusters (Supplement Fig. 3). There is no compelling evidence for a higher number of clusters. To test if the four clusters from each cohort have the same expression profiles, a published centroid clustering method was followed (10). The centroid clustering shows a four group structure, where each cohort is in each group, with only one cohort absent in one group (Supplement Fig. 4). Therefore, the four clusters (“mRNA expression subtypes”) found in the five discovery cohorts have consistent expression profiles. To derive the optimal subtype for each patient, a multi-cohort centroid classification was used to assign each patient to a subtype, similar to published methods (28). A centroid clustering based on these optimal subtypes again shows a four group structure and complete, unambiguous cross-cohort correspondence (Fig. 1). The cross-cohort clustering is statistically significant (Sigclust (31) p-values in Fig. 1). Interestingly, the subtypes have approximately the same prevalence among the discovery cohorts (Table 1). Using biological characteristics described below, the lung SCC mRNA expression subtypes are named: primitive, classical, secretory, and basal.
Figure 1. Discovery cohort correlation matrix and dendrogram.

Cells are labeled by discovery cohort and adjusted centroid where A-D are from Supplement Fig. 4. Cells in the matrix represent the 1 – Pearson correlation coefficient between two discovery cohort adjusted centroids by shading according to the scale above (B). For example, BildA and RoepmanA have highly similar expression profiles, have a large Pearson correlation coefficient, a small 1 – Pearson correlation coefficent value and have a cell darkly shaded. The matrix is ordered by columns and rows by the dendrogram at the top of the matrix (A). The dendrogram is the result of an agglomerative, average-linkage, hierarchical clustering using this correlation matrix. The four expression subtypes are marked (C). Statistical significance of the three binary divisions leading to the four subtypes is shown by Sigclust (31) p-values in the dendrogram at the corresponding binary split.
Table 1. Clinical characteristics of lung SCC expression subtypes.
Percent values indicate the proportion of the samples in a particular subtype with a particular variable (e.g. 36% of the primitive subtype samples came from female patients in the discovery cohort). Some percents may not total 100% due to rounding. Overall survival percents are Kaplan-Meier estimates. Gray shading indicates data unavailability.
| Discovery cohorts | Validation cohort (UNC) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Primitive | Classical | Secretory | Basal | Total | Primitive | Classical | Secretory | Basal | Total | ||
| Number of Patients | Bild et al | 7 | 20 | 15 | 10 | 52 | |||||
| Lee et al | 14 | 30 | 22 | 9 | 75 | ||||||
| Expo | 4 | 15 | 11 | 6 | 36 | ||||||
| Raponi et al | 20 | 41 | 32 | 34 | 127 | ||||||
| Roepman et al | 15 | 35 | 19 | 23 | 92 | ||||||
| Total | 60 | 141 | 99 | 82 | 382 | ||||||
| 9 | 21 | 14 | 12 | 56 | |||||||
| Age | Median | 68 | 64 | 66 | 67 | 66 | 65 | 68 | 64 | 72 | 67 |
| Gender | % Female | 36 | 19 | 26 | 29 | 26 | 67 | 33 | 43 | 42 | 43 |
| % Male | 64 | 81 | 74 | 71 | 74 | 33 | 67 | 57 | 58 | 57 | |
| Smoking | % Non-smoker | 8 | 1 | 3 | 2 | 3 | 0 | 0 | 0 | 0 | 0 |
| Mean pack years | 64 | 72 | 60 | 68 | 66 | 43 | 74 | 62 | 46 | 60 | |
| Stage | % I | 66 | 58 | 66 | 55 | 61 | 56 | 57 | 57 | 75 | 61 |
| % II | 25 | 26 | 20 | 37 | 26 | 44 | 33 | 36 | 25 | 34 | |
| % III | 5 | 17 | 14 | 9 | 13 | 0 | 10 | 7 | 0 | 5 | |
| % IV | 3 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Grade | % Poor | 39 | 15 | 21 | 16 | 21 | 56 | 24 | 43 | 33 | 36 |
| % Moderate | 58 | 82 | 76 | 76 | 75 | 44 | 76 | 57 | 67 | 64 | |
| % Well | 3 | 2 | 3 | 8 | 4 | 0 | 0 | 0 | 0 | 0 | |
| Overall Survival | # Patients | 42 | 96 | 66 | 67 | 271 | 8 | 19 | 13 | 11 | 51 |
| % 1 year survival | 64 | 89 | 84 | 88 | 84 | 88 | 100 | 82 | 90 | 92 | |
| % 3 year survival | 47 | 63 | 59 | 71 | 62 | 15 | 38 | 48 | 60 | 41 | |
| % Tumor | Median | 90 | 80 | 80 | 93 | 90 | |||||
| Interquartile range | 75-94 | 60-100 | 70-90 | 73-96 | 60-95 | ||||||
| % Necrosis | Median | 5 | 5 | 5 | 4 | 5 | |||||
| % Fibrosis | Median | 15 | 13 | 18 | 10 | 10 | |||||
| Lymphocytes | % Marked | 33 | 31 | 40 | 40 | 36 | |||||
SCC subtype independent validation
While the four SCC subtypes were “cross-cohort” validated in that they were repeatedly found in five cohorts, this validation was not independent because discovery co-occurred with validation. For an independent validation, we tested the hypothesis that the SCC subtypes will exist in a new, discovery-independent cohort. To test this hypothesis, a subtype predictor was built using the discovery cohorts, which consisted of 208 genes and had 94% leave-one-out cross validation accuracy. Using this predictor, subtype classifications were made for microarrays from a new cohort of 56 lung SCC tumors collected at UNC. All four subtypes were predicted in the UNC cohort and in approximately the same prevalence as the discovery cohorts (Fig. 2; Table 1), which supports subtype reproducibility. To confirm the validity of the predictions, a comparison of expression characteristics between the discovery and UNC cohorts was completed similar to a recent related study (32). We compiled a large validation gene set of the discovery cohorts’ top 100 genes overexpressed and underexpressed per subtype (Fig. 2A), which yielded 1,117 unique genes. Subtype expression patterns are highly concordant between the discovery and UNC cohorts across the validation gene set (Fig. 2A, 2B), confirming the large scale expression patterns are consistent beyond the predictor gene set. In addition, the UNC cohorts’ subtypes are a statistically significant partition of its mRNA expression (SWISSMADE (33) subtypes vs. random classes, P < 0.001). We conclude that the predefined SCC subtypes exist in the UNC cohort and are, therefore, independently validated.
