

Abstract
For infectious prion protein (designated PrPSc) to act as a template to convert normal cellular protein (PrPC) to its distinctive pathogenic conformation, the two forms of prion protein (PrP) must interact closely. The neuronal receptor that rapidly endocytoses PrPC is the low-density lipoprotein receptor-related protein 1 (LRP1). We show here that on sensory neurons LRP1 is also the receptor that binds and rapidly endocytoses smaller oligomeric forms of infectious prion fibrils, and recombinant PrP fibrils. Although LRP1 binds two molecules of most ligands independently to its receptor clusters 2 and 4, PrPC and PrPSc fibrils bind only to receptor cluster 4. PrPSc fibrils out-compete PrPC for internalization. When endocytosed, PrPSc fibrils are routed to lysosomes, rather than recycled to the cell surface with PrPC. Thus, although LRP1 binds both forms of PrP, it traffics them to separate fates within sensory neurons. The binding of both to ligand cluster 4 should enable genetic modification of PrP binding without disrupting other roles of LRP1 essential to neuronal viability and function, thereby enabling in vivo analysis of the role of this interaction in controlling both prion and LRP1 biology.
Keywords: Endocytosis, LRP1, Lysosome, Neuron, Prion protein
Introduction
The causative event in prion disease is thought to be a change in conformation of the predominantly α-helical C-terminal domain of cellular prion protein (PrPC) that increases its β-pleated sheet content as the protein converts to the infectious PrPSc isoform (Prusiner, 1998). This conversion rarely occurs de novo as a single molecule reaction. Pre-existing PrPSc oligomers powerfully stimulate the conversion (Silveira et al., 2005), not only recruiting adjacent PrPC to their amyloidogenic conformation and thereby propagating infection, but also acting as template to specify the new conformation. For there is not a single conformation that characterises PrPSc fibrils, nor a single set of pathological characteristics that define prion diseases, but rather multiple different strains of disease that differ in their molecular and pathogenic characteristics (Aguzzi, 1998; Bruce et al., 1994). These different strains are stably propagated through many generations in animals or cell lines, and require intimate contact between PrPSc as template, and PrPC as substrate, to dictate the precise conformation adopted on the growing amyloid seed. Interaction between PrPC and PrPSc is also required for neurotoxicity, since PrPSc in the brain kills only PrPC-expressing neurons by a mechanism that remains to be elucidated (Brandner et al., 1998; Chesebro et al., 2005; Mallucci et al., 2003; Mallucci et al., 2007).
Where do the infectious and cellular forms of PrP meet? A range of evidence indicates the infectious interaction occurs either at the cell surface or in some post-surface organelle to which the proteins are trafficked (Caughey and Baron, 2006; Krammer et al., 2009; Marijanovic et al., 2009; Morris et al., 2006). PrPC, being glycosylphosphatidylinositol (GPI)-anchored to the surface membrane, occupies lipid ‘rafts’ (Brügger et al., 2004; Taraboulos et al., 1995). On neurons and N2a cells, PrPC rapidly leaves these ordered lipid domains to enter disordered membrane and then coated pits, from where it is recycled back to the surface, the whole process taking only a few minutes (Morris et al., 2006; Shyng et al., 1994; Shyng et al., 1993; Sunyach et al., 2003). The fate of infectious PrPSc on the cell surface has not been followed, although it appears to be corralled in heavier rafts than PrPC (Naslavsky et al., 1997; Vey et al., 1996).
We have recently shown that the endocytic partner of PrPC on sensory neurons is LRP1 (low-density lipoprotein receptor-related protein 1) (Parkyn et al., 2008). This enormous (600 kDa) receptor is derived from four linear repeats of the LDL receptor (Fig. 1), and retains in two repeats (clusters 2 and 4) high affinity binding sites for a range of ligands. These include the proteins that mediate lipid and sterol uptake in neurons (apo-lipoprotein E and α2macroglobulin), regulate the extracellular adhesive and proteolytic environments, and remove Aβ amyloid fibrils from the surrounding cerebrospinal fluid (Deane et al., 2004; May and Herz, 2003). Expression of ‘minireceptors’ composed of individual ligand binding clusters 2 or 4 (Fig. 1) has facilitated the analysis of ligand binding to LRP1 and shown that most ligands bind to both clusters (Obermoeller-McCormick et al., 2001). LRP1 (often shortened to LRP in the literature) should not be confused with the laminin receptor precursor (also abbreviated as LRP), a multifunctional dual cytoplasmic-surface protein of 37/67 kDa that has been identified as a receptor for normal and infectious PrP (Gauczynski et al., 2006; Gauczynski et al., 2001; Pflanz et al., 2009).
Fig. 1.

Schematic comparison of the modular structure of LRP1, its minireceptors and PrPC. The domains of LRP1 [adapted from Li et al. and Springer (Li et al., 2000; Springer, 1998)], the minireceptors mLRP2 and mLRP4 containing, respectively, ligand-binding clusters 2 and 4 (Obermoeller-McCormick et al., 2001), and PrPC, for which the flexible N- and structured C-terminal domains, and GPI anchor, are indicated; grey lines represent the surface membrane. LRP1 domains and the site of furin cleavage are indicated (see legend); Roman numerals indicate the four repeats of the ligand-binding complement-like domains; endocytic motifs (Li et al., 2000) are indicated in the cytoplasmic domain. EGF, epidermal growth factor receptor; HA,’flu haemagglutinin.
The N-terminal domain of PrPC is necessary and sufficient for endocytosis of this protein, and in particular the basic motif (KKRPKP) at its immediate N-terminus is essential (Sunyach et al., 2003). This N-terminal domain binds to human LRP1 with a Kd of approximately 20 nM (Parkyn et al., 2008). The assembly of amyloid fibrils of both native and recombinant PrP by interactions between their C-terminal domains leaves their N-terminal domains accessible for antibody binding (Jeffrey et al., 1997; Novitskaya et al., 2006; Safar et al., 1998). Should these N-terminal domains bind LRP1, each receptor molecule could bring together the template (PrPSc) and substrate (PrPC) for the conformational conversion that produces prion disease (Morris et al., 2006).
The binding of infectious PrPSc to cells has been studied using fluorescently tagged fibrils proteolytically digested with proteinase K (Horonchik et al., 2005; Magalhaes et al., 2005). This step degrades non-PrP protein within the fibrils to ensure that only the protease resistant PrPRes is labelled, thereby validating the use of the fluorescent tag to follow PrPSc uptake by the cells (Magalhaes et al., 2005); but it also removes much of the N-terminal domain of PrP that is present in freshly isolated infectious fibrils (Hope et al., 1986).
We have followed Baron et al. (Baron et al., 2006) and Greil et al. (Greil et al., 2008) in studying a native (non-proteolysed) fibril preparation with the N-terminal domain of PrP intact. We have isolated PrPSc fibrils from mouse brain infected with ME7 strain of scrapie and tagged them with Alexa Fluor 594 fluorochrome. Although PrP is the dominant labelled protein in this preparation, it is difficult to exclude the possibility that it is some fluorescent contaminant, unrelated to prion infection, that is bound and endocytosed by the neurons. We have therefore tested our main conclusions with fluorescently labelled pure recombinant PrP fibrils, and by detection of PrPSc fibrils by their protease resistance (PrPRes) in immunoblotting.
Results
Production of PrPSc fibrils that bind to sensory neurons
PrPSc fibrils, isolated from mouse brain terminally infected with ME7 strain of scrapie, showed the appropriate PrP bands on immunoblots, before and after digestion with proteinase K (supplementary material Fig. S1). These same bands were those primarily labelled in the fibrils by either biotin or Alexa Fluor 700 using N-hydroxysuccinimide linkage to couple the label to the fibrils. The same linkage was used to couple Alexa Fluor 488 and 594 to fibrils to follow their cellular trafficking.
