

Abstract
Granulocyte colony-stimulating factor (GCSF) is currently in clinical trials to treat neurodegenerative diseases and stroke. Here, we tested whether LIM domain only 4 protein (LMO4), a hypoxia-inducible gene that protects neurons from ischemic injury, could modulate the neuroprotective effect of GCSF. We showed that GCSF treatment acetylates and phosphorylates Stat3, activates expression of a Stat3-dependent anti-apoptotic gene, p27, and increases neuron survival from ischemic injury. LMO4 participates in Stat3 signaling in hepatocytes and associates with histone deacetylase 2 (HDAC2) in cancer cells. In the absence of LMO4, GCSF fails to rescue neurons from ischemic insults. In wild-type neurons, inhibition of HDAC promoted Stat3 acetylation and the antiapoptotic effect of GCSF. In LMO4 null cortical neurons, expression of wild-type but not HDAC-interaction-deficient LMO4 restored GCSF-induced Stat3 acetylation and p27 expression. Thus, our results indicate that LMO4 enhances GCSF-induced Stat3 signaling in neurons, in part by sequestering HDAC.
Keywords: GCSF, LMO4, Stat3, HDAC, Neurons, Ischemia, Cell death, Acetylation
Introduction
The use of growth factors as neuroprotective agents for various types of neurological disorders has been under investigation for many years. Growth factors are able to act directly on the CNS to regulate survival, maturation, and sprouting of developing neuronal cells. Almost all growth factors display endogenous neuroprotective and neurotrophic effects on mature neurons [1]. One of the few growth factors currently approved for clinical use is the granulocyte colony-stimulating factor (GCSF), routinely prescribed to treat neutropenia (low neutrophil count in the blood) [2]. In addition, GCSF can cross the blood–brain barrier, and recent experimental studies have shown that the administration of GCSF is neuroprotective both for in vitro cultured cortical neurons and in vivo focal cerebral ischemia [3–7]. GCSF upregulates expression of several anti-apoptotic genes, including B-cell lymphoma 2 protein (Bcl2), B-cell lymphoma-extra large protein (Bcl-xl), cellular inhibitor of apoptosis protein 2 (ciap2) and p27 [6, 8] (see review by Solaroglu et al. [9]). Here, we explored factors that could enhance the neuroprotective effect of GCSF.
Binding of GCSF to the cell surface receptor GCSFR activates the JAK2/Stat3 signaling pathway in neurons and is anti-apoptotic [3–5]. GCSF induces a robust and sustained phosphorylation and activation of Stat3. In addition to phosphorylation at the tyrosine 705 residue, Stat3 activity is also modulated by acetylation at the Lysine685 residue [10]. Acetylation of Lys685 is critical for Stat3 dimerization, DNA binding and transcription activation of target genes. Mutation of Lys685 that prevents acetylation renders Stat3 inactive but does not prevent cytokine-induced Stat3 phosphorylation. Thus, acetylation and phosphorylation are necessary for Stat3-dependent gene activation. Cytokine treatment induces histone acetyl-transferase p300-mediated Stat3 Lys 685 acetylation, and this is reversed by type I histone deacetylase (including HDAC 1–3). Treatment of T antigen-transformed human kidney (293T) cells with trichostatin A (TSA), a broad inhibitor of HDACs, further augments cytokine-induced Stat3 acetylation and activation of a Stat3-responsive promoter [10].
LMO4 is expressed in cortical neurons and plays a pivotal role in the nervous system. LMO4 interacts with transcription factors, including LIM homeodomain proteins, basic Helix-Loop-Helix (bHLH) proteins, peroxisome proliferator-activated receptor gamma (PPARγ), and cAMP response element-binding protein (CREB), to activate gene expression in the nervous system [11–15]. Germline deletion of LMO4 leads to embryonic lethality and failure of the cranial neural tube to close, resulting in exencephaly [16, 17]. Ablation of LMO4 at a late embryonic stage disrupts the patterning of thalamocortical connections [15]. These studies highlight the importance of LMO4 in neuronal development.
The expression levels of LMO4 are tightly regulated in neurons. We showed previously that signals like extracellular ATP, hypoxia, and depolarization stabilize LMO4 mRNA and protein [11, 18, 19]. Ablation of LMO4 increases neuron susceptibility to ischemic injury, resulting in a larger infarction in an experimental model of stroke [11]. Hypoxia-induced expression of LMO4 promotes protein expression of the superoxide dismutase SOD2, an antioxidant gene that scavenges free radicals and reduces oxidative stress [11].
LMO4 was previously reported as a novel regulator of Stat3 signaling in hepatocytes [20]. Over-expression of LMO4 enhanced the transcriptional activity and target gene expression of Stat 3. Furthermore, silencing LMO4 expression in stable cell lines expressing small interfering RNA of LMO4 decreased Stat3 activity [20].
On the other hand, LMO4 interacts with transcription inhibitors and HDACs and negatively regulates gene expression in non-neuronal cells. LMO4 interacts with metastasis tumor antigen 1 (MTA1) and HDAC2 that inhibit estrogen receptor α (ERα) transactivation function in breast cancer cells [21]. High levels of LMO4 expression associate with the development of ERα-negative phenotypes and aggressive breast cancer growth in MDA-MB-231 cells [22]. However, over-expression of LMO4 activates the BMP7 promoter and regulates mammary gland development, likely through its interaction and sequestration of HDAC2 proteins from the bone morphogenic protein 7 (BMP7) promoter [23]. It remains to be tested whether LMO4 modulates HDAC function in neurons to favor or inhibit the expression of survival genes.
Here, we determined how LMO4 might influence GCSF anti-apoptotic signaling through Stat3. We found that blocking HDAC activity with TSA augments the anti-apoptotic effect of GCSF. We also found that GCSF does not protect LMO4 null cortical neurons from ischemia-like injuries. Our study is the first to demonstrate that LMO4 is required for Stat3 acetylation and signaling in neurons in response to GCSF, and that this function depends on LMO4 interaction with HDAC2.
