

Abstract
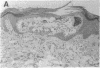
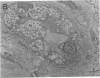
Recently a novel case of angiokeratoma corporis diffusum with glycoaminoaciduria was described in a 46-yr-old Japanese woman. Known causes of the cutaneous manifestation were eliminated by enzyme analyses, and further characterization of the accumulated urinary O-linked sialopeptides revealed identity to those excreted by patients with an infantile neuroaxonal dystrophy due to lysosomal alpha-N-acetylgalactosaminidase deficiency. Investigation of the alpha-N-acetylgalactosaminidase activity and protein in the proband revealed less than 2% of normal activity and the absence of detectable immunoreactive enzyme protein, findings comparable to those in the patients with infantile neuroaxonal dystrophy and alpha-N-acetylgalactosaminidase deficiency. In addition, the proband's unaffected offspring had half-normal levels of alpha-N-acetylgalactosaminidase activity, consistent with this enzymatic deficiency being the primary metabolic defect in this autosomal recessive trait. Ultrastructural examination of skin and blood cells from the adult proband revealed the presence of prominent lysosomal inclusions containing diffuse amorphous and filamentous material. In contrast, these morphologic findings were not observed in the nonneural tissues from patients with infantile neuroaxonal dystrophy and alpha-N-acetylgalactosaminidase deficiency. These studies document the occurrence of two forms of alpha-N-acetylgalactosaminidase deficiency and sialopeptiduria, a severe infantile-onset form of neuroaxonal dystrophy without angiokeratoma or visceral lysosomal inclusions and an adult-onset form characterized by angiokeratoma, extensive lysosomal accumulation of sialoglycopeptides and the absence of detectable neurologic involvement.
Full text
PDF




Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bishop D. F., Desnick R. J. Affinity purification of alpha-galactosidase A from human spleen, placenta, and plasma with elimination of pyrogen contamination. Properties of the purified splenic enzyme compared to other forms. J Biol Chem. 1981 Feb 10;256(3):1307–1316. [PubMed] [Google Scholar]
- Böhlen P., Stein S., Dairman W., Udenfriend S. Fluorometric assay of proteins in the nanogram range. Arch Biochem Biophys. 1973 Mar;155(1):213–220. doi: 10.1016/s0003-9861(73)80023-2. [DOI] [PubMed] [Google Scholar]
- Epinette W. W., Norins A. L., Drew A. L., Zeman W., Patel V. Angiokeratoma corporis diffusum with alpha-L-fucosidase deficiency. Arch Dermatol. 1973 May;107(5):754–757. [PubMed] [Google Scholar]
- Fabbro D., Desnick R. J., Grabowski G. A. Gaucher disease: genetic heterogeneity within and among the subtypes detected by immunoblotting. Am J Hum Genet. 1987 Jan;40(1):15–31. [PMC free article] [PubMed] [Google Scholar]
- Gehler J., Sewell A. C., Becker C., Hartmann J., Spranger J. Clinical and biochemical delineation of aspartyl-glycosaminuria as observed in two members of an Italian family. Helv Paediatr Acta. 1981;36(2):179–189. [PubMed] [Google Scholar]
- Hirabayashi Y., Matsumoto Y., Matsumoto M., Toida T., Iida N., Matsubara T., Kanzaki T., Yokota M., Ishizuka I. Isolation and characterization of major urinary amino acid O-glycosides and a dipeptide O-glycoside from a new lysosomal storage disorder (Kanzaki disease). Excessive excretion of serine- and threonine-linked glycan in the patient urine. J Biol Chem. 1990 Jan 25;265(3):1693–1701. [PubMed] [Google Scholar]