When isolated, the fibrils were large (~10 μm) aggregates (Fig. 2A,B) that did not bind to the sensory neurons. Sonication released many smaller oligomers of ~20 nm (Fig. 2C-E) and increased by 100-fold the proportion of Alexa-Fluor-594-labelled fibrils that bound to the neurons (supplementary material Table S1). The smaller oligomers remained in the supernatant after the short centrifugation used to pellet the large unsonicated aggregates (Fig. 2E) and accounted for most of the binding to neurons (supplementary material Table S1). Binding of the sonicated fibrils to the neurons was saturable (supplementary material Fig. S2). The whole sonicated fraction (without size separation) was used in these studies and is referred to as PrPSc fibrils. Stored aliquots of labelled fibrils were re-sonicated for 90 minutes immediately before addition to cells.
Fig. 2.

Electron micrographs of prion fibrils. Negatively stained samples of PrPSc fibrils before (A,B) and after (C-E) sonication are shown. Those in C and D have not been further centrifuged (and are typical of the fibrils added to the neuronal cultures). (E) The supernatant left after a sonicated fraction was centrifuged at 14,000 g for 2 minutes. Arrows in C-E point to smaller oligomers of about 20 nm released by sonication.
Binding and uptake of PrPSc by sensory neurons
Adult sensory neurons in culture spread their axons over the substrate with their large (15-50 μm diameter) cell bodies protruding into the medium above. Their uptake of surface-labelled proteins was analysed in optical sections of ~3 μm taken through 5-12 cells, reconstructed from deconvolved serial sections taken every 100 nm in the Z-axis towards the middle of the cell body [5-20 μm above the substrate (Parkyn et al., 2008)]. Sonicated Alexa-Fluor-594-labelled fibrils were pre-incubated with neurons for 30 minutes at 12-15°C to allow binding to the cell surface; unbound fibrils were removed by washing, and the cells transferred to 37°C to allow the trafficking of this pulse of surface-bound PrPSc fibrils to be followed. Other surface proteins simultaneously labelled with Alexa-Fluor-488-coupled ligands were Thy-1 (which is not endocytosed and so delineates the cell surface at 37°C) and PrPC, both labelled with Fab antibody fragments (Sunyach et al., 2003); transferrin (Tf; binding to the Tf receptor) and activated α2-macroglobulin (α2M*, binding to LRP1), both endocytosed rapidly via coated pits.
Alexa-Fluor-594-PrPSc fibrils were rapidly endocytosed, with 96.2±2.6% internalized by 2 minutes (Fig. 3A,B), significantly more (P=0.02) than the 85.6±13.5% of surface-labelled PrPC internalized by a separate population of neurons (supplementary material Table S2). Endocytosed PrPSc accumulated in perinuclear tubular structures, where it colocalized at 2 minutes with Tf (Fig. 3C; of internalized protein, 28.4±20.2% of PrPSc colocalized with Tf, and 72.2±28.3% Tf colocalized with PrPSc) and particularly with the LRP1 ligand, α2 M* (Fig. 3D; 66.7±18.1% of PrPSc colocalized with α2M*, and 69.6±14.6% of α2 M* colocalized with PrPSc; supplementary material Table S3). PrPSc uptake in the presence of 1 μM α2M* was identical (95.8±3.4%; P=0.8) to its uptake in the absence of this LRP1 ligand.
Fig. 3.

Endocytosis of sonicated prion fibrils by neurons. Binding and uptake of Alexa-Fluor-594-labelled PrPSc fibrils (red) by sensory neurons is compared with other ligands labelled with Alexa Fluor 488 (green). The chromatin was labelled with DAPI (blue) throughout this and following figures, which show cells with typical (average) levels of labelling. Scale bars: 5 μm. (A,B) PrPSc fibrils, and Alexa Fluor 488-Fab for Thy-1, on neurons placed at 37°C for 30 (A) and 120 (B) seconds. (C,D) PrPSc fibrils, and Alexa-Fluor-488-labelled transferrin (C) or α2M* (D), on neurons placed at 37°C for 2 minutes. In C, Tf but not PrPSc fibrils was re-added to the medium at 37°C to enable Tf binding to delineate the surface. (E,F) PrPSc fibril binding and uptake at the level of substrate cells. E shows Alexa-Fluor-594-PrPSc fibrils (arrows) on an axon emerging from a neuronal cell body. F shows a cluster of Alexa-Fluor-594-PrPSc below the lowest extremity of the neuronal nucleus (arrow; the majority of this cell and its nucleus lies above this plane); adjacent substrate cells (nuclei visible) have not bound the fibrils. These are the lowest images (normally not used) collected in the Z-axis series for neurons allowed to endocytose PrPSc fibrils for 2 minutes. (G,H) PrPSc fibrils out-compete PrPC for internalization. Neurons were prelabelled at 12-15°C with Alexa-Fluor-488-conjugated Fab to label PrPC, and with Alexa-Fluor-594-labelled PrPSc fibrils; the cells were washed and transferred to 37°C and fixed after 2 (G) or 4 (H) minutes.
When imaged at substrate level, virtually all Alexa-Fluor-594-PrPSc fibrils were on neurons, including their processes (Fig. 3E) with the substrate cells themselves not binding labelled fibrils (Fig. 3F).
Trafficking of endogenous PrPC (prelabelled with Alexa-Fluor-488-Fab) and exogenous Alexa-Fluor-594-PrPSc on the same neurons was examined. After 2 minutes at 37°C, 91.6±7.7% of PrPSc (red), but only 38.4±9.3% of PrPC (green) was internalized (Fig. 3G), compared with 82.4±4.9% of PrPC in control cells not exposed to PrPSc (P<0.001 for PrPC endocytosed). Even after 4 minutes at 37°C (Fig. 3H), only 60.9±27.0% of PrPC had been internalized (compared with >98% by control cells without additional PrPSc fibrils). This displacement of PrPC endocytosis by PrPSc suggests that the two forms of PrP compete for the same binding site, in contrast to the lack of competitive inhibition by PrPSc of α2M* endocytosis.
Inhibition of LRP1 lowers binding and uptake of PrPSc
Receptor associated protein (RAP), the specific inhibitor of LRP receptors (Iadonato et al., 1993; Moestrup et al., 1993) strongly inhibited the binding and uptake of PrPSc by wild-type (WT) neurons (PrP+/+; Fig. 4A,B and Table 1). This binding was independent of PrPC expression since knockout (KO; PrP−/−) neurons bound and internalized PrPSc, and this was inhibited by 80 nM RAP (Fig. 4C,D and Table 1).
Fig. 4.

Inhibition of LRP1 inhibits the uptake of PrPSc fibrils by neurons. (A,B) Neurons were pre-incubated at 37°C for 2 hours with vehicle only (A) or 80 nM RAP (B), and then for 30 minutes at 12-15°C with Alexa-Fluor-594-laelled PrPSc (red) before being returned to 37°C for 2 minutes. Cells were fixed and surface immunolabelled for LRP1 (green). (C,D) KO (PrP−/−) neurons were incubated with vehicle (C) or 80 nM RAP (D) and then with PrPSc fibrils as above, with Tf (green) added as an endocytic control; cells were placed for 2 minutes at 37°C before fixing and processing. (E,F) WT (PrP+/+) neurons were preincubated for 2 hours at 37°C with vehicle (E) or 80 nM RAP (F), and then at 12-15°C with recombinant PrP fibrils labelled with Alexa Fluor 594 (red) and Alexa-Fluor-488-Tf (green), then placed at 37°C for 2 minutes. (G-J) WT neurons were pre-incubated for 2 hours with siRNAControl (G) or siRNALRP1.1-3 (H-J) and then PrPSc bound for 30 minutes at 12-15°C before being returned to 37°C for 2 minutes. Alexa Fluor-488-Tf labelling, done as a positive control, is not shown so as not to obscure the reduction of PrPSc binding. Scale bars: 5 μm.
Table 1.