Figure 2. Independent validation of lung SCC expression subtypes.
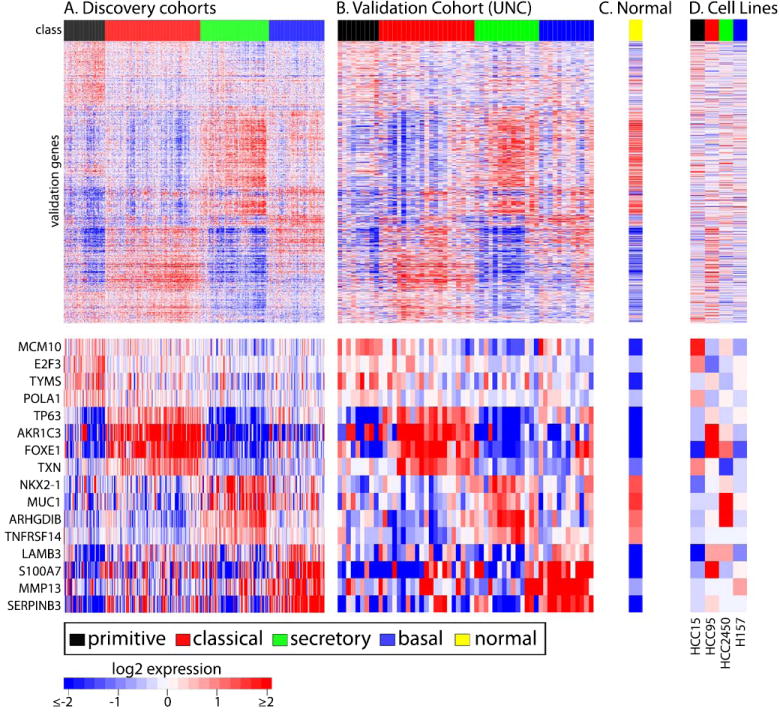
Heatmaps depict mRNA expression of discovery cohorts (A), the validation cohort (B), a normal lung centroid (C), and SCC cell lines (D). Microarrays are columns and are labeled with their class. Genes are rows and are ordered by a discovery cohort hierarchical clustering. The normal lung centroid is scaled to the validation cohort for visualization. Manually-selected, lung-relevant, validation genes are displayed separately for viewability.
To preliminarily evaluate if clinically-applicable biomarkers can distinguish the subtypes, we selected one overexpressed gene per subtype (basal – S100A8; classical – TP63; secretory – KRT7; primitive – MCM6) for immunohistochemical protein expression comparison using a tissue microarray subset of the UNC cohort (N=38). All antibodies targeting these genes except MCM6 had sufficient staining for analysis. Protein expression clustering using basal, classical and secretory samples revealed three essentially mutually exclusive groups with one marker defining each group (Supplement Fig. 5). These groups were significantly associated with tumor subtype (Fisher exact P = 0.007). This suggests that SCC subtypes can also be distinguished by IHC and future work may find the optimal panel of IHC antibodies.
Subtypes exhibit distinct biological processes
In order to discern biological processes associated with each subtype, subtype mRNA expression was evaluated for enrichment in gene ontology, pathway, transcription factor binding site, and cytoband gene sets by the Gene Set Enrichment Analysis (30). Because of the inherent redundancy in biology, we have collapsed these processes into functional themes (Table 2). Here, subtypes are described in terms of overexpression relative to the other subtypes.
Table 2. Subtype biological functional themes.