Materials and methods
Reagents
Tyrphostin AG490, a Jak-2 protein tyrosine kinase inhibitor, potassium cyanide (KCN), TSA, a Streptomyces metabolite that specifically inhibits mammalian histone deacetylase at a nanomolar concentration, and GCSF were purchased from Sigma–Aldrich Canada (Oakville, ON, Canada).
Antibodies
Antibodies to Stat3, Stat3 phosphorylated at Tyr705 (P-Stat3), acetylated-lysine were obtained from Cell Signaling Technology (Danvers, MA, USA), to actin, myc and FLAG from Sigma–Aldrich Canada, to Gal4 from Abcam (Cambridge, MA, USA) and to p27 and GCSF receptor, from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Animals
LMO4 hemizygous mice were obtained from Terence Rabbits (University of Leeds, UK) and genotyped as described [17]. LMO4 homozygous null (KO) and littermate wild-type (WT) embryos were used for primary cultures of cortical neurons.
Primary neuronal cultures, ischemic insults and cell survival assay
Primary cultures of cortical neurons were prepared from LMO4 KO or littermate WT control 14.5-day-old mouse embryos as we described previously and maintained for 12–14 days in vitro [18]. For oxygen and glucose deprivation (OGD)/reperfusion, cultures were washed twice with a balanced salt solution at room temperature with the following composition: 140 mM NaCl, 3.5 mM KCl, 0.4 mM KH2PO4, 5 mm NaHCO3, 1.3 mM CaCl2, 1.2 mM MgSO4, and 10 mM HEPES, pH 7.4. Cultures were subjected to an anaerobic environment of 95% N2/5% CO2 for 3 h, returning the stored medium to the cells and maintained at normal atmospheric O2 with 5% CO2, at 37°C. Chemical hypoxia was induced by 1 mM KCN for 3 h followed by reperfusion with stored culture medium. GCSF (100 ng/ml) was added to the cells 3 h prior to and immediately following reperfusion. For HDAC inhibition, TSA (0.66 μM) was added during the KCN or OGD treatment and reperfusion. For Stat3 inhibitor treatment, AG490 (50 μM) was added in the presence of GCSF. For immunoblot analysis, cells were harvested after 6 h reperfusion. For cells not subject to hypoxic injury, cells were harvested after 12 h of GCSF/TSA treatment. Cell survival was measured 16 h after treatments by the lactate dehydrogenase (LDH) assay, as described previously [18]. Student's t test was used to compare means and P < 0.05 was considered significant. All experiments were performed in quadruplicate and reported as mean ± SEM.
Plasmids, siRNA and transfection
F11 cells (a chimeric cell line of the mouse neuroblastoma cell line N18TG-2 fused with embryonic rat dorsal-root ganglion neurons) were transiently transfected as described previously [18]. In brief, cells were transfected with 400 ng of luciferase-expressing vector and 100 ng of indicated expression constructs or appropriate empty vectors (e.g., cDNA3) and 200 ng of pCMVβgal to normalize transfection efficiency per well of a 12-well plate. Transfection was carried out with a 1.5 ratio of total plasmid/Lipofectamine2000 reagent (Invitrogen) in 500 μl Opti-MEM (Invitrogen) for 4–6 h, after which medium was supplemented with serum-containing medium (final 5 or 0.5% fetal bovine serum) for 12–16 h. The next day, cells were treated with GCSF (100 ng/ml), TSA (0.66 μM), or AG490 (50 μM) and were harvested 24 h after treatment. Luciferase and β-galactosidase assays were performed [24]. For all experiments, assays were done in triplicate and repeated as indicated.
For the mammalian two-hybrid assay, the ActLMO4(WT), ActLMO4(C23S), ActLMO4(C87S), and pG5 luciferase reporter were described previously [18]. The Gal4-HDAC2 FL and Gal4-HDAC2 286–489 were obtained from Diane Hayward (Johns Hopkins School of Medicine) [25]. A Gal4-HDAC2 1–285 was obtained by digesting a PCR fragment amplified using 5′-AAGGATCCTGATGGCGTACAGTCAAG-3′ and 5′-CCTCTAGAACATTTAGCATGACCTTTGACAG-3′ with BamHI and XbaI into the pBIND vector (Promega, Madison, Wisconsin, USA).
For immunoprecipitation assays, pcDNA3-mycLMO4, myc-LMO4C23S, and myc-LMO4C87S expression vectors were obtained from Anirvan Ghosh (University of California, San Diego) [15]. pcDNA3-Flag-LMO4 was obtained from Tetsu Akiyama (University of Tokyo) [26].
Transfection of primary neuronal cultures was carried out at 8 days in vitro (DIV8) using Lipofectamine 2000 reagent (Invitrogen Canada, Burlington, ON, Canada) in defined medium, as described [18]. Two days after transfection, cells were subjected to GCSF treatment for 12 h and harvested for immunoblot analysis.
The Stealth LMO4siRNA (Invitrogen) [18] contains the sense RNA 5′-CCGGGAGAUCGGUUUCACUACAUCA-3′ and anti-sense RNA 5′-UGAUGUAGUGAAACCGAUCUCCCGG-3′. Control siRNA contains the “sense” RNA 5′-CCGUAGAUGGCACUUCAUCAGGUCA-3′ and the “antisense” RNA 5′-UGACCUGAUGAAGUGCCAUCUACGG-3′. Stealth siRNA was applied at a concentration of 5 nM per well. After transfection, F11 cells were cultured in 0.5% FBS-DMEM for 24 h before harvesting for luciferase assay.