Inhibition of LRP1 ligand binding (RAP) or expression level (siRNA) reduces binding of PrPSc, or recombinant PrPRec, fibrils to sensory neurons
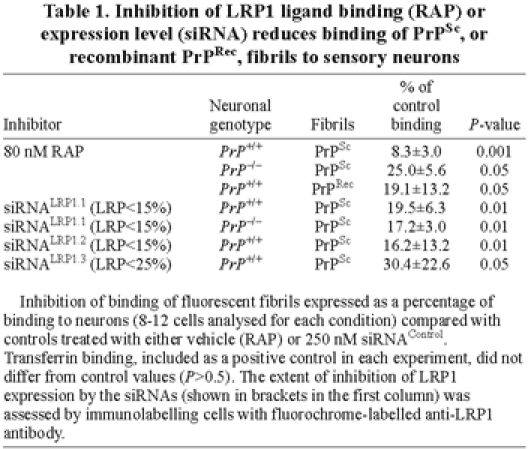
Amyloid fibrils composed purely of recombinant PrP (PrPRec) (Bocharova et al., 2005) bound to sensory neurons and were >95% internalized within 2 minutes at 37°C; this binding was inhibited by 80 nM RAP (Fig. 4E,F and Table 1).
LRP1 protein levels were transiently (for several hours) reduced to 10-25% of normal levels by delivering three independent siRNAs specific for LRP1 [siRNALRP1.1-3 (Parkyn et al., 2008)] using the membrane-crossing peptide penetratin-1, and assayed 2 hours after applying the siRNAs to avoid the multiple effects of longer term suppression of neuronal LRP1 (Parkyn et al., 2008). All three siRNALRP1 inhibited the binding and uptake of PrPSc (Fig. 4G-J, Table 1 and supplementary material Table S4).
PrPC and PrPSc are internalized by binding to ligand binding cluster 4 but not 2
The LRP1-binding activity of stably transfected N2a cells expressing low levels of endogenous LPR1 (control cells transfected with empty vector), or expressing the minireceptors mLRP2 (M2 cells) or mLRP4 (M4 cells), was assessed by incubating them with trace (1 nM) levels of Alexa-Fluor-488-labelled RAP, and Alexa-Fluor-594-PrPSc fibrils (Table 2). The cells expressing mLRP2 had six, and the cells expressing mLRP4 had thirty, times the level of RAP binding compared with the parental control cells. Only the M4 cells bound Alexa Fluor 594-PrPSc fibrils significantly above controls, and ten times more than the M2 cells (Table 2).
Table 2.
Binding of PrPSc and RAP (A), and endocytosis of surface PrPC (B), by control N2a cells and stable transfectants expressing LRP1 minireceptors

To assess the interaction between the endogenous PrPC expressed by these cells and the LRP1 minireceptors, the surface levels of PrPC, and the amount internalized within 5 minutes at 37°C, were measured (Table 2). Surface levels of PrPC on M4 cells were nearly three times the level on control cells, and four times that on M2 cells. Over 50% of the high level of PrPC on the M4 cells was endocytosed within 5 minutes, compared with only a third of the PrPC on control cells (i.e. M4 cells endocytosed five times more PrPC than the control cells). The low amount of surface PrPC on M2 cells was barely endocytosed, ten times less than the M4 cells. Together, these results indicate that the fourth but not second ligand-binding cluster of LRP1 binds both PrPSc and PrPC.
As an independent test of the interaction of PrPSc and PrPC with the minireceptors, the later were immunoprecipitated from detergent lysates of the N2a cell lines after allowing PrPSc to bind. Immunoprecipitates of the minireceptors were digested with proteinase K (PK) to remove PrPC from the sample, enabling PrPSc to be identified by antibody binding to the residual protease-resistant PrPRes, which was detected only in the mLRP4 cells (Fig. 5A,B).
Fig. 5.

Co-immunoprecipitation of PrPSc and PrPC with mLRP4 (M4) but not mLRP2 (M2) minireceptor of LRP1. (A,B) Cells were incubated with (A) or without (B) PrPSc fibrils, the cell lysates were sonicated then minireceptors immunoprecipitated by their HA epitope. PrP was detected after proteinase K digestion (500 μg/ml, 30 minutes, 37°C) by immunoblotting with SAF83 antibody (Demart et al., 1999). PrPRes is seen in A; proteinase K removes all trace of PrPC expressed by the cells (B). (C) Total PrPC in detergent lysates of the cells detected by immunoblotting using SAF 83 (actin levels shown in G). (D) Immunoblot of LRP1 (MMMM antibody specific for the cytoplasmic domain shared by LRP1 and minireceptors) in the cell lysates. (E) Immunoblot of PrPC co-immunoprecipitated with the minireceptors via the HA epitope. (F) Immunoprecipitate of minireceptors (via their HA epitope), detected by immunoblotting for the LRP cytoplasmic domain. (G) Actin immunoblots from cell lysates used in C-F.
To determine the interaction of PrPC with the minireceptors, the control and minireceptor cells were found to express comparable levels of total PrPC, as seen in immunoblots (Fig. 5C, actin control in G), despite their different levels of the surface protein (Table 2). Full-length LRP1 is proteolytically cleaved in trans Golgi by furin to yield an 85 kDa cytoplasmic plus transmembrane fragment and a 550 kDa extracellular fragment that contained all ligand binding clusters (Fig. 1). The 550 kDa chain of native LRP1 could only be detected on these cells using more sensitive detection, confirming their very low level of endogenous LRP1 expression. Furin cleavage of full-length minireceptors (190 kDa) produces two fragments, both with a molecular mass of 85 kDa (Obermoeller-McCormick et al., 2001). In the cell lysate, the uncleaved 190 kDa form of mLRP2 predominates, whereas furin-cleaved mLRP4 is more prominent (Fig. 5D), showing that more mLRP4 than mLRP2 progresses from the ER through the Golgi to the cell surface, in keeping with the fivefold higher surface level of mLRP4 (Table 2). Immunoprecipitation of the minireceptors via their HA tag preferentially selected for the furin-cleaved 85 kDa forms (Fig. 5F). mLPR4 but not mLRP2 co-immunoprecipitated PrPC (Fig. 5E).
Endocytosed PrPSc is routed to lysosomes, not recycled back via endosomes
Not only do PrPSc and PrPC bind to LRP1 on the neuronal surface, their initial endocytic trafficking is to the same perinuclear endosomal compartments (Fig. 3H). However, PrPC is recycled back to the neuronal surface (Morris et al., 2006; Sunyach et al., 2003), whereas labelled PrPSc fibrils were not similarly recycled, at least within the time scale of these studies. An extended experiment was set up in which sensory neurons were pre-labelled with PrPSc fibrils (30 minutes at 12-15°C), which for this experiment were not removed by washing but remained in the medium for 0.5-30 minutes incubation at 37°C before fixation and immunolabelling for LAMP2, a marker of late endosomes and lysosomes (Cuervo and Dice, 1996).
LAMP2 marked tubular compartments located initially in the cortical regions of the cell past which the endocytosed PrPSc fibrils rapidly moved to concentrate in deeper, perinuclear compartments (Fig. 6A,B). By 10-30 minutes, however, the LAMP2-labelled lysosomes had moved into the cell interior and the proportion of labelled PrPSc fibrils that colocalized with LAMP2 steadily rose (Fig. 6C,D, Fig. 7). The total amount of Alexa Fluor 594 fluorescence associated with PrPSc fibrils bound to or internalized by the neurons increased over the first 10 minutes of incubation at 37°C, then decreased by 30 minutes (Figs 6 and 7).
Fig. 6.

PrPSc fibrils are routed to lysosomes in sensory and hippocampal neurons. (A-D) Adult sensory neurons prelabelled at 12-15°C with Alexa-Fluor-594-PrPSc fibrils (red) and held at 37°C for 0.5, 2, 10 and 30 minutes, respectively, before being fixed, permeabilized and immunolabelled (green) for LAMP2. Images collected in a Z-axis series (34 sections at 100 nm intervals) have been deconvolved and reassembled in 3D; chromatin was labelled with DAPI (blue). (E) Cultured hippocampal neurons at 37°C were incubated for 15 minutes with Lysotracker (red) and Alexa-Fluor-488-PrPSc fibrils (green; note the change in fluorochrome), then washed extensively with artificial cerebrospinal fluid and photographed in the same buffer. Arrowhead indicates a pyramidal neuron cell body with colocalized Lysotracker and PrPSc fibrils; small arrows indicate similar colocalization in ‘spots’ along neurites. (F) Stills from a film of neurites in the hippocampal neuron culture shown in E, focused on four spots of Alexa-Fluor-488-PrPSc fibrils within lysosomes (red Lysotracker channel not shown for clarity), two of which (arrows) loose their fluorescence within the 15 second period shown. Scale bars: 5 μm.
