

Abstract
RNA editing that converts adenosine to inosine in double-stranded RNA (dsRNA) is mediated by adenosine deaminases acting on RNA (ADAR). ADAR1 and ADAR2 form respective homodimers, and this association is essential for their enzymatic activities. In this investigation, we set out experiments aiming to determine whether formation of the homodimer complex is mediated by an amino acid interface made through protein-protein interactions of two monomers or via binding of the two subunits to a dsRNA substrate. Point mutations were created in the dsRNA binding domains (dsRBDs) that abolished all RNA binding, as tested for two classes of ADAR ligands, long and short dsRNA. The mutant ADAR dimer complexes were intact, as demonstrated by their ability to co-purify in a sequential affinity-tagged purification and also by their elution at the dimeric fraction position on a size fractionation column. Our results demonstrated ADAR dimerization independent of their binding to dsRNA, establishing the importance of protein-protein interactions for dimer formation. As expected, these mutant ADARs could no longer perform their catalytic function due to the loss in substrate binding. Surprisingly, a chimeric dimer consisting of one RNA binding mutant monomer and a wild type partner still abolished its ability to bind and edit its substrate, indicating that ADAR dimers require two subunits with functional dsRBDs for binding to a dsRNA substrate and then for editing activity to occur.
RNA editing mediated by adenosine deaminases acting on RNA (ADAR)2 involves adenosine-to-inosine (A → I) changes in double-stranded RNA (dsRNA) (1-4). A → I RNA editing can alter the protein coding sequence of several genes to create various isoforms, such as in the glutamate receptor ion channel subunits (5, 6), serotonin 2C subtype receptors (7), and Kv1.1 potassium channel (8). The association of malfunctioning A → I editing mechanisms and certain human disease, such as neurodegeneration in amyotrophic lateral sclerosis and depression in suicide victims has been implicated (9, 10). Bioinformatic approaches have revealed numerous A → I editing sites within non-coding sequences in introns and untranslated regions harboring repetitive elements such as Alu and LINE (11-15). Furthermore, recent evidence has revealed the intersection of ADAR with the RNA interference pathway, indicating a much broader role for A → I RNA editing (16-21). A → IRNA editing is mediated by ADAR (1-4). In vertebrates, three separate ADAR family members have been identified, and they are conserved in their C-terminal deaminase region as well as in their N-terminal double-stranded RNA binding domains (dsRBDs) (22-29). ADARs are also present in invertebrates, such as a single Drosophila member (dADAR) that is similar to the mammalian ADAR2 (30), as well as two less conserved Caenorhabditis elegans members (c.e.ADR1 and c.e.ADR2) (22, 31).
The common structural features shared by mammalian ADARs include dsRBDs repeated two or three times that are located in the N-terminal region and the C-terminal deaminase domain that provide the catalytic action (22, 23, 32). Evidence suggests that dsRBDs consisting of 65–70 amino acids provide general binding with little sequence selectivity, although the number and distance between dsRBDs may provide some specificity for its substrates and recognize distinct structural determinants within dsRNA (27, 33-35). Two reported dsRBD structures for Xenopus laevis RNA-binding protein A (Xlrbpa) and the Drosophila Staufen protein interacting with dsRNA reveal that the domain makes important contacts across the major groove of a dsRNA helix (36, 37). These dsRBD contacts span 16 base pairs of dsRNA and interact with the phosphate oxygen backbone and not specific nucleotides (36). A common structural theme of these domains appears to be the interaction of a cluster of lysines in a KKXXK motif (X is any amino acid), and mutagenesis of these residues reduces binding to dsRNA (36, 37). Likewise, ADARs contain this KKXXK motif in their dsRBDs, and deletions or site-directed mutagenesis also indicates that they are important for function (33, 34, 38-41). More recently, the NMR structures of the two dsRBDs of ADAR2 modeled with dsRNA verify the magnitude to which these lysines are significant (33).
Recent findings indicate that ADARs act as a dimer in mammals and flies (40, 42). In vitro, mammalian ADAR1 and ADAR2 form homodimers, which is required for A → I editing activity (42). It is proposed that this dimer interaction allows for the proper formation of active site alignment to deaminate the adenosine moiety (40, 42). Currently, it is not known whether the interplay between the monomers act cooperatively with respect to their dsRBDs. By use of fluorescence or bioluminescence resonance energy transfer, an examination of ADAR dimerization revealed that the N-terminal dsRBDs are providing much of the interface for the monomer subunits to interact in mammalian cells (43, 44).
Although several publications by our