Significantly enriched gene sets that are overexpressed in a subtype (GSEA pre-ranked, FDR < 0.05) and genes representative of the set are shown. Pathways and biological processes are organized into functional themes, indicated by italics. Transcription factor binding site refers to gene sets having a predicted transcription factor binding site. Cellular component refers to gene sets having a particular cellular location. Drug targets are defined as overexpressed in all pairwise subtype comparisons (FDR < 0.01).
| Primitive subtype |
| Proliferation: cell cycle, DNA replication, pyrimidine metabolism, purine metabolism, mitosis, cell division, DNA replication, cell cycle |
| MCM10, MCM3, MCM6, BUB1, TIMELESS, POLA1, TYMS, ATIC, PRIM1, CKAP5, CDK2, E2F3, E2F8, CHEK1 |
| RNA processing: mRNA processing, rRNA processing, tRNA processing, Nuclear mRNA splicing via spliceosome |
| LSM2, SNRPA, CPSF1, EXOSC5, WDR3, PTBP2, TRMT11 |
| DNA repair: base excision repair, nucleotide excision repair, mismatch repair, DNA repair, response to DNA damage stimulus |
| LIG1, PARP1, UNG, SSRP1, RECQL4, KIF22, FANCA, BARD1, GTF2H4 |
| Cellular component: nucleoplasm, spliceosome, nucleolus |
| Transcription factor binding sites: E2F, NRF |
| Drug targets: TYMS, DNMT1, BCL2, CDK2. |
| Published signatures: Cellular proliferation (34) |
| Classical subtype |
| Energy metabolism: oxidative phosphorylation, citrate cycle, electron transport chain |
| COX5B, NDUFB5, ATP5G3, COX7B, DLD, SDHD, TXN, ATP6V1F |
| Xenobiotics metabolism: metabolism of xenobioitics by cytochrome p450, glutathione metabolism |
| ODC1, GSTA4, GSTM4, GSTO1, GPX2, ALDH3A1, AKR1C3, EPHX1, ADH7, G6PD |
| Cellular component: mitochondrial inner membrane, respiratory chain |
| Cytobands. 3q27-28: TP63, BCL6, ABCC5 |
| Published signatures: Lung cell culture 24 hour smoke exposure (38) |
| Secretory subtype |
| Immune response: complement and coagulation cascade, antigen processing and presentation, natural killer cell mediated cytotoxicity, leukocyte transendothelial migration, B cell receptor signaling, T cell receptor signaling, toll-like receptor signaling, immune response, inflammatory response, innate immune response, cellular defense response, defense response, humoral immune response, T cell activation |
| SERPINA1, C2, C5, VAV1, GZMB, ITGAM, NFKBIE, ARHGDIB, TNFRSF14, HLA-DPA1, IL32, ALOX5, AIF1, DPP4, TCIRG1, TLR2, TLR4, IRF7 |
| Positive regulation of I-kappaB kinase/NF-kappaB cascade: |
| RIPK2, CASP1, RHOA, TGM2, MYD88, APOL3, |
| Transcription factor binding sites: PEA3, NFKB, AML, IRF1 |
| Cellular component: external side of plasma membrane, lysosome |
| Drug targets: C1QA, CSF2RA, CSF2RB, IL3RA, ALOX5, DPYD, SOAT1, TCN2 |
| Basal subtype |
| Cell adhesion: ECM recepor interaction, focal adhesion, cell adhesion, cell matrix adhesion, homophillic adhesion |
| ITGB4, LAMB3, COL11A1, COL17A1, LAMC2, RAC1, ACTN1, PGF, ITGB5, TNFAIP6, CLDN1, HES1, CLSTN1 |
| Epidermal development: epidermis development, keratinocyte differentiation |
| GJB5, S100A7, KRT5, FABP5, COL17A1, LAMC2 |
| Cellular component: proteinaceous extracellular membrane, basement membrane, collagen |
| Drug targets: TCN1, MMP3 |
The distinctive functional theme of the primitive subtype is cellular proliferation, which includes genes such as minichromosome maintenance 10 – MCM10, E2F transcription factor 3 – E2F3, thymidylate synthetase – TYMS and polymerase alpha 1 – POLA1; and a published proliferation signature (34). This proliferation theme is overexpressed in the most rapidly growing breast cancer cell lines (35) and in the most poorly differentiated, poor survival tumors from various organ sites (34). Complementary to the cellular proliferation functional theme, target genes of the E2F transcription factor, a known proliferation modulator (36), are overexpressed in this subtype as well as two members of the E2F family, E2F3 and E2F8. Other primitive subtype functional themes are RNA processing and DNA repair, which could be a consequence of the proliferation theme or an independent process.
The classical subtype exhibits the distinctive functional theme of xenobiotics metabolism, which detoxifies foreign chemicals. One study showed overexpression of this theme in smokers’ versus nonsmokers’ airway transcriptomes including genes such as GPX2 and ALDH3A1 (37). Furthermore, this subtype is enriched with a gene signature derived from lung cell lines exposed to cigarette smoke, including genes such as AKR1C3 (38). Interestingly, the classical subtype has the greatest concentration of smokers and the heaviest smokers among the subtypes. This theme including genes (GPX2, AKR1C1, TXNRD1, GSTM3) was noted as overexpressed in one head and neck squamous cell carcinoma subtype (group 4 in (39)), suggesting a possible relative to the lung SCC classical subtype. The classical subtype overexpresses TP63, a transcription factor essential for stratified squamous epithelium development (40) that is more commonly overexpressed and amplified in lung SCC compared to other histological types (41). Cytoband gene overexpression, a proxy for underlying genomic DNA amplification, suggests 3q27-28, which contains TP63, is amplified in the classical subtype. This study’s microarrays do not have enough resolution to measure TP63 isoform-specific expression, but this may be a goal of future investigations.