Fig. 7.

Fate of endocytosed Alexa-Fluor-594-PrPSc fibrils. Variation in intensity of fluorescence of PrPSc fibrils (black bars) and of its percentage overlap with LAMP2 immunolabelling (grey bars), as a function of time of incubation at 37°C with sensory neurons.
To gain a better view of the fate of labelled PrPSc fibrils following endocytosis, we examined differentiated hippocampal organ cultures in which the pyramidal cell neurites, flattened on the substrate, could be viewed continuously in a single plane of focus.
Hippocampal neurons rapidly took up labelled PrPSc fibrils similarly to the sensory neurons and transported them to lysosomes within their cell bodies and neurites (Fig. 6E; 50.9±10.2% of labelled PrPSc fibrils colocalized with Lysotracker, and 69.7±5.7% of Lysotracker colocalized with labelled PrPSc fibrils). Lysosomes in neurites that had taken up PrPSc fibrils did not move perceptibly during 60 minutes of observation, allowing the fate of their ingested Alexa Fluor 594-fibrils to be followed. Within 5-15 minutes after focusing on neurites, lysosomes that fluoresced green with PrPSc fibrils rapidly (with 15 seconds) lost their fluorescence (Fig. 6F shows two examples), and by 60 minutes very little green fluorescence remained in the culture.
Since the endocytosis of PrPC on hippocampal neurons has not previously been described, we note here that endogenous surface PrPC labelled with Alexa Fluor 488-Fab (Parkyn et al., 2008) was rapidly endocytosed and transported retrogradely along processes without going into lysosomes. This movement was significantly faster, and over longer distances, than the transport of PrPSc to proximal lysosomes (supplementary material Table S5).
Binding of PrPSc fibrils to LRP1, detected by protease resistance (PrPRes)
The protease resistance of PrPSc fibrils was used as an independent check on their binding by sensory neurons. We refer to the fibrils so detected as PrPRes, although this includes any endogenous PrPC that has been converted by the initial exogenous fibrils.
PrPRes was bound by both WT and KO neurons, and was inhibited by RAP and siRNALRP1.1 (Fig. 8A), confirming LRP1 as the receptor involved.
Fig. 8.

Fate of ingested PrPSc fibrils, determined as PrPRes, in long-term cultures of sensory neurons. All samples were sonicated for 90 minutes before splitting into samples, one-tenth for non-proteolysed analysis and nine-tenths for PK treatment with PrPRes, detected by SAF83. (A) PrPRes, with actin as a loading control, in wild-type (WT) and knockout (KO) cultures, treated for 1.5 hours with vehicle or 80 nM RAP, or siRNAControl or siRNALRP1.1 before PrPSc fibrils were incubated with the cells for 30 minutes at 37°C. (B) PrPSc fibrils, incubated for 30 minutes with sensory neurons from WT, KO or Tg20 mice, were maintained in long-term culture for the weeks shown. To keep the signal within a linear range of intensity, ECL Plus development of the peroxidase signal was used rather than the more sensitive ECL Advanced used in A and D. WT and KO PrPRes samples were run on the same gel to enable direct comparison (week 12* for WT is a one-tenth dilution of the 12-week PrPRes sample). Tg20 samples were run separately. Both monomer and dimer bands of PrPRes are shown. (C) PrPRes recovered in cultures after the times shown were measured by scanning the bands and normalized against actin levels (mean ± s.d., n≥ 3; supplementary material Table S6). Asterisks indicate that the value differs from the KO value at that time point with *P<0.05 or *P<0.01. (D) Cultures were pre-treated with siRNAControl or siRNALRP1.1 for 1.5 hours before incubating for 0.5 hours with PrPSc fibrils, then washed and maintained in culture for 4 weeks. The left panel shows that less PrPRes was bound initially by the cells incubated with siRNALRP1.1, whereas by 4 weeks the level of PrPRes in both control and LRP1-inhibited cells was comparable (middle panel). PrPC levels (right panel) were, however, strongly stimulated in the cultures in which LRP1 levels were originally suppressed. Actin levels are shown in lower inset on the right.
To test the long-term fate of bound fibrils, cultures initially incubated with PrPSc fibrils were maintained for up to 12 (Fig. 8B) and 15 weeks (supplementary material Fig. S3A). In WT cultures, PrPRes persisted at reduced levels (30-50% of the initial level) for 6 weeks, and then rose steeply to be more than seven times the initial level by 12 weeks (determined by scanning the gels; Fig. 8C; the increase was tenfold when estimated by comparison of a 1:10 dilution with the 0 week sample; supplementary material Table S6). KO levels of PrPRes remained below that of the initial inoculum, whereas cultures from Tg20 mice, whose sensory neurons express 14-fold higher levels of surface PrPC than WT neurons (Parkyn et al., 2008), showed a slightly faster increase in PrPRes than WT neurons by 8 weeks (Fig. 8B,C; 2.7-fold over the initial value by scanning of gels, or fourfold by comparing a 1:10 dilution with the 0 week sample; supplementary material Fig. S3B and Table S6).
The effect of transient inhibition of LRP1 during the initial incubation with PrPSc fibrils, upon longer term development of PrPRes, was analysed after 4 weeks (Fig. 8D). Inhibition of initial fibril binding in these cultures by siRNALRP1.1was indicated by the higher level of PrPRes remaining in the medium after that incubation (Fig. 8D). Four weeks later, the level of PrPRes in the control and LRP1-suppressed cultures was very similar. The major difference was in total PrP, and so of PrPC, which was distinctly higher in the siRNALRP1.1-treated cultures.
Discussion
Identification of a neuronal surface receptor for PrPSc is of interest primarily because it must play a role in determining whether and how PrPSc interacts with PrPC on neurons. Since this study, and a previous investigation of the neuronal endocytic partner for PrPC, both led to the well characterized, rapidly endocytosed neuronal surface receptor LRP1, we focus here on the interaction between PrP and LRP1. Recent reviews (e.g. Caughey and Baron, 2006; Krammer et al., 2009; Linden et al., 2008) provide a wider perspective on interactions proposed for PrP with other receptors, and their implication for prion infection.
We found it necessary to sonicate PrPSc fibrils isolated from infected brain to reduce them to smaller (~20 nm) oligomers before they would bind and be endocytosed by sensory neurons. The initial large aggregates presumably results from the isolation procedure, since ME7 strain of scrapie has diffuse, sub-light microscopic (<1 μm) fibrils deposited in infected brain (Jeffrey et al., 1997). Previous studies with unsonicated PrPSc fibrils observed they were not taken up by neurons until they were slowly broken down into smaller units by biological processes at the cell surface (Baron et al., 2006; Magalhaes et al., 2005); with fibrils of varied size, smaller oligomers are taken up faster (Greil et al., 2008). Sonication has been used to break PrPSc fibrils of the RML strain into their most infectious units, which are oligomers of 17-27 nm corresponding to 14-28 PrP molecules (Silveira et al., 2005; Caughey et al., 2009).
Sonicated PrPSc fibrils were internalized even faster than PrPC, presumably because they were pre-bound to their receptor at 12-15°C, allowing endocytosis to commence immediately when the temperature was raised. By contrast, only a portion of surface PrPC is bound to LRP1 at any time, the remainder being contained within lipid ‘rafts’ which exclude LRP1 (Chen et al., 2009). PrPSc binding was inhibited by RAP, indicating the receptor to be an LRP-family member, where LRP1 is the only member detected on sensory neurons (Parkyn et al., 2008), and the only member endocytosed this rapidly (Li et al., 2001). Three different siRNAs that lower the expression of LPR1 proportionately inhibited the binding of PrPSc fibrils. Together, these results strongly implicate LRP1 as the neuronal receptor that binds and endocytoses the smaller oligomeric fibrils of PrPSc.