Western blot analysis
Cortical neurons were harvested in RIPA buffer with 1 mM sodium orthovanadate and proteinase inhibitor (Leupeptin, 0.5 μg/ml; Aprotinin 2 μg/ml; PMSF 1 mM) (Sigma–Aldrich Canada). Then, 50–100 μg of protein extracts were loaded onto 12% SDS-polyacrylamide gels and transferred to PVDF membranes (GE Healthcare, Baie d’Urfe, Quebec, Canada) as described previously [27, 28]. Blots were blocked with 5% non-fat milk in TBST at room temperature for 1 h and probed with primary antibody at 4°C overnight. The blots were washed five times for 5 min each in TBST and probed with appropriate horseradish peroxidase (HRP) conjugated secondary antibodies in blocking solution. SuperBlock (Fisher Scientific Canada, Ottawa, ON, Canada) was used instead of blocking solution at 1:10 dilutions when probed with phosphorylated Stat3-specific antibody. HRP signals were detected with ECL (GE Healthcare). Blots were stripped with 62.5 mM Tris (pH 6.8), 2% SDS, and 100 mM β-mercaptoethanol for 30 min at 50°C before being reprobed.
Immunoprecipitation and anti-acetylated-lysine immunoblot was used to analyze Stat3 acetylation as described [10]. In brief, the cell extracts were immunoprecipitated with anti-Stat3 antibody and protein A/G beads (Santa Cruz Biotechnology). The beads were washed with 0.5 ml of RIPA buffer three times and eluted by SDS sample buffer (62.5 mM Tris–HCl pH 6.8, 10% Glycerol, 2% SDS, 0.01% bromophenol blue). The samples were further analyzed by western blot with anti-acetylated lysine or anti-Stat3 antibodies.
Statistical analysis
Data are presented as mean ± standard deviation, and analysis of variance (ANOVA) followed by post-hoc tests using Fisher’s least significant difference (LSD) was used to compare means. Significance was determined at P < 0.05 level.
Results
Effect of GCSF-induced Stat3 signaling on cortical neuron survival from hypoxia reperfusion injuries
We characterized the neuroprotective effect of GCSF using in vitro models of neuronal ischemic injury-induced cell death. In these models, mouse embryonic (E14.5) cortical neurons were cultured for 14 days in vitro (DIV = 14) to allow neurons to mature and express glutamate receptor just like neurons in the postnatal brain. These DIV 14 cortical neurons were then subjected to various oxidative stresses to mimic ischemic insults, including 3 h treatment with 1 mM KCN, an inhibitor of the mitochondrial respiratory chain [29], or 3 h OGD (oxygen glucose deprivation) followed by reperfusion for both treatments. Either KCN or OGD/reperfusion induces widespread cell death. Optimal doses and durations of GCSF treatment were determined empirically (data not shown). Treatment of cortical neuron cultures with GCSF (100 ng/ml culture medium) significantly increased neuron survival from hypoxic insult (Fig. 1a).
Fig. 1.

Cortical neuron survival of ischemia-like injuries is promoted by GCSF. a In wild-type cortical neurons, either KCN or OGD/reperfusion caused significant cell death by 16 h after treatment. Treating with GCSF rescued cortical neurons from cell death and AG490 abolished this effect. HDAC inhibitor trichostatin A (TSA) added during reperfusion further promotes neuron survival. b In LMO4 null cortical neurons, GCSF no longer protects against ischemia-like insults whereas TSA promotes neuron survival. *P < 0.05, ns not significant, n = 8 experiments
Granulocyte colony stimulating factor exerts its neuroprotective effect through activation of the Jak/Stat signaling pathway, namely by activating Stat3 signaling. Treatment of DIV 14 cortical neurons with GCSF induced robust Stat3 phosphorylation and activation [30]. Blocking Stat signaling with inhibitor AG490 abrogated the anti-apoptotic effect of GCSF (Fig. 1a).
On the other hand, histone acetyltransferase acetylates Stat3 which leads to Stat3 dimerization, a prerequisite to activate Stat3-dependent target genes. Type I HDACs (HDAC1, 2, 3) terminate Stat3 signaling by deacetylating Stat3, and inhibition of HDAC maintains Stat3 acetylated and activated [10, 31]. Thus, we hypothesized that inhibition of HDAC activity might fortify the neuroprotective effect of GCSF by enhancing Stat3 signaling. Although TSA was somewhat toxic to neurons, we found that the survival of GCSF-treated cortical neuron after KCN-induced ischemia-like injury increased further if the HDAC inhibitor TSA was added over the time course of KCN-induced injury (Fig. 1a). These results suggest that HDAC attenuates GCSF signaling and its anti-apoptotic effect.
Effect of LMO4 ablation on GCSF-dependent protection of cortical neurons
Since LMO4 plays an important function in cortical neuron development, we asked whether Stat3 signaling by GCSF would be affected in cortical neurons that lack LMO4. GCSF did not protect LMO4 null cortical neurons from the ischemia-like insults of KCN or OGD/reperfusion (Fig. 1b). This result suggests that LMO4 participates in the GCSF-mediated anti-apoptotic pathway.
Effect of LMO4 ablation on GCSF induced Stat3 phosphorylation and acetylation
To confirm that the effect of LMO4 was mediated through Stat3 signaling, we measured phosphorylated Stat3 by immunoblot analysis. Whereas wild-type cortical neurons activated phospho-Stat3 in response to GCSF (compare lanes 1 and 2, Fig. 2a), no such activation was observed in LMO4 null cortical neurons (compare lanes 5 and 6, Fig. 2a). Stat3 phosphorylation was reduced by the Jak/Stat inhibitor AG490 in wild-type neurons (compare lanes 2 and 4, Fig. 2a). GCSF treatment induced Stat3 acetylation in wild-type but not in LMO4 null cortical neurons (compare lanes 2 and 6, Fig. 2a). The presence of GCSF receptors was confirmed in both wild-type and LMO4 null cortical neurons (Fig. 2b), demonstrating that the deficit in GCSF-induced acetylation of Stat3 is not due to a lack of GCSF receptors in LMO4 null neurons. Together, these results suggest that LMO4 enhances GCSF-induced Stat3 signaling by promoting Stat3 phosphorylation and by inhibiting deacetylation of Stat3. The HDAC inhibitor TSA increased the level of Stat3 acetylation in wild-type, but also in LMO4 null cortical neurons in the presence of GCSF, demonstrating that there is no intrinsic defect in acetylation in LMO4 null cortical neurons (Fig. 2, lanes 3 and 7).