Immune response is the major distinctive functional theme of the secretory subtype and includes genes such as Rho GDP dissociation inhibitor beta - ARHGDIB and tumor necrosis factor receptor 14 – TNFRSF14. Consistent with this theme, the secretory subtype has a NF-kappaB regulation theme and NF-kappaB target gene overexpression. This subtype also overexpresses the lung secretory cell markers: mucin – MUC1, pulmonary surfactant proteins - SFTPC, SFTPB, SFTPD (7, 42). Interestingly, thyroid transcription factor 1 – NKX2-1/TTF1, known to be highly expressed in adenocarcinoma (43), is overexpressed in the secretory subtype relative to the other SCC subtypes. This commonality could be a result of adenocarcinoma’s glandular cell structure, which perhaps has secretory properties similar to the SCC secretory subtype. A UNC normal lung centroid shows very similar expression pattern to the secretory subtype over the independent validation gene list, which was selected without considering normal samples (Fig. 2C). To evaluate any possible difference between the secretory subtype samples and normal samples, an unsupervised clustering was completed using only these microarrays (Supplement Fig. 6). Secretory and normal microarrays clustered with their group in essentially all cases; suggesting that the secretory subtype and normal lung are distinct mRNA expression groups.
The basal subtype expression profile shows a cell adhesion functional theme including genes such as the laminins - LAMB3, LAMC2; collagens – COL11A1, COL17A1; integrins – ITGB4, ITGB5; and claudin 1 – CLDN1. Additionally, this subtype has an epidermal development theme, including as keratin 5 - KRT5, psoriasin - S100A7, and gap junction protein beta 5 - GJB5. Several of the basal subtype’s genes, such as COL17A1, LAMC2, and CDH3, are common with a HNSCC subtype (Group I in (39)) and a breast cancer subtype (basal-like in (9)) suggesting these different organ site subtypes may share biological properties. The basal subtype overexpresses several S100 family genes: S100A2, S100A3, S100A7, S100A8, S100A9, S100A12, S100A14. S100A8 and S100A9 are highly expressed in the basal layer in psoriatic epidermal tissue (44). S100A2 is a marker specific for the basal layer of the lung epithelium and SCC (45). KRT5 is a basal layer marker in epithelial tissue (46). The basal subtype is enriched with genes whose products are localized in the basement membrane.
In parallel to differential biological functions are patterns of mRNA expression with implications for pharmacologic intervention (Table 2). For example, TYMS, a target of antifolates including Pemetrexed, is overexpressed in the primitive subtype. The antifolate metabolism pathway is differentially expressed among SCC subtypes with the secretory subtype showing underexpression and similarity to adenocarcinoma (Supplement Fig. 7). Overexpression of TYMS has been shown to be related to Pemetrexed resistance in a dose-dependent manner in lung cancer cell culture (47). Also PARP1, a target of several drugs in development is overexpressed in the primitive subtype.
SCC subtype tumor morphologic and patient characteristics
The subtypes’ morphologic and patient characteristics are displayed in Table 1. Grade is significantly associated with subtype (Fisher exact test P = 0.024). The primitive-subtype has an overrepresentation of poorly differentiated tumors and the basal subtype has an overrepresentation of well differentiated tumors. Tumor stage is not appreciably different among subtypes, although we note that the classical and secretory subtypes have increased proportions of stage III tumors. The surgical cohorts oversample early stages and possibly greater sampling of late stage patients may find additional subtype-stage associations. Specimen quality metrics of percent tumor, percent necrosis, and percent lymphocyte infiltration are not appreciably different among the subtypes, arguing against sampling artifacts as the source of the subtypes. Two cases of WHO morphologic SCC subclass were definitively called by pathologist review (one basaloid in primitive and classical subtypes) suggesting that these SCC morphologic subclasses are rare.
Patient sex approaches statistically significant association with subtype (Fisher exact test P = 0.058). Females are overrepresented in the primitive subtype and males in the classical subtype. Consistent with the classical subtype’s smoking expression profile, the classical subtype has the greatest mean pack years, 73, (Kruskal-Wallis test P = 0.319) and the lowest proportion of non-smokers, 1% (Fisher exact test P = 0.214), although these observations do not meet statistical significance.
SCC subtypes have different patient survival outcomes
Overall and relapse-free survival outcomes are significantly different among SCC subtypes (Fig. 3). The primitive subtype has worse overall and relapse-free survival compared to the other subtypes in all stages and in stage I (Fig. 3), while the basal, secretory and classical appear to have similar outcomes. Considering the UNC cohort alone, the primitive subtype outcome is also worse compared to the other subtypes over all stages (logrank test OS P = 0.066, RFS P = 0.004) and stage I (logrank test OS P = 0.057, RFS P = 0.007). In the UNC cohort, 7/18 recurrences were extrapulmonary and the basal subtype had the lowest number and proportion, 0/3. In order to evaluate the independent contribution of SCC subtype to patient risk in light of known prognostic factors, univariate and multivariate Cox proportional hazard models were constructed (Supplement Table 2). Significant univariate predictors were primitive subtype for overall survival and relapse-free survival and tumor stage for overall survival. Patient age and tumor grade were not significant predictors of either outcome. In multiple variable models, only subtype retained significance for overall survival and relapse-free survival. Tumor stage’s non-significant prediction may be due to the under-representation of late stage patients across the cohorts.