In fact, PrPSc fibrils competed with endogenous PrPC for endocytosis and so presumably for binding to LRP1. The ligand binding sites on LRP1 have been analysed by the use of minireceptors (Mikhailenko et al., 2001; Obermoeller-McCormick et al., 2001) or recombinant proteins (Neels et al., 1999) that express the four ligand-binding clusters singly and in combination. Most endogenous ligands of LRP1 bind separately to both clusters 2 and 4, so the receptor is divalent for these ligands (Neels et al., 1999; Obermoeller-McCormick et al., 2001). Clusters 1 and 3 retain only vestigial binding domains. RAP binds with high affinity to clusters 2 and 4, and weakly to cluster 3, the only LRP ligand known to do so (Neels et al., 1999). Also unique is the binding of α2M* to a site spanning clusters 1 and 2, and not to cluster 4 (Mikhailenko et al., 2001), which explains its failure to compete with PrPSc for binding and internalization. Pseudomonas exotoxin A binds only to cluster 4 (Obermoeller-McCormick et al., 2001). The binding of both PrPC and PrPSc fibrils only to cluster 4, and not 2, is therefore unparalleled for an endogenous ligand of LPR1. This property should allow the selective inactivation of the PrP binding site in cluster 4 to enable an otherwise functional LRP1 to be made. This should allow stable inhibition of PrP-LRP1 interactions in otherwise healthy neurons.
Currently, the standard approach of stably inhibiting the expression of native LRP1 to study its role in prion infection is not possible because of its multiple roles essential for neuronal viability (May and Herz, 2003; May et al., 2004). The limitations of even transient inhibition of full-length LRP1 for studying long-term effects upon prion infectivity were evident in this study. Transient knockdown of LRP1 during infection lowered the uptake of fibrils, but even at 4 weeks stimulated PrPC expression and thus possibly the rate of conversion of PrPC to PrPRes to compensate for the lower level of PrPSc bound initially. The interdependence of levels of PrPC and LRP1 on the cell surface (and not the total amount expressed by neurons) has been noted previously [inhibition of LRP1 led to accumulation of PrPC in biosynthetic compartments at the expense of cell surface levels; PrPC-over-expressing Tg20 mice had 1.8-fold higher levels of surface LRP1 (Parkyn et al., 2008)]. The same interdependent biosynthetic trafficking is presumably responsible for the elevated levels of surface (not total) PrPC and minireceptor mLRP4 on transfected N2a cells, compared with the levels on mLRP2-expressing cells, since only the former minireceptor binds PrPC. A stable experimental system in which neurons respond only to prion infection, rather than to interdependent changes in surface expression of PrPC and LRP1, is needed to elucidate the role of LRP1 in prion infection.
The adult sensory neuron cultures did serve to demonstrate the persistence of PrPRes in the neurons for weeks after the fluorescent tag was hydrolysed in the lysosomes. Importantly, the marked accumulation of PrPRes after a lag phase of 6 weeks demonstrated that prion infection, as indicated by conversion of PrPC to PrPRes, had occurred. Furthermore, Tg20 neurons, despite their very high levels of PrPC expression, increased their PrPRes levels only slightly faster than the WT neurons. Although the course of prion disease is more than twice as fast in Tg20 compared to WT mice, in the end stages of the disease the Tg20 mice have half the levels of PrPRes compared to WT animals (Fischer et al., 1996). It will be interesting in this system to identify when prion-dependent neuronal death occurs; in preliminary experiments to date we have failed to maintain infected Tg20 neurons for 12-15 weeks. These sensory neuron cultures contain, in addition to neurons, fibroblasts, Schwann cells and some satellite cells that together we refer to as substrate cells. Their failure to bind PrPSc fibrils, or to express PrPC (Parkyn et al., 2008) suggests they are unlikely to play a direct role in the conversion process, a view that is borne out by the lack of PrPRes in cultures in which the neurons had died (supplementary material Fig. S3A).
Why is PrPC recycled back to the cell surface whereas PrPSc fibrils are routed to lysosomes? A possible factor is size — PrPC is clustered (Brügger et al., 2004; Sunyach et al., 2003) but not known to be physically aggregated at the cell surface, whereas the size of PrPSc oligomers could signal their transfer to lysosomes. Infectivity of PrPSc is critically linked to size, with oligomers up to hexomers being non-infectious, those of 14-28 subunits being maximally infectious, and then diminishing in proportion to increasing oligomer size (Riesner et al., 1996; Silveira et al., 2005). It will be of interest to determine the lower size limit that triggers routing of PrPSc to lysosomes. There is also an upper limit of 120 nm on the size of cargo that can be endocytosed via coated pits (Conner and Schmid, 2003), so that only oligomers of intermediate size are likely to be endocytosed by LRP1 and trafficked to lysosomes.
Despite the rapid quenching of fluorescence in the lysosomes, the fibrils detected as PrPRes persist for weeks in the cells. Possibly it is only the PrPSc fibrils that are conveyed to the lysosome that become infectious. Although in every endocytic cycle most PrPC is returned to the surface, a small proportion is routed to lysosomes for degradation, ensuring overall that PrPC has a short half-life (Shyng et al., 1993). Given the long persistence of the fibrils, the lysosome may trap PrPSc as in a filter through which all PrPC is funnelled for degradation, thus maximizing over several weeks the opportunity for interaction between the two forms of PrP.
This proposal is at variance with the suggestion, arising from studies on stably infected GT1 and N2a cell lines, that neither early nor late endosomes are the site of prion conversion, but rather it is the recycling endosome (Marijanovic et al., 2009). For stable infection of proliferating cell lines, the rate of PrPSc production must match that of cell division (doubling every day or so) and not be cytotoxic, neither of which are characteristics of prion infection either in vivo or in the cultured sensory neurons. Differentiated adult neurons never divide; their endocytic trafficking of both forms of PrP occurs considerably more rapidly, and their production of PrPRes is very much slower, than the cell lines. We see no recycling of fibrils that would give them access, in sensory neurons, to the recycling endosomes identified as the likely site of conversion in rapidly dividing cell lines.
While the primary role of LRP1 is lipid uptake, it also plays a significant role as a scavenger receptor, removing protease and matrix complexes from extracellar space (May and Herz, 2003). PrPC is emerging as an active partner for LRP1 in the scavenger role. PrPC through its His-containing octapeptide repeats can bind and endocytose pathological levels of Cu2+ (Wells et al., 2006) and the toxic metabolite hemin (Lee et al., 2007). PrPC has recently been shown to be a high-affinity ligand for Aβ oligomers (Lauren et al., 2009), suggesting that the endocytotic trafficking of PrPC mediated by LRP1 may be important in removing these neurotoxic oligomers in the brain. LRP1 further plays a complex role in controlling the metabolism of Alzheimer precursor protein (APP) (Bu, 2009; Marzolo and Bu, 2009), so this receptor may be the link between PrPC expression and APP metabolism (Parkin et al., 2007).
That a single receptor should mediate such a diverse range of roles is unusual, but LRP1 is most unusual, being 5-10 times larger than most receptors, with 31 repeats of its ligand-binding domain extracellularly, and with intracellular motifs that bind a range of adaptor proteins linking LRP1 to neuronal proteins including APP, and neurotransmitter and growth factor receptors (May and Herz, 2003). Heparan sulphate and lactoferrin are key regulators of LRP1 activity (e.g. Mahley and Ji, 1999; Nathan et al., 2002; Wang et al., 2004), which may explain their roles in modulating PrPC trafficking and PrPSc fibril binding to cells (Ben-Zaken et al., 2003; Hijazi et al., 2005; Horonchik et al., 2005; Iwamaru et al., 2008; Paquet et al., 2007). Deciphering the individual roles of this massive, multi-faceted receptor is complex. PrP, despite its small size, is proving to have its own complex biology and pathology that includes a unique relationship to LRP1 during biosynthesis and endocytic trafficking of the normal, and pathogenic, forms of PrP. Understanding the functional consequences of this interaction will not be straightforward, but promises to be interesting.