- Kanzaki T., Yokota M., Mizuno N., Matsumoto Y., Hirabayashi Y. Novel lysosomal glycoaminoacid storage disease with angiokeratoma corporis diffusum. Lancet. 1989 Apr 22;1(8643):875–877. doi: 10.1016/s0140-6736(89)92867-5. [DOI] [PubMed] [Google Scholar]
- Latham T., Grabowski G. A., Theophilus B. D., Smith F. I. Complex alleles of the acid beta-glucosidase gene in Gaucher disease. Am J Hum Genet. 1990 Jul;47(1):79–86. [PMC free article] [PubMed] [Google Scholar]
- Levran O., Desnick R. J., Schuchman E. H. Niemann-Pick disease: a frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc Natl Acad Sci U S A. 1991 May 1;88(9):3748–3752. doi: 10.1073/pnas.88.9.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden H. U., Klein R. A., Egge H., Peter-Katalinic J., Dabrowski J., Schindler D. Isolation and structural characterization of sialic-acid-containing glycopeptides of the O-glycosidic type from the urine of two patients with an hereditary deficiency in alpha-N-acetylgalactosaminidase activity. Biol Chem Hoppe Seyler. 1989 Jul;370(7):661–672. doi: 10.1515/bchm3.1989.370.2.661. [DOI] [PubMed] [Google Scholar]
- Miyatake T., Atsumi T., Obayashi T., Mizuno Y., Ando S., Ariga T., Matsui-Nakamura K., Yamada T. Adult type neuronal storage disease with neuraminidase deficiency. Ann Neurol. 1979 Sep;6(3):232–244. doi: 10.1002/ana.410060310. [DOI] [PubMed] [Google Scholar]
- Schindler D., Bishop D. F., Wolfe D. E., Wang A. M., Egge H., Lemieux R. U., Desnick R. J. Neuroaxonal dystrophy due to lysosomal alpha-N-acetylgalactosaminidase deficiency. N Engl J Med. 1989 Jun 29;320(26):1735–1740. doi: 10.1056/NEJM198906293202606. [DOI] [PubMed] [Google Scholar]
- Schindler D., Kanzaki T., Desnick R. J. A method for the rapid detection of urinary glycopeptides in alpha-N-acetylgalactosaminidase deficiency and other lysosomal storage diseases. Clin Chim Acta. 1990 Sep;190(1-2):81–91. doi: 10.1016/0009-8981(90)90282-w. [DOI] [PubMed] [Google Scholar]
- Wang A. M., Bishop D. F., Desnick R. J. Human alpha-N-acetylgalactosaminidase-molecular cloning, nucleotide sequence, and expression of a full-length cDNA. Homology with human alpha-galactosidase A suggests evolution from a common ancestral gene. J Biol Chem. 1990 Dec 15;265(35):21859–21866. [PubMed] [Google Scholar]
- Wang A. M., Desnick R. J. Structural organization and complete sequence of the human alpha-N-acetylgalactosaminidase gene: homology with the alpha-galactosidase A gene provides evidence for evolution from a common ancestral gene. Genomics. 1991 May;10(1):133–142. doi: 10.1016/0888-7543(91)90493-x. [DOI] [PubMed] [Google Scholar]
- Wang A. M., Schindler D., Desnick R. Schindler disease: the molecular lesion in the alpha-N-acetylgalactosaminidase gene that causes an infantile neuroaxonal dystrophy. J Clin Invest. 1990 Nov;86(5):1752–1756. doi: 10.1172/JCI114901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger D. A., Sattler M., Mueller O. T., Myers G. G., Schneiman R. S., Nixon G. W. Adult GM1 gangliosidosis: clinical and biochemical studies on two patients and comparison to other patients called variant or adult GM1 gangliosidosis. Clin Genet. 1980 May;17(5):323–334. doi: 10.1111/j.1399-0004.1980.tb00158.x. [DOI] [PubMed] [Google Scholar]
- Wenger D. A., Sujansky E., Fennessey P. V., Thompson J. N. Human beta-mannosidase deficiency. N Engl J Med. 1986 Nov 6;315(19):1201–1205. doi: 10.1056/NEJM198611063151906. [DOI] [PubMed] [Google Scholar]
- van Diggelen O. P., Schindler D., Willemsen R., Boer M., Kleijer W. J., Huijmans J. G., Blom W., Galjaard H. alpha-N-acetylgalactosaminidase deficiency, a new lysosomal storage disorder. J Inherit Metab Dis. 1988;11(4):349–357. doi: 10.1007/BF01800424. [DOI] [PubMed] [Google Scholar]