Fig. 2.

GCSF activates Stat3 in wild-type (WT) but not in LMO4 null cortical (KO) neurons. a Stat3 phosphorylation and acetylation was analyzed in whole cell extracts prepared from cortical neurons treated with and without GCSF and TSA. Lysates prepared from these cells were immunoprecipitated with anti-Stat3 antibody and revealed with antibodies to acetylated lysine, phospho-Stat3, and Stat3 antibody. Representative blot of three experiments. b Western blot of GCSF receptor from wild-type and LMO4 null cortical cortical neurons
Effect of GCSF on the expression of the cell cycle regulator p27
The cyclin-dependent kinase inhibitor p27 is a known target of Stat3 signaling [32] and is important to prevent inappropriate cell cycle entry and cell death in differentiated cortical neurons [33, 34]. GCSF upregulated the expression of p27 in wild-type but not in LMO4 null cortical neurons (Fig. 3a), consistent with our observation that GCSF activates Stat3 signaling in wild-type but not LMO4 null cortical neurons. In wild-type cortical neurons, activation of p27 was further increased by treating the cells with the HDAC inhibitor TSA together with GCSF (compare lanes 2 and 3, Fig. 3a). In LMO4 null cortical neurons, TSA also upregulated p27 (compare lanes 5 and 6, Fig. 3a), but to a much lower level than in wild-type neurons. Thus, in the absence of LMO4, the effect of combined GCSF and TSA treatment is limited.
Fig. 3.
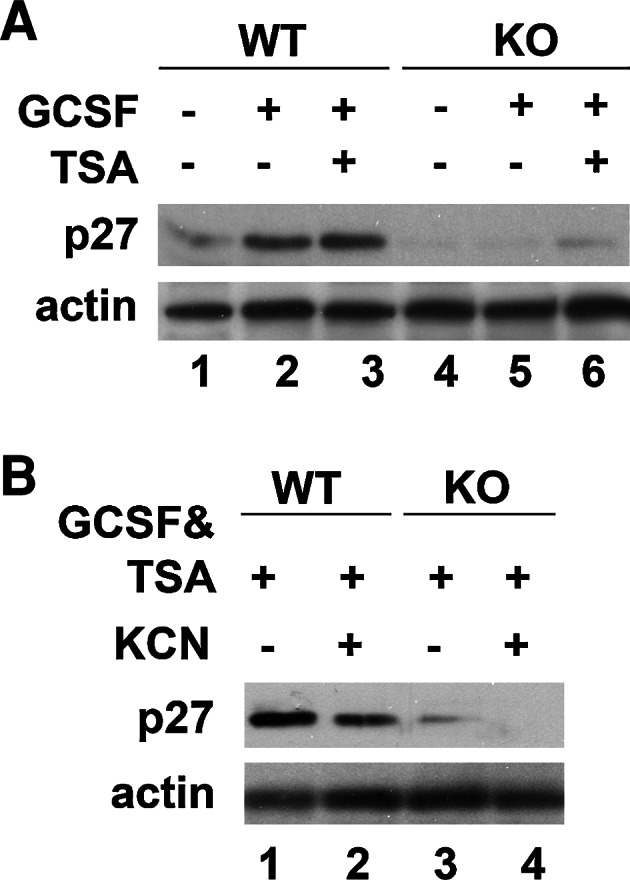
GCSF robustly upregulates p27 expression in wild-type (WT) but not in LMO4 null cortical (KO) neurons. a Addition of the HDAC inhibitor TSA with GCSF further increased p27 expression in LMO4 null but not in wild-type neurons. b GCSF/TSA treatment maintains p27 expression in wild-type but not in LMO4 null cortical neurons following KCN insult. Actin served as a loading control. Representative blots of three experiments
Ischemic injury reduces p27 expression and activates inappropriate cell cycle reentry as an early sign of neuronal cell death [33]. GCSF and TSA maintained p27 expression in wild-type but not in LMO4 null cortical neurons in the face of ischemia-like insult caused by KCN exposure (Fig. 3b).
To test whether p27 upregulation occurs at the level of transcription, a p27 luciferase reporter was tested in the F11 neuronal cell line in response to GCSF. p27 promoter activity was upregulated by GCSF, and this upregulation was blocked by Stat3 signaling inhibitor AG490. siRNA to LMO4, but not the control siRNA vector, reduced but did not completely abolish the activation of the p27 promoter by GCSF (Fig. 4). On the other hand, over-expression of LMO4 enhanced GCSF-induced activation of p27 promoter. In addition, co-treatment of GCSF with the HDAC inhibitor TSA further upregulated p27 promoter activity to the same extent as the over-expression of LMO4, suggesting that LMO4, when over-expressed, might sequester and inhibit HDAC.
Fig. 4.

GCSF activates the p27 promoter. F11 neuronal cells were transfected with p27 luciferase reported and treated with GCSF alone or with the Jak/Stat inhibitor AG490. siRNA to LMO4, but not the control siRNA, attenuated p27 promoter activation by GCSF. The HDAC inhibitor TSA further augmented the activation of the p27 promoter in the presence of GCSF. Over-expression of LMO4 also augments GCSF-mediated p27 promoter activation. n = 8 experiments
Mapping the LMO4 interaction domain with HDAC2
To further characterize the interaction of LMO4 with HDAC2, FLAG-tagged LMO4 was cotransfected with Gal4-tagged HDAC2 in F11 cells. Both full-length or the C-terminal 286–489 amino acids of HDAC2 were co-immunoprecipitated with LMO4, but not a Gal4-tagged HDAC2 containing the N-terminal 1–285 amino acids (Fig. 5a). Thus, the C-terminal domain of HDAC2 interacts with LMO4.