Materials and Methods
General methods
Fluorochrome labelling of reagents and their use to follow endocytosis; siRNA inhibition; immunoprecipitation; immunoblotting and protein determination have been described previously (Parkyn et al., 2008; Sunyach et al., 2003). The 2S anti-PrP Fab [against amino acids 142-160 in the C-terminal domain of mouse PrP (Ford et al., 2002)] used to label PrPC failed to detect intact PrPSc fibrils, shown by the failure of the Fab to react at all with PrP0/0 neurons to which PrPSc fibrils were bound (not shown). Alexa-Fluor-700-labelled PrPSc fibrils were visualized using an ODYSSEY Infrared Imaging system (LI-COR Biosciences).
PrPSc Fibrils
PrPSc fibrils were prepared as described previously (Hope et al., 1986). Briefly, an homogenate of five ME7-infected mouse brains was solubilized at 12-15°C in 10% Sarkosyl, a low speed (30 minutes at 10,000 g, 15°C) pellet was removed and the supernatant pelleted at 205,000 g for 160 minutes. This pellet, resuspended in 0.6 M KI plus 6 mM Na2S2O3, was centrifuged through a 20% sucrose cushion at 255,000 g for 90 minutes. The pellet was dispersed in water by short (15 minutes) sonication in a cuphorn sonicator (Sonics and Materials VCX500 ultrasonic processor with a 53 mm cuphorn, pulsing for 5 seconds with 1-second intervals to deliver 10,000 joules/minute, water cooled to 23°C), with subsequent steps done aseptically. Protein concentration, determined by the Bio-Rad Protein Assay on an aliquot of fibrils solubilized in 1 M NaOH then neutralized with 1 M HCl, was adjusted to 2 mg/ml and 200 μl aliquots stored at 4°C. For labelling with Alexa fluorochromes or biotin, thawed samples were pelleted (14,000 g, 2 minutes), resuspended in 0.1 M Na2HCO3 buffer pH 8.0, and labelled with N-hydroxysuccinimide ester of fluorochromes (Molecular Probes) or biotin (Pierce) at half the manufacturer's recommended level to limit reaction with lysine residues important for the endocytosis of PrPC (Sunyach et al., 2003). Fibrils were dialysed for 2 days against sterile PBS; immediately before use they were sonicated as above for 90 minutes and added at 5 μg/ml to tissue culture supernatant for labelling cells. For these experiments, four different preparations of fibrils were made, three from archived stocks and one from mice infected with aliquots from the first batch of sonicated fibrils. The fibrils used here have also been used in a companion study of fibril uptake by mouse colon in vivo, in which the appearance, over the space of months, of PrPRes in lymphoreticular tissue and then brain is followed by the onset of terminal scrapie in the mice (R.C.M., unpublished data).
Mouse full-length recombinant PrP encompassing residues 23-230 was expressed and purified as described previously (Bocharova et al., 2005) with modifications (Breydo et al., 2005) and converted to amyloid fibrils according to the procedure of Bocharova et al. (Bocharova et al., 2005). It was fluorochrome labelled as above, and sonicated for 1 minute before use.
Electron microscopy (EM)
Cell suspension (2 μl) was dropped into Pioloform-coated EM grids and allowed to settle for 2 minutes. Excess solution was drawn off carefully by blotting the edge of the grid with the cut edge of a piece of hardened filter paper. The preparations were negative stained by dropping 2 μl 1% aqueous uranyl acetate onto the grids, and this was then removed after 90 seconds by blotting as described above. The preparation was allowed to dry before examination in a FEI Tecnai12 electron microscope; images were captured with a Gatan BioScan camera.
Cell culture
Primary sensory neurons were routinely cultured from the dorsal root ganglia of 4- to 5-week-old C57Bl6 mice. Cells from each mouse, dispersed evenly on ten poly-D-lysine- and laminin-coated 13 mm glass coverslips were cultured in a 24-well plate at 37°C with 5.0% CO2 in phenol-red-free neurobasal medium (Gibco) with B-27 serum-free supplement, plus 7.5 pg/ml nerve growth factor (NGF), 200 μM GlutaMAX-1, and 1 μg/ml cholesterol as low-density lipoproteins (Sigma Aldrich) for 3-7 days prior to commencing experiments (Parkyn et al., 2008; Sunyach et al., 2003). For detection of PrPRes after long-term culture, mice of 129/0la (PrP+/+), NPU PrP0/0 KO strain (Manson et al., 1994), and Tg20 mice over-expressing PrPC (Fischer et al., 1996) were used (99 mice in total). Dissociated ganglia of each single mouse were applied to a 16 mm coverslip to provide sufficient material for assay. Mice were processed in two groups (differing in genotype) of three animals, on consecutive days. Three days after the second group was introduced to culture, all wells were incubated with sonicated fibrils at 37°C, washed thoroughly three times, then returned to culture with one-third of the medium changed twice a week. On harvesting, cells were solubilized in 0.5% Brij 96-PBS with 400 Kunitz unit/ml of DNase, sonicated for 30 minutes to ensure PrPRes was dispersed evenly, and one-tenth volume removed to determine (and equalize) protein concentration and immunoblot for PrPC and actin, with the remainder used to determine PrPRes. PK digestion of PrPSc fibrils was for 1 hour at 37°C with 50 μg PK/ml for PrPSc fibrils, and 500 μg/ml protein of cell lysate. Peroxidase reaction was developed with ECL Advanced or (for more accurate quantification) ECL Plus (both GE Healthcare), and bands measured by scanning into ImageJ software at a resolution of 1200 dpi using a Canon Canoscan 3200F.
Postnatal day 4 or 5 rat organotypic hippocampal cultures were maintained for a week in DMEM with 20% horse serum before use; lysosomes were labelled and imaged as described previously (McGuinness et al., 2007). N2a cells, stably transfected with empty vector or the LRP1 minigene containing cluster 2 or 4 ligand binding sites were produced as described for CHO-LRP null cells (Obermoeller-McCormick et al., 2001). We previously noted that N2a cells and sensory neurons should not be chilled below 12-15°C during the pre-incubation to bind ligands prior to endocytosis. Here it was found that if hippocampal pyramidal neurons were chilled even to 20°C they were slow to recover vesicle trafficking; they were incubated at 37°C with 5 μg/ml PrPSc fibrils or anti-PrP Fab, and 75 nM Lysotracker Red DND99 (Molecular Probes, Invitrogen).
N2a control and minireceptor lines were fragmented by N2 bomb cavitation (Chen et al., 2009); the post-nuclear membrane fraction was solubilized in lysis buffer (Obermoeller-McCormick et al., 2001) and cleared by ultracentrifugation (100,000 g, 1 hour) and minireceptors immunoprecipitated using the Dynal Protein G Immunoprecipitation Kit (Invitrogen); the appropriate antibody was pre-bound to the beads before addition to the lysates [anti-HA was monoclonal 12CA5 from Babco; affinity purified sheep 2S antibody for PrP (Ford et al., 2002) and rabbit MMMM antiserum for LRP1 cytoplasmic domain (Zerbinatti et al., 2004)].