Figure 3. Survival outcomes of SCC subtypes.
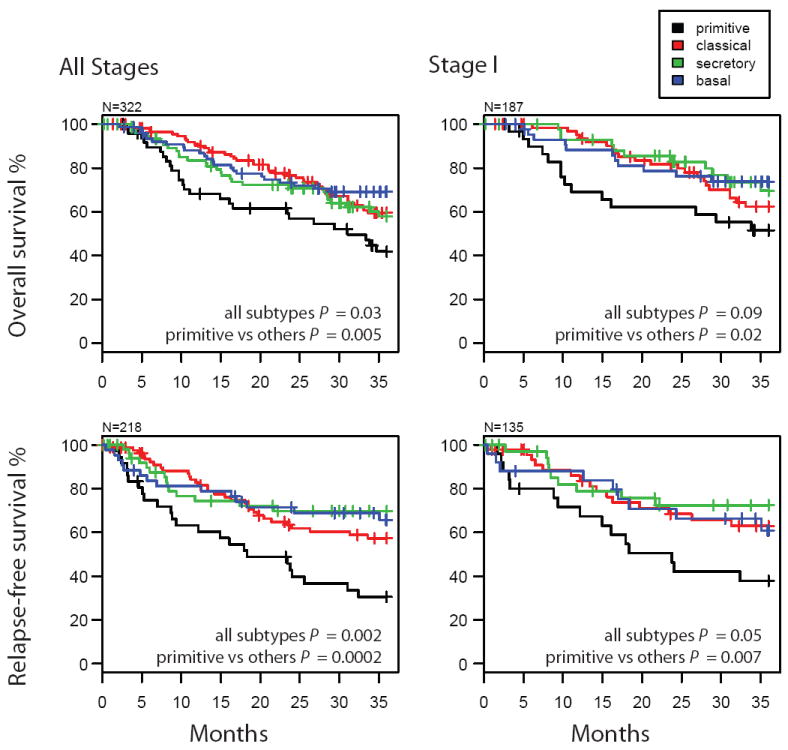
Survival was estimated by the Kaplan-Meier method using all cohorts’ available data. The sample sizes (N) are different than the overall study sample size due to data availability (OS: Bild et al, Raponi et al, Roepman et al, and UNC cohorts; RFS: Lee et al, Roepman et al, and UNC cohorts). P-values are from log rank tests evaluating the independence of survival and subtype.
Raponi et al reported two SCC mRNA expression subtypes with a survival difference and provided a list of differentially expressed genes where high expression of the “majority of the genes were down-regulated in the high-risk group” (13). Comparison of Raponi et al’s microarrays by their gene list and the subtypes discovered in this study shows two clear subtype groups: underexpression (primitive and secretory) and overexpression (basal and classical) (Supplement Fig. 8). Therefore, the four subtypes discovered in this study map to prior results and this study has divided each of the prior subtypes into two new ones and improves the SCC mRNA expression subtype granularity. Interestingly, the Raponi et al poor survival subtype totals 43% of their patients where the poor survival subtype of this study (primitive) is 16% of their patients. It appears that a fraction of Raponi et al’s high risk subtype shows poor survival outcome relative to the remainder of SCC.
SCC subtypes are similar to different normal lung cell types and SCC cell lines
To evaluate the hypothesis that SCC subtypes are derived from different cell types present in the normal lung, SCC subtypes were compared by mRNA expression to three published model systems. The first model, “Mouse lung development”, is a time series of mouse lungs extracted from embryonic stages to adult (21). Expression similarity is defined as high positive Pearson correlation between an SCC subtype and time points within the model. The primitive subtype shows expression similarity to early stage mouse lung and the secretory subtype shows similarity to late stage mouse lung (Fig. 4A). The second model, “Human bronchial epithelial cell air liquid interface culture” (HBEC-ALIC), is a time series of cultured normal, healthy, human bronchial epithelial cells in which the early time points consist of stratified basal cells and later time points include secretory and ciliated cells (22). The basal subtype showed expression similarity to the early time points during which basal cells are predominant (Fig. 4B). The primitive and secretory subtypes show expression similarity to the later time points at which there are secretory and ciliated cells. The third model system, “Human microdissected lung cell compartments” (HMLCC), was laser capture microdissected cells contained in surface epithelium and in submucosal glands of normal healthy lung (20). The secretory subtype overexpresses genes that are overexpressed in submucousal glands (Fig. 4C). The basal subtype overexpresses genes that are overexpressed in surface epithelia. The classical subtype does not show appreciable similarity to any specific lung model, is the only subtype to have this property, and could be most similar to multiple or unobserved cell types. Therefore, by the combination of all three lung models, 3 of the 4 SCC subtypes have unique similarities to different, normal lung cell types.
Figure 4. SCC subtypes compared to lung cell type models.