Fig. 5.

LMO4 interacts with HDAC2. a Co-immunoprecipitation of Gal4-HDAC2 full length (FL) and Gal4-HDAC2 286-489 but not Gal4-HDAC2 1-285 with Flag-LMO4. b Co-immunoprecipitation of Gal4-HDAC2 with wild-type LMO4 and LMO4 with mutated second LIM domain (C87S). Mutation of the first LIM domain (C23S) reduced the interaction with HDAC2. n = 3 experiments. c Functional protein interaction revealed by a mammalian two-hybrid assay. Gal4-dependent pG5-luciferase reporter is activated by co-expression of Gal4HDAC2 286 (containing amino acids 286–489) and ActLMO4, demonstrating functional protein interaction. EV Empty vector. n = 8 experiments
LMO4 contains 2 LIM protein interaction domains. Each LIM domain consists of two zinc-finger motifs. Mutation of cysteine 23 to serine or cysteine 87 to serine were shown to disrupt the function of the first or second LIM domain of LMO4, respectively [15]. We used these mutated forms of LMO4 for co-immunoprecipitation assays and found that disruption of the first LIM domain (C23S) significantly reduced HDAC2 interaction with LMO4, whereas the second LIM domain mutant retained strong interaction with HDAC2 (Fig. 5b).
We confirmed the interaction of LMO4 with HDAC2 using the mammalian two-hybrid assay in F11 neuronal cells. In this assay, the DNA binding domain of the yeast transcription factor Gal4 is fused in frame to the HDAC2 sequence. LMO4 is fused to the activation domain of herpes virus viral protein 16 (Act). Interaction of these two chimeric proteins will reconstitute a functional transcription factor and activate a luciferase reporter construct bearing five copies of the Gal4 response element (pG5-luciferase). Consistent with the co-immunoprecipitation results, LMO4 bound to the C-terminal domain of HDAC2, and this interaction was disrupted by mutation of the first LIM domain (Fig. 5c).
Effect of restoring LMO4 in LMO4 null cortical neurons on the GCSF response
Cortical neurons from LMO4 null mouse embryos were cultured and transfected with expression vectors for the wild-type or the C23S mutant form of LMO4. GCSF did not induce Stat3 signaling nor activate p27 expression in cortical neurons transfected with empty vector (Fig. 6, lanes 1 and 2). However, over-expression of LMO4 rescued GCSF signaling, increasing Stat3 acetylation and phosphorylation and elevating p27 expression (Fig. 6, lane 3). The C23S mutant form of LMO4, that shows impaired interaction with HDAC2 only partially rescued GCSF signaling, but did not activate p27 expression.
Fig. 6.
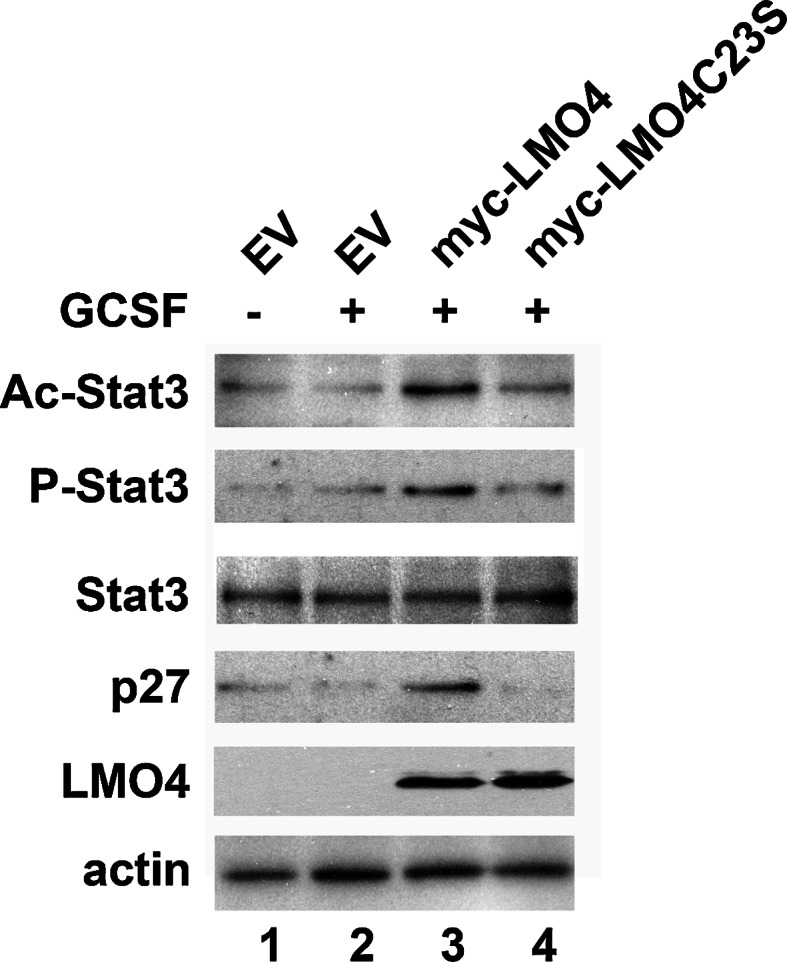
Restoring LMO4 expression in LMO4 null cortical neurons rescues GCSF signaling. Neurons were transfected with empty vector (EV) or myc-tagged wild-type or C23S mutant LMO4 expression vectors. Representative blots from two experiments
Discussion
GCSF signaling activates both neuroprotection and stem cell mobilization [35]. Exploiting these strategies may potentially be useful to treat ischemic stroke. Here, we showed that LMO4 is required for the neuroprotective effect of GCSF.