Supplementary Material
Acknowledgments
We thank Chris Birkett and Andy Gill (Institute for Animal Health, Compton), and Oduolo Abiolo and Steve Whatley (Institute of Psychiatry, King's College London) for donating three different batches of mouse brains terminally infected with ME7 scrapie strain. This work was supported by BBSRC grants 18/BS516350 and BB/C506680/1, and an MRC doctoral training studentship to C.J.P. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/2/246/DC1
References
- Aguzzi A. (1998). Protein conformation dictates prion strain. Nat. Med. 4, 1125-1126 [DOI] [PubMed] [Google Scholar]
- Baron G. S., Magalhaes A. C., Prado M. A., Caughey B. (2006). Mouse-adapted scrapie infection of SN56 cells: greater efficiency with microsome-associated versus purified PrP-res. J. Virol. 80, 2106-2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zaken O., Tzaban S., Tal Y., Horonchik L., Esko J. D., Vlodavsky I., Taraboulos A. (2003). Cellular heparan sulfate participates in the metabolism of prions. J. Biol. Chem. 278, 40041-40049 [DOI] [PubMed] [Google Scholar]
- Bocharova O. V., Breydo L., Parfenov A. S., Salnikov V. V., Baskakov I. V. (2005). In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J. Mol. Biol. 346, 645-659 [DOI] [PubMed] [Google Scholar]
- Brandner S., Isenmann S., Kuhne G., Aguzzi A. (1998). Identification of the end stage of scrapie using infected neural grafts. Brain Pathol. 8, 19-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breydo L., Bocharova O. V., Makarava N., Salnikov V. V., Anderson M., Baskakov I. V. (2005). Methionine oxidation interferes with conversion of the prion protein into the fibrillar proteinase K-resistant conformation. Biochemistry 44, 15534-15543 [DOI] [PubMed] [Google Scholar]
- Bruce M., Chree A., McConnell I., Foster J., Pearson G., Fraser H. (1994). Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Phil. Trans. R. Soc. Lond. B 343, 405-411 [DOI] [PubMed] [Google Scholar]
- Brügger B., Graham C. H., Leibrecht I., Mombelli E., Jen A., Wieland F. T., Morris R. J. (2004). The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J. Biol. Chem. 279, 7530-7536 [DOI] [PubMed] [Google Scholar]
- Bu G. (2009). Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B., Baron G. S. (2006). Prions and their partners in crime. Nature 443, 803-810 [DOI] [PubMed] [Google Scholar]
- Caughey B., Baron G. S., Chesebro B., Jeffrey M. (2009). Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 78, 177-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Jen A., Warley A., Lawrence M. J., Quinn P. J., Morris R. J. (2009). Isolation at physiological temperature of detergent-resistant membranes with properties expected of lipid rafts: the influence of buffer composition. Biochem. J. 417, 525-533 [DOI] [PubMed] [Google Scholar]
- Chesebro B., Trifilo M., Race R., Meade-White K., Teng C., LaCasse R., Raymond L., Favara C., Baron G., Priola S., et al. (2005). Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308, 1435-1439 [DOI] [PubMed] [Google Scholar]
- Conner S. D., Schmid S. L. (2003). Regulated portals of entry into the cell. Nature 422, 37-44 [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Dice J. F. (1996). A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501-503 [DOI] [PubMed] [Google Scholar]
- Deane R., Wu Z., Sagare A., Davis J., Du, Yan S., Hamm K., Xu F., Parisi M., LaRue B., Hu H. W., et al. (2004). LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43, 333-344 [DOI] [PubMed] [Google Scholar]
- Demart S., Fournier J.-G., Creminon C., Frobert Y., Lamoury F., Marce D., Lasmézas C., Dormont D., Grassi J., Deslys J.-P. (1999). New insight into abnormal prion protein using monoclonal antibodies. Biochem. Biophys. Res. Comm. 265, 652-627 [DOI] [PubMed] [Google Scholar]
- Fischer M., Rülicke T., Raeber A., Sailer A., Moser M., Oesch B., Brandner S., Aguzzi A., Weissmann C. (1996). Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 15, 1255-1264 [PMC free article] [PubMed] [Google Scholar]
- Ford M. L., Burton L. J., Li H., Graham C. H., Frobert Y., Grassi J., Hall S. M., Morris R. J. (2002). A marked disparity between the expression of prion protein and its message by neurons of the central nervous system. Neuroscience 111, 533-551 [DOI] [PubMed] [Google Scholar]
- Gauczynski S., Peyrin J. M., Haik S., Leucht C., Hundt C., Rieger R., Krasemann S., Deslys J. P., Dormont D., Lasmezas C. I., et al. (2001). The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 20, 5863-5875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauczynski S., Nikles D., El-Gogo S., Papy-Garcia D., Rey C., Alban S., Barritault D., Lasmezas C. I., Weiss S. (2006). The 37-kDa/67-kDa laminin receptor acts as a receptor for infectious prions and is inhibited by polysulfated glycanes. J. Infect. Dis. 194, 702-709 [DOI] [PubMed] [Google Scholar]
- Greil C. S., Vorberg I. M., Ward A. E., Meade-White K. D., Harris D. A., Priola S. A. (2008). Acute cellular uptake of abnormal prion protein is cell type and scrapie-strain independent. Virology 379, 284-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijazi N., Kariv-Inbal Z., Gasset M., Gabizon R. (2005). PrPSc incorporation to cells requires endogenous glycosaminoglycan expression. J. Biol. Chem. 280, 17057-17061 [DOI] [PubMed] [Google Scholar]
- Hope J., Morton L. J., Farquhar C. F., Multhaup G., Beyreuther K., Kimberlin R. H. (1986). The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein (PrP). EMBO J. 5, 2591-2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horonchik L., Tzaban S., Ben-Zaken O., Yedidia Y., Rouvinski A., Papy-Garcia D., Barritault D., Vlodavsky I., Taraboulos A. (2005). Heparan sulfate is a cellular receptor for purified infectious prions. J. Biol. Chem. 280, 17062-17067 [DOI] [PubMed] [Google Scholar]
- Iadonato S. P., Bu G., Maksymovitch E. A., Schwartz A. L. (1993). Interaction of a 39 kDa protein with the low-density-lipoprotein-receptor-related protein (LRP) on rat hepatoma cells. Biochem. J. 296, 867-875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamaru Y., Shimizu Y., Imamura M., Murayama Y., Endo R., Tagawa Y., Ushiki-Kaku Y., Takenouchi T., Kitani H., Mohri S., et al. (2008). Lactoferrin induces cell surface retention of prion protein and inhibits prion accumulation. J. Neurochem. 107, 636-646 [DOI] [PubMed] [Google Scholar]
- Jeffrey M., Goodsir C. M., Bruce M. E., McBride P. A., Fraser J. R. (1997). In vivo toxicity of prion protein in murine scrapie: ultrastructural and immunogold studies. Neuropathol. Appl. Neurobiol. 23, 93-101 [PubMed] [Google Scholar]
- Krammer C., Vorberg I., Schatzl H. M., Gilch S. (2009). Therapy in prion diseases: from molecular and cellular biology to therapeutic targets. Infect. Disord. Drug Targets 9, 3-14 [DOI] [PubMed] [Google Scholar]
- Lauren J., Gimbel D. A., Nygaard H. B., Gilbert J. W., Strittmatter S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128-1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. S., Raymond L. D., Schoen B., Raymond G. J., Kett L., Moore R. A., Johnson L. M., Taubner L., Speare J. O., Onwubiko H. A., et al. (2007). Hemin interactions and alterations of the subcellular localization of prion protein. J. Biol. Chem. 282, 36525-36533 [DOI] [PubMed] [Google Scholar]
- Li Y., Marzolo M. P., van Kerkhof P., Strous G. J., Bu G. (2000). The YXXL motif, but not the two NPXY motifs, serves as the dominant endocytosis signal for low density lipoprotein receptor-related protein. J. Biol. Chem. 275, 17187-17194 [DOI] [PubMed] [Google Scholar]
- Li Y., Lu W., Marzolo M. P., Bu G. (2001). Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J. Biol. Chem. 276, 18000-18006 [DOI] [PubMed] [Google Scholar]
- Linden R., Martins V. R., Prado M. A., Cammarota M., Izquierdo I., Brentani R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673-728 [DOI] [PubMed] [Google Scholar]
- Magalhaes A. C., Baron G. S., Lee K. S., Steele-Mortimer O., Dorward D., Prado M. A., Caughey B. (2005). Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J. Neurosci. 25, 5207-5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley R. W., Ji Z. S. (1999). Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res. 40, 1-16 [PubMed] [Google Scholar]