The relationship of relative SCC subtype expression differences to relative expression differences of published lung model systems. The models “Mouse lung development” (21) and “Human bronchial epithelial cell air liquid interface culture (HBEC-ALIC)” (22) are microarray time series, where time is indicated on the horizontal axis (A,B). Points mark Pearson correlation coefficients of SCC subtype centroids to model time points using the top 1,000 genes having the greatest Pearson correlation coefficient with time. Bars represent 95% confidence intervals. Lines connect points corresponding to the same subtype. Large positive correlations indicate mRNA expression similarity while large negative correlations indicate dissimilarity. In (A), ‘e’ refers to embryonic day and ‘p’ refers to postnatal day. The model “Human microdissected lung cell compartments (HMLSCC)” (20) is compared to SCC subtypes via a heatmap of genes that are overexpressed in submucosal glands and in surface epithelium as rows and subtype centroids in columns (C).
In addition to the cell type models, SCC subtypes may correspond to different SCC cell lines which could establish additional manipulatable models for future investigations into subtype biology. To ascertain if SCC cell lines correspond to different SCC tumor subtypes by mRNA expression, four published SCC cell line microarrays (23) were given subtype classifications by the nearest-centroid predictor. Interestingly, the four cell lines were predicted to be different subtypes (Fig. 2D). Expression of the subtypes between the cell lines and tumors are consistent over the validation gene set (Fig. 2A,D). For example, genes are consistent and mutually exclusive in the cell lines as predicted (HCC15 – primitive and MCM10; HCC95 – classical and AKR1C3; HCC2450 – secretory and MUC1; H157 – basal and MMP13).
DISCUSSION
The principal novel hypotheses tested in this study is that lung SCC expression subtypes exist, are reproducible, clinically relevant, and exhibit patterns that correlate with unique cell types in the normal lung. These subtypes (primitive, basal, secretory, and classical) were identified in an unbiased and objective manner and are supported by cross-cohort validation using five training cohorts and by independent validation using a sixth cohort, which together total 438 patients. The expression subtypes were also found in a wide variety of patient populations from the United States, Asia and Europe, in a wide variety of cohort sizes from 36 to 127. All cohorts showed approximately the same subtype proportions, overall: primitive – 16%, classical – 37%, secretory – 26%, basal – 21%. These subtypes were associated with tumor differentiation and patient sex. Survival outcomes are significantly different among the subtypes and subtype is an independent predictor of survival. Possible limitations of our analysis include possible sample quality artifacts or patient behavior, such as smoking immediately prior to surgery; however, all six cohorts showed the same results so any limitation would have to occur in six large, independently collected, cohorts.
The SCC expression subtypes are biologically distinct and show similarities to distinct normal lung cell populations. These biological characteristics serve as the basis for the SCC nomenclature. The basal subtype exhibits many characteristics of lung basal cells such as: cell adhesion and epidermal development functional themes, S100A2 and KRT5 basal cell markers, overexpression of genes whose products are localized in the basement membrane, similarity to basal cells in the HBEC-ALIC model, and similarity to surface epithelia in the HMLCC model. The secretory subtype has many features of lung secretory cells such as: surfactant and mucin overexpression, similarity to secretory cells in the HBEC-ALIC model, and similarity to submucosal glands in the HMLCC. The primitive subtype has a cellular proliferation functional theme, the worst survival outcome, an overabundance of female patients, the most nonsmokers, and an overabundance of poorly differentiated tumors. This subtype is similar to early embryonic mouse lungs, where primitive, less differentiated cells may be predominant and would be consistent with the poorly differentiated nature of these tumors. The primitive subtype also has similarity to late stage HBEC-ALIC, which could be explained by lung “transient expression” in which differentiation markers are expressed during early lung formation and again in the developed lung (48). Alternatively, a late-emerging and late-active cell type in HBEC-ALIC may be most similar to the embryonic mouse lung. The classical subtype, exhibits features representative of typical lung SCC including the highest prevalence at 37%, overabundance of males, greatest patient smoking behavior, overexpression of TP63, and putative amplification of the TP63-containing locus 3q27-28.
The distinct SCC subtype to cell population similarities could be explained by the SCC subtypes having different ancestor cells. These different ancestor cells could be cell types of distinct lineages or cellular differentiation stages such as proposed in breast cancer (49). This scenario provides a reason why the SCC subtypes have dramatically different mRNA expression. The subtypes could arise by genetic mutation from different ancestors that have different mRNA expression and this ancestral mRNA expression could persist in progeny tumor cells. This putative subtype ancestral cell information could be utilized in developing SCC subtype pharmacologic interventions that exploit differences in the ancestral cell types. A caveat to our interpretation of SCC subtype to cell population similarity is that the similarity could be caused by coincidence and expression similarities could reflect similar biology and not similar origin. The lung has multiple proposed cellular development pathways and future studies that describe the molecular profiles of the lung cell types or lung cancer stem cells would further clarify the putative ancestral cells of the SCC subtypes (50).
The SCC subtypes may have applications in patient care and in cancer research. For instance, patients with the primitive subtype could be treated more aggressively because of this subtype’s poor survival expectation or could be given a more accurate prognosis than by using traditional prognostic factors alone. Basic cancer research could be conducted using the subtype model system partners described in this study. The SCC subtypes could be useful for therapy benefit studies and possibly serve as a foundation for clinical trial selection.