In neurons, LMO4 promotes GCSF-induced Stat3 anti-apoptotic signaling likely via two mechanisms: one, by promoting Stat3 phosphorylation, and the other, by promoting Stat3 acetylation. LMO4 is primarily localized in the nucleus with low levels also detected in the cytoplasm in several cell types, including hepatocytes and F11 neuronal cells [19, 20]. Although LMO4 does not physically interact with Stat3 ([20], and Chen et al., data not shown), LMO4 interacts with several proteins involved in Stat3 signaling, including the interleukin-6 (IL-6) receptor glycoprotein 130 (gp130), Janus kinase 1 (Jak1), SH2 domain containing tyrosine phosphatase 2 (Shp2) and suppressor of cytokine signaling 3 (SOCS3) [20]. Downregulation of LMO4 shortens gp130 half-life, reduces Stat3 tyrosine phosphorylation and Stat3-dependent gene activation [20]. We observed that GCSF treatment increased Stat3 phosphorylation in wild-type but not in LMO4 null cortical neurons, supporting the notion that LMO4 is required for GCSF-induced Stat3 phosphorylation and activation.
Extracellular signals like the IL-6 family of cytokines activate Stat3-dependent gene expression by increasing Stat3 phosphorylation and acetylation. Acetylation of Stat3 is required for Stat3 dimerization and binding to its target sequences to activate transcription [10]. Here, we also found that GCSF increases Stat3 acetylation in wild-type cortical neurons but not in LMO4 null cortical neurons. Moreover, restoring LMO4 expression in LMO4-null cortical neurons also restored GCSF-induced Stat3 phosphorylation and acetylation. Ours is the first study to identify a role for LMO4 in promoting GCSF signaling by maintaining Stat3 phosphorylation and acetylation. The finding that LMO4 promotes acetylation of Stat3 is novel but not surprising. LMO4 has been shown to associate with and modulate HDAC2 function [21, 23]. LMO4 over-expression was reported to prevent HDAC2 recruitment to the BMP7 promoter, thereby allowing transcription activation of the BMP7 gene [23]. Here, we showed that LMO4 interacts with the C-terminal domain of HDAC, the same domain that interacts with Stat3 [10]. Thus, one plausible mechanism whereby LMO4 might promote GCSF-mediated Stat3 signaling is by sequestering HDAC, preventing it from interacting with and deacetylating Stat3.
Ischemic injuries activate cell cycle genes, and inappropriate re-entry to the cell cycle triggers apoptosis in differentiated neurons. Glutamate excitotoxicity activates calpain protease that downregulates the expression of the cyclin-dependent kinase inhibitor p27 [34]. Elevated expression of p27 promotes survival of sympathetic neurons from DNA-damage-induced injury [36], whereas ablation of p27 induces cell death of cultured cortical neurons [33] and enhances kainate-induced seizures and hippocampal degeneration [37]. One of the possible mechanisms attributed to a anti-apoptotic role of p27 may be to prevent inappropriate cell cycle reentry (see review in [38]). Here, we found that GCSF upregulates luciferase expression from the p27 gene promoter. The promoter sequences of p27 contain a Stat3-responsive element [32]. Treatment with the Jak/Stat signaling inhibitor AG490 blocks GCSF-induced p27 promoter activity, demonstrating that GCSF signals through Stat3 to activate the p27 promoter. Our finding is in agreement with a previous study showing that expression of dominant negative Stat3 abolishes GCSF-induced p27 expression [32]. GCSF was ineffective at inducing p27 expression in LMO4 null cortical neurons further supporting that LMO4 contributes to GCSF-induced Stat3 signaling and gene activation. Similarly, in F11 neuronal cells, LMO4 siRNA attenuated the upregulation of the p27 promoter following GCSF stimulation. Moreover, restoring LMO4 to LMO4 null cortical neurons also rescued GCSF induction of p27.
HDAC inhibitors can be toxic to neurons, de-repressing cell death genes like myeloblastosis protein B (B-myb) and the Bcl2-interacting protein Bim [39, 40]. However, maintaining acetylation of certain transcription factors (like Stat3, NFkB, and SP1) with HDAC inhibitors can activate genes involved in neuronal survival [41, 42]. Several studies have shown that HDAC inhibitors significantly decrease neuronal injury and improve functional outcomes in multiple preclinical models of focal cerebral ischemia (see review by Langley [41]). Therefore, selecting the correct type and dose of HDAC inhibitor could be useful in the treatment of cerebral ischemia following stroke [42]. Our study suggests that combining the neuroprotective effect of GCSF with the gene-activating function of HDAC inhibitors might alleviate the negative effects of HDAC inhibitors. It remains to be tested in vivo whether, in combination with GCSF, low doses of HDAC inhibitors would be beneficial for the recovery from cerebral ischemia.
Acknowledgments
We are grateful to Dr. Alexandre Stewart for the Gal4 and anti-acetylated lysine antibodies and for helpful comments on this manuscript, and for Jin Xu’s excellent technical support. H-H Chen is supported by grants from the Canadian Institutes of Health Research (NSB 179197), the Heart and Stroke Foundation of Canada (Gant-in-Aid, NA6301), and the Heart and Stroke Foundation of Ontario Centre for Stroke Recovery. H-H Chen is the recipient of a Henry J. M. Barnett Research Scholarship award and New Investigator Award from the Heart and Stroke Foundation of Canada, and an Early Researcher Award from Ontario Ministry of Research and Innovation, and receives infrastructure support from the Canada Foundation for Innovation and the Ontario Research Fund.