- Mallucci G., Dickinson A., Linehan J., Klohn P. C., Brandner S., Collinge J. (2003). Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302, 871-874 [DOI] [PubMed] [Google Scholar]
- Mallucci G. R., White M. D., Farmer M., Dickinson A., Khatun H., Powell A. D., Brandner S., Jefferys J. G., Collinge J. (2007). Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 53, 325-335 [DOI] [PubMed] [Google Scholar]
- Manson J. C., Clarke A. R., Hooper M. L., Aitchison L., McConnell I., Hope J. (1994). 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 8, 121-127 [DOI] [PubMed] [Google Scholar]
- Marijanovic Z., Caputo A., Campana V., Zurzolo C. (2009). Identification of an intracellular site of prion conversion. PLoS Pathog. 5, e1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzolo M. P., Bu G. (2009). Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer's disease. Semin. Cell Dev. Biol. 20, 191-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P., Herz J. (2003). LDL receptor-related proteins in neurodevelopment. Traffic 4, 291-301 [DOI] [PubMed] [Google Scholar]
- May P., Rohlmann A., Bock H. H., Zurhove K., Marth J. D., Schomburg E. D., Noebels J. L., Beffert U., Sweatt J. D., Weeber E. J., et al. (2004). Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol. Cell. Biol. 24, 8872-8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness L., Bardo S. J., Emptage N. J. (2007). The lysosome or lysosome-related organelle may serve as a Ca2+ store in the boutons of hippocampal pyramidal cells. Neuropharmacology 52, 126-135 [DOI] [PubMed] [Google Scholar]
- Mikhailenko I., Battey F. D., Migliorini M., Ruiz J. F., Argraves K., Moayeri M., Strickland D. K. (2001). Recognition of alpha 2-macroglobulin by the low density lipoprotein receptor-related protein requires the cooperation of two ligand binding cluster regions. J. Biol. Chem. 276, 39484-39491 [DOI] [PubMed] [Google Scholar]
- Moestrup S. K., Nielsen S., Andreasen P., Jorgensen K. E., Nykjaer A., Roigaard H., Gliemann J., Christensen E. I. (1993). Epithelial glycoprotein-330 mediates endocytosis of plasminogen activator-plasminogen activator inhibitor type-1 complexes. J. Biol. Chem. 268, 16564-16570 [PubMed] [Google Scholar]
- Morris R. J., Parkyn C. J., Jen A. (2006). Traffic of prion protein between different compartments on the neuronal surface, and the propagation of prion disease. FEBS Lett. 580, 5565-5571 [DOI] [PubMed] [Google Scholar]
- Naslavsky N., Stein R., Yanai A., Friedlander G., Taraboulos A. (1997). Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J. Biol. Chem. 272, 6324-6331 [DOI] [PubMed] [Google Scholar]
- Nathan B. P., Jiang Y., Wong G. K., Shen F., Brewer G. J., Struble R. G. (2002). Apolipoprotein E4 inhibits, and apolipoprotein E3 promotes neurite outgrowth in cultured adult mouse cortical neurons through the low-density lipoprotein receptor-related protein. Brain Res. 928, 96-105 [DOI] [PubMed] [Google Scholar]
- Neels J. G., van Den Berg B. M., Lookene A., Olivecrona G., Pannekoek H., van Zonneveld A. J. (1999). The second and fourth cluster of class A cysteine-rich repeats of the low density lipoprotein receptor-related protein share ligand-binding properties. J. Biol. Chem. 274, 31305-31311 [DOI] [PubMed] [Google Scholar]
- Novitskaya V., Makarava N., Bellon A., Bocharova O. V., Bronstein I. B., Williamson R. A., Baskakov I. V. (2006). Probing the conformation of the prion protein within a single amyloid fibril using a novel immunoconformational assay. J. Biol. Chem. 281, 15536-15545 [DOI] [PubMed] [Google Scholar]
- Obermoeller-McCormick L. M., Li Y., Osaka H., FitzGerald D. J., Schwartz A. L., Bu G. (2001). Dissection of receptor folding and ligand-binding property with functional minireceptors of LDL receptor-related protein. J. Cell Sci. 114, 899-908 [DOI] [PubMed] [Google Scholar]
- Paquet S., Daude N., Courageot M. P., Chapuis J., Laude H., Vilette D. (2007). PrPc does not mediate internalization of PrPSc but is required at an early stage for de novo prion infection of Rov cells. J. Virol. 81, 10786-10791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin E. T., Watt N. T., Hussain I., Eckman E. A., Eckman C. B., Manson J. C., Baybutt H. N., Turner A. J., Hooper N. M. (2007). Cellular prion protein regulates beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc. Natl. Acad. Sci. USA 104, 11062-11067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkyn C. J., Vermeulen E. G., Mootoosamy R. C., Sunyach C., Jacobsen C., Oxvig C., Moestrup S., Liu Q., Bu G., Jen A., et al. (2008). LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J. Cell Sci. 121, 773-783 [DOI] [PubMed] [Google Scholar]
- Pflanz H., Vana K., Mitteregger G., Renner-Muller I., Pace C., Kuchenhoff H., Kretzschmar H. A., Wolf E., Weiss S. (2009). Scrapie-infected transgenic mice expressing a laminin receptor decoy mutant reveal a prolonged incubation time associated with low levels of PrPres. J. Mol. Biol. 388, 721-729 [DOI] [PubMed] [Google Scholar]
- Prusiner S. B. (1998). Prions. Proc. Natl. Acad. Sci. USA 95, 13363-13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesner D., Kellings K., Post K., Wille H., Serban H., Groth D., Baldwin M. A., Prusiner S. B. (1996). Disruption of prion rods generates 10-nm spherical particles having high alpha-helical content and lacking scrapie infectivity. J. Virol. 70, 1714-1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar J., Wille H., Itri V., Groth D., Serban H., Torchia M., Cohen F. E., Prusiner S. B. (1998). Eight prion strains have PrP(Sc) molecules with different conformations [In Process Citation]. Nat. Med. 4, 1157-1165 [DOI] [PubMed] [Google Scholar]
- Shyng S.-L., Huber M. T., Harris D. A. (1993). A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J. Biol. Chem. 268, 15922-15928 [PubMed] [Google Scholar]
- Shyng S.-L., Heuser J. E., Harris D. A. (1994). A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J. Cell Biol. 125, 1239-1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira J. R., Raymond G. J., Hughson A. G., Race R. E., Sim V. L., Hayes S. F., Caughey B. (2005). The most infectious prion protein particles. Nature 437, 257-261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer T. A. (1998). An extracellular beta-propeller module predicted in lipoprotein and scavenger receptors, tyrosine kinases, epidermal growth factor precursor, and extracellular matrix components. J. Mol. Biol. 283, 837-862 [DOI] [PubMed] [Google Scholar]
- Sunyach C., Jen A., Deng J., Fitzgerald K., Frobert Y., McCaffrey M., Morris R. J. (2003). The mechanism of internalisation of GPI anchored prion protein. EMBO J. 22, 3591-3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboulos A., Scott M., Semenov A., Avrahami D., Laszlo L., Prusiner S. B., Avraham D. (1995). Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 129, 121-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vey M., Pilkuhn S., Wille H., Nixon R., DeArmond S. J., Smart E. J., Anderson R. G., Taraboulos A., Prusiner S. B. (1996). Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl. Acad. Sci. USA 93, 14945-14949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Herndon M. E., Ranganathan S., Godyna S., Lawler J., Argraves W. S., Liau G. (2004). Internalization but not binding of thrombospondin-1 to low density lipoprotein receptor-related protein-1 requires heparan sulfate proteoglycans. J. Cell Biochem. 91, 766-776 [DOI] [PubMed] [Google Scholar]
- Wells M. A., Jelinska C., Hosszu L. L., Craven C. J., Clarke A. R., Collinge J., Waltho J. P., Jackson G. S. (2006). Multiple forms of copper (II) co-ordination occur throughout the disordered N-terminal region of the prion protein at pH 7.4. Biochem. J. 400, 501-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti C. V., Wozniak D. F., Cirrito J., Cam J. A., Osaka H., Bales K. R., Zhuo M., Paul S. M., Holtzman D. M., Bu G. (2004). Increased soluble amyloid-beta peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc. Natl. Acad. Sci. USA 101, 1075-1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}