In conclusion, we identified four, robust, expression subtypes of lung SCC using a multi-cohort discovery and validation strategy. The subtypes are clinically and phenotypically different, suggesting different therapies.
Supplementary Material
Acknowledgments
Financial support by author initials: NIH F32CA142039 from the National Cancer Institute (MDW)
Lineberger Comprehensive Cancer Center Translational Small Grants Program (DNH)
NIH K12-RR023248 from the National Center for Research Resources (DNH)
Thomas G. Labrecque Foundation, through Joan’s Legacy Foundation (DNH)
Footnotes
Potential conflicts of interest: DNH, CMP, and PSB hold a provisional patent that is related to work described in this manuscript but there is no current financial interest. All other authors have no conflicts of interest.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Koyi H, Hillerdal G, Branden E. A prospective study of a total material of lung cancer from a county in Sweden 1997-1999: gender, symptoms, type, stage, and smoking habits. Lung Cancer. 2002;36:9–14. doi: 10.1016/s0169-5002(01)00451-2. [DOI] [PubMed] [Google Scholar]
- 3.Visbal AL, Williams BA, Nichols FC, 3rd, et al. Gender differences in non-small-cell lung cancer survival: an analysis of 4,618 patients diagnosed between 1997 and 2002. Ann Thorac Surg. 2004;78:209–15. doi: 10.1016/j.athoracsur.2003.11.021. discussion 15. [DOI] [PubMed] [Google Scholar]
- 4.Travis WD World Health Organization. Histological typing of lung and pleural tumours. Berlin: Springer; 1999. [Google Scholar]
- 5.Auerbach O, Garfinkel L, Parks VR. Histologic type of lung cancer in relation to smoking habits, year of diagnosis and sites of metastases. Chest. 1975;67:382–7. doi: 10.1378/chest.67.4.382. [DOI] [PubMed] [Google Scholar]
- 6.Jones DR, Detterbeck FC. Surgery for Stage I Non-small Cell Lung Cancer. In: Detterbeck FC, Socinski MA, Rivera MP, Rosenman JG, editors. Diagnosis and Treatment of Lung Cancer. First. Philadelphia: W.B Saunders Company; 2001. pp. 177–90. [Google Scholar]
- 7.Churg AM, Myers JL, Tazelaar HD, Wright JL. Thurlbeck’s Pathology of the Lung. Third. New York: Thieme Medical Publishers, Inc; 2005. [Google Scholar]
- 8.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 9.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 10.Hayes DN, Monti S, Parmigiani G, et al. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J Clin Oncol. 2006;24:5079–90. doi: 10.1200/JCO.2005.05.1748. [DOI] [PubMed] [Google Scholar]
- 11.Inamura K, Fujiwara T, Hoshida Y, et al. Two subclasses of lung squamous cell carcinoma with different gene expression profiles and prognosis identified by hierarchical clustering and non-negative matrix factorization. Oncogene. 2005;24:7105–13. doi: 10.1038/sj.onc.1208858. [DOI] [PubMed] [Google Scholar]
- 12.Larsen JE, Pavey SJ, Passmore LH, et al. Expression profiling defines a recurrence signature in lung squamous cell carcinoma. Carcinogenesis. 2007;28:760–6. doi: 10.1093/carcin/bgl207. [DOI] [PubMed] [Google Scholar]
- 13.Raponi M, Zhang Y, Yu J, et al. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006;66:7466–72. doi: 10.1158/0008-5472.CAN-06-1191. [DOI] [PubMed] [Google Scholar]
- 14.Hu Z, Troester M, Perou CM. High reproducibility using sodium hydroxide-stripped long oligonucleotide DNA microarrays. Biotechniques. 2005;38:121–4. doi: 10.2144/05381MT02. [DOI] [PubMed] [Google Scholar]
- 15.Ritchie ME, Silver J, Oshlack A, et al. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007;23:2700–7. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- 16.Garrett-Mayer E, Parmigiani G, Zhong X, Cope L, Gabrielson E. Cross-study validation and combined analysis of gene expression microarray data. Biostatistics. 2008;9:333–54. doi: 10.1093/biostatistics/kxm033. [DOI] [PubMed] [Google Scholar]
- 17.Bild AH, Yao G, Chang JT, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–7. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 18.Lee ES, Son DS, Kim SH, et al. Prediction of recurrence-free survival in postoperative non-small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clin Cancer Res. 2008;14:7397–404. doi: 10.1158/1078-0432.CCR-07-4937. [DOI] [PubMed] [Google Scholar]
- 19.Roepman P, Jassem J, Smit EF, et al. An immune response enriched 72-gene prognostic profile for early-stage non-small-cell lung cancer. Clin Cancer Res. 2009;15:284–90. doi: 10.1158/1078-0432.CCR-08-1258. [DOI] [PubMed] [Google Scholar]
- 20.Fischer AJ, Goss KL, Scheetz TE, Wohlford-Lenane CL, Snyder JM, McCray PB., Jr Differential gene expression in human conducting airway surface epithelia and submucosal glands. Am J Respir Cell Mol Biol. 2009;40:189–99. doi: 10.1165/rcmb.2008-0240OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mariani TJ, Reed JJ, Shapiro SD. Expression profiling of the developing mouse lung: insights into the establishment of the extracellular matrix. Am J Respir Cell Mol Biol. 2002;26:541–8. doi: 10.1165/ajrcmb.26.5.2001-00080c. [DOI] [PubMed] [Google Scholar]