Footnotes
M. Gomez-Smith, Z. Qin, and X. Zhou contributed equally to this work.
References
- 1.Mattson MP, Scheff SW. Endogenous neuroprotection factors and traumatic brain injury: mechanisms of action and implications for therapy. J Neurotrauma. 1994;11:3–33. doi: 10.1089/neu.1994.11.3. [DOI] [PubMed] [Google Scholar]
- 2.Metcalf D. The colony stimulating factors. Discovery, development, and clinical applications. Cancer. 1990;65:2185–2195. doi: 10.1002/1097-0142(19900515)65:10<2185::AID-CNCR2820651005>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 3.Schabitz WR, Kollmar R, Schwaninger M, Juettler E, Bardutzky J, Scholzke MN, Sommer C, Schwab S. Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischemia. Stroke. 2003;34:745–751. doi: 10.1161/01.STR.0000057814.70180.17. [DOI] [PubMed] [Google Scholar]
- 4.Schneider A, Kruger C, Steigleder T, Weber D, Pitzer C, Laage R, Aronowski J, Maurer MH, Gassler N, Mier W, Hasselblatt M, Kollmar R, Schwab S, Sommer C, Bach A, Kuhn HG, Schabitz WR. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest. 2005;115:2083–2098. doi: 10.1172/JCI23559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komine-Kobayashi M, Zhang N, Liu M, Tanaka R, Hara H, Osaka A, Mochizuki H, Mizuno Y, Urabe T. Neuroprotective effect of recombinant human granulocyte colony-stimulating factor in transient focal ischemia of mice. J Cereb Blood Flow Metab. 2006;26:402–413. doi: 10.1038/sj.jcbfm.9600195. [DOI] [PubMed] [Google Scholar]
- 6.Solaroglu I, Tsubokawa T, Cahill J, Zhang JH. Anti-apoptotic effect of granulocyte-colony stimulating factor after focal cerebral ischemia in the rat. Neuroscience. 2006;143:965–974. doi: 10.1016/j.neuroscience.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byts N, Samoylenko A, Woldt H, Ehrenreich H, Siren AL. Cell type specific signalling by hematopoietic growth factors in neural cells. Neurochem Res. 2006;31:1219–1230. doi: 10.1007/s11064-006-9149-0. [DOI] [PubMed] [Google Scholar]
- 8.Huang HY, Lin SZ, Kuo JS, Chen WF, Wang MJ. G-CSF protects dopaminergic neurons from 6-OHDA-induced toxicity via the ERK pathway. Neurobiol Aging. 2007;28:1258–1269. doi: 10.1016/j.neurobiolaging.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 9.Solaroglu I, Cahill J, Jadhav V, Zhang JH. A novel neuroprotectant granulocyte-colony stimulating factor. Stroke. 2006;37:1123–1128. doi: 10.1161/01.STR.0000208205.26253.96. [DOI] [PubMed] [Google Scholar]
- 10.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 11.Schock SC, Xu J, Duquette PM, Qin Z, Rai PS, Lewandowski AJ, Thompson CS, Seifert EL, Harper ME, Chen HH. Rescue of neurons from ischemic injury by PPARgamma requires a novel essential cofactor LMO4. J Neurosci. 2008;28:12433–12444. doi: 10.1523/JNEUROSCI.2897-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi K, Lee S, Lee B, Lee JW, Lee SK. LMO4 controls the balance between excitatory and inhibitory spinal V2 interneurons. Neuron. 2009;61:839–851. doi: 10.1016/j.neuron.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song MR, Sun Y, Bryson A, Gill GN, Evans SM, Pfaff SL. Islet-to-LMO stoichiometries control the function of transcription complexes that specify motor neuron and V2a interneuron identity. Development. 2009;136:2923–2932. doi: 10.1242/dev.037986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manetopoulos C, Hansson A, Karlsson J, Jonsson JI, Axelson H. The LIM-only protein LMO4 modulates the transcriptional activity of HEN1. Biochem Biophys Res Commun. 2003;307:891–899. doi: 10.1016/S0006-291X(03)01298-1. [DOI] [PubMed] [Google Scholar]
- 15.Kashani AH, Qiu Z, Jurata L, Lee SK, Pfaff S, Goebbels S, Nave KA, Ghosh A. Calcium activation of the LMO4 transcription complex and its role in the patterning of thalamocortical connections. J Neurosci. 2006;26:8398–8408. doi: 10.1523/JNEUROSCI.0618-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hahm K, Sum EY, Fujiwara Y, Lindeman GJ, Visvader JE, Orkin SH. Defective neural tube closure and anteroposterior patterning in mice lacking the LIM protein LMO4 or its interacting partner Deaf-1. Mol Cell Biol. 2004;24:2074–2082. doi: 10.1128/MCB.24.5.2074-2082.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tse E, Smith AJ, Hunt S, Lavenir I, Forster A, Warren AJ, Grutz G, Foroni L, Carlton MB, Colledge WH, Boehm T, Rabbitts TH. Null mutation of the Lmo4 gene or a combined null mutation of the Lmo1/Lmo3 genes causes perinatal lethality, and Lmo4 controls neural tube development in mice. Mol Cell Biol. 2004;24:2063–2073. doi: 10.1128/MCB.24.5.2063-2073.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen HH, Schock SC, Xu J, Safarpour F, Thompson CS, Stewart AF. Extracellular ATP-dependent upregulation of the transcription cofactor LMO4 promotes neuron survival from hypoxia. Exp Cell Res. 2007;313:3106–3116. doi: 10.1016/j.yexcr.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 19.Chen HH, Xu J, Safarpour F, Stewart AF. LMO4 mRNA stability is regulated by extracellular ATP in F11 cells. Biochem Biophys Res Commun. 2007;357:56–61. doi: 10.1016/j.bbrc.2007.03.113. [DOI] [PubMed] [Google Scholar]
- 20.Novotny-Diermayr V, Lin B, Gu L, Cao X. Modulation of the interleukin-6 receptor subunit glycoprotein 130 complex and its signaling by LMO4 interaction. J Biol Chem. 2005;280:12747–12757. doi: 10.1074/jbc.M500175200. [DOI] [PubMed] [Google Scholar]