- 22.Ross AJ, Dailey LA, Brighton LE, Devlin RB. Transcriptional profiling of mucociliary differentiation in human airway epithelial cells. Am J Respir Cell Mol Biol. 2007;37:169–85. doi: 10.1165/rcmb.2006-0466OC. [DOI] [PubMed] [Google Scholar]
- 23.Zhou BB, Peyton M, He B, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10:39–50. doi: 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gollub J, Sherlock G. Clustering microarray data. Methods Enzymol. 2006;411:194–213. doi: 10.1016/S0076-6879(06)11010-1. [DOI] [PubMed] [Google Scholar]
- 25.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418–23. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning. 2003;52:91–118. [Google Scholar]
- 27.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Z, Fan C, Oh DS, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006;7:96. doi: 10.1186/1471-2164-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi JK, Yu U, Kim S, Yoo OJ. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics. 2003;19(Suppl 1):i84–90. doi: 10.1093/bioinformatics/btg1010. [DOI] [PubMed] [Google Scholar]
- 30.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu YH, Hayes DN, Nobel A, Marron JS. Statistical Significance of Clustering for High Dimension Low Sample Size Data. Journal of the American Statistical Association. 2007;103 [Google Scholar]
- 32.Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cabanski CR, Qi Y, Yin X, et al. SWISS MADE: Standardized WithIn Class Sum of Squares to evaluate methodologies and dataset elements. PLoS One. 5:e9905. doi: 10.1371/journal.pone.0009905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitfield ML, George LK, Grant GD, Perou CM. Common markers of proliferation. Nat Rev Cancer. 2006;6:99–106. doi: 10.1038/nrc1802. [DOI] [PubMed] [Google Scholar]
- 35.Perou CM, Jeffrey SS, van de Rijn M, et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A. 1999;96:9212–7. doi: 10.1073/pnas.96.16.9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polager S, Ginsberg D. E2F - at the crossroads of life and death. Trends Cell Biol. 2008;18:528–35. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Spira A, Beane J, Shah V, et al. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A. 2004;101:10143–8. doi: 10.1073/pnas.0401422101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jorgensen E, Stinson A, Shan L, Yang J, Gietl D, Albino AP. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer. 2008;8:229. doi: 10.1186/1471-2407-8-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung CH, Parker JS, Karaca G, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 2004;5:489–500. doi: 10.1016/s1535-6108(04)00112-6. [DOI] [PubMed] [Google Scholar]
- 40.King KE, Weinberg WC. p63: defining roles in morphogenesis, homeostasis, and neoplasia of the epidermis. Mol Carcinog. 2007;46:716–24. doi: 10.1002/mc.20337. [DOI] [PubMed] [Google Scholar]
- 41.Massion PP, Taflan PM, Jamshedur Rahman SM, et al. Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res. 2003;63:7113–21. [PubMed] [Google Scholar]
- 42.Gaillard D, Puchelle E. Differentiation and maturation of airway epithelial cells: role of extracellular matrix and growth factors. In: Gaultier C, Bourbon JR, Post M, editors. Lung Development. Oxford: Oxford University Press; 1999. pp. 46–76. [Google Scholar]
- 43.Garber ME, Troyanskaya OG, Schluens K, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98:13784–9. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broome AM, Ryan D, Eckert RL. S100 protein subcellular localization during epidermal differentiation and psoriasis. J Histochem Cytochem. 2003;51:675–85. doi: 10.1177/002215540305100513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith SL, Gugger M, Hoban P, et al. S100A2 is strongly expressed in airway basal cells, preneoplastic bronchial lesions and primary non-small cell lung carcinomas. Br J Cancer. 2004;91:1515–24. doi: 10.1038/sj.bjc.6602188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu PG, Weiss LM. Keratin expression in human tissues and neoplasms. Histopathology. 2002;40:403–39. doi: 10.1046/j.1365-2559.2002.01387.x. [DOI] [PubMed] [Google Scholar]
- 47.Ozasa H, Oguri T, Uemura T, et al. Significance of thymidylate synthase for resistance to pemetrexed in lung cancer. Cancer Sci. 101:161–6. doi: 10.1111/j.1349-7006.2009.01358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wuenschell CW, Sunday ME, Singh G, Minoo P, Slavkin HC, Warburton D. Embryonic mouse lung epithelial progenitor cells co-express immunohistochemical markers of diverse mature cell lineages. J Histochem Cytochem. 1996;44:113–23. doi: 10.1177/44.2.8609367. [DOI] [PubMed] [Google Scholar]
- 49.Prat A, Perou CM. Mammary development meets cancer genomics. Nat Med. 2009;15:842–4. doi: 10.1038/nm0809-842. [DOI] [PubMed] [Google Scholar]
- 50.Snyder JC, Teisanu RM, Stripp BR. Endogenous lung stem cells and contribution to disease. J Pathol. 2009;217:254–64. doi: 10.1002/path.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.