- 21.Singh RR, Barnes CJ, Talukder AH, Fuqua SA, Kumar R. Negative regulation of estrogen receptor alpha transactivation functions by LIM domain only 4 protein. Cancer Res. 2005;65:10594–10601. doi: 10.1158/0008-5472.CAN-05-2268. [DOI] [PubMed] [Google Scholar]
- 22.Sum EY, Segara D, Duscio B, Bath ML, Field AS, Sutherland RL, Lindeman GJ, Visvader JE. Overexpression of LMO4 induces mammary hyperplasia, promotes cell invasion, and is a predictor of poor outcome in breast cancer. Proc Natl Acad Sci USA. 2005;102:7659–7664. doi: 10.1073/pnas.0502990102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang N, Lin KK, Lu Z, Lam KS, Newton R, Xu X, Yu Z, Gill GN, Andersen B. The LIM-only factor LMO4 regulates expression of the BMP7 gene through an HDAC2-dependent mechanism, and controls cell proliferation and apoptosis of mammary epithelial cells. Oncogene. 2007;26:6431–6441. doi: 10.1038/sj.onc.1210465. [DOI] [PubMed] [Google Scholar]
- 24.Ou XM, Jafar-Nejad H, Storring JM, Meng JH, Lemonde S, Albert PR. Novel dual repressor elements for neuronal cell-specific transcription of the rat 5-HT1A receptor gene. J Biol Chem. 2000;275:8161–8168. doi: 10.1074/jbc.275.11.8161. [DOI] [PubMed] [Google Scholar]
- 25.Zhou S, Fujimuro M, Hsieh JJ, Chen L, Hayward SD. A role for SKIP in EBNA2 activation of CBF1-repressed promoters. J Virol. 2000;74:1939–1947. doi: 10.1128/JVI.74.4.1939-1947.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Setogawa T, Shinozaki-Yabana S, Masuda T, Matsuura K, Akiyama T. The tumor suppressor LKB1 induces p21 expression in collaboration with LMO4, GATA-6, and Ldb1. Biochem Biophys Res Commun. 2006;343:1186–1190. doi: 10.1016/j.bbrc.2006.03.077. [DOI] [PubMed] [Google Scholar]
- 27.Chen HH, Kong WP, Zhang L, Ward PL, Roos RP. A picornaviral protein synthesized out of frame with the polyprotein plays a key role in a virus-induced immune-mediated demyelinating disease. Nat Med. 1995;1:927–931. doi: 10.1038/nm0995-927. [DOI] [PubMed] [Google Scholar]
- 28.Kochanek S, Clemens PR, Mitani K, Chen HH, Chan S, Caskey CT. A new adenoviral vector: replacement of all viral coding sequences with 28 kb of DNA independently expressing both full-length dystrophin and beta-galactosidase. Proc Natl Acad Sci USA. 1996;93:5731–5736. doi: 10.1073/pnas.93.12.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arai M, Imai H, Koumura T, Yoshida M, Emoto K, Umeda M, Chiba N, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase plays a major role in preventing oxidative injury to cells. J Biol Chem. 1999;274:4924–4933. doi: 10.1074/jbc.274.8.4924. [DOI] [PubMed] [Google Scholar]
- 30.Hasegawa T, Suzuki K, Sakamoto C, Ohta K, Nishiki S, Hino M, Tatsumi N, Kitagawa S. Expression of the inhibitor of apoptosis (IAP) family members in human neutrophils: up-regulation of cIAP2 by granulocyte colony-stimulating factor and overexpression of cIAP2 in chronic neutrophilic leukemia. Blood. 2003;101:1164–1171. doi: 10.1182/blood-2002-05-1505. [DOI] [PubMed] [Google Scholar]
- 31.Sun Y, Chin YE, Weisiger E, Malter C, Tawara I, Toubai T, Gatza E, Mascagni P, Dinarello CA, Reddy P. Cutting edge: negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J Immunol. 2009;182:5899–5903. doi: 10.4049/jimmunol.0804388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Koning JP, Soede-Bobok AA, Ward AC, Schelen AM, Antonissen C, van Leeuwen D, Lowenberg B, Touw IP. STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27(Kip1) Oncogene. 2000;19:3290–3298. doi: 10.1038/sj.onc.1203627. [DOI] [PubMed] [Google Scholar]
- 33.Akashiba H, Matsuki N, Nishiyama N. p27 small interfering RNA induces cell death through elevating cell cycle activity in cultured cortical neurons: a proof-of-concept study. Cell Mol Life Sci. 2006;63:2397–2404. doi: 10.1007/s00018-006-6194-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akashiba H, Matsuki N, Nishiyama N. Calpain activation is required for glutamate-induced p27 down-regulation in cultured cortical neurons. J Neurochem. 2006;99:733–744. doi: 10.1111/j.1471-4159.2006.04100.x. [DOI] [PubMed] [Google Scholar]
- 35.Yamashita T, Deguchi K, Sehara Y, Lukic-Panin V, Zhang H, Kamiya T, Abe K. Therapeutic strategy for ischemic stroke. Neurochem Res. 2009;34:707–710. doi: 10.1007/s11064-008-9842-2. [DOI] [PubMed] [Google Scholar]
- 36.Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueyama C, Akashiba H, Nakayama K, Nakayama KI, Nishiyama N, Matsuki N. Ablation of p27 enhance kainate-induced seizure and hippocampal degeneration. Neuroreport. 2007;18:1781–1785. doi: 10.1097/WNR.0b013e3282f16df6. [DOI] [PubMed] [Google Scholar]
- 38.Rashidian J, Iyirhiaro GO, Park DS. Cell cycle machinery and stroke. Biochim Biophys Acta. 2007;1772:484–493. doi: 10.1016/j.bbadis.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 39.Biswas SC, Liu DX, Greene LA. Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci. 2005;25:8349–8358. doi: 10.1523/JNEUROSCI.1570-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu DX, Biswas SC, Greene LA. B-myb and C-myb play required roles in neuronal apoptosis evoked by nerve growth factor deprivation and DNA damage. J Neurosci. 2004;24:8720–8725. doi: 10.1523/JNEUROSCI.1821-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langley B, Brochier C, Rivieccio MA. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke. 2009;40:2899–2905. doi: 10.1161/STROKEAHA.108.540229. [DOI] [PubMed] [Google Scholar]
- 42.Langley B, D’Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008;28:163–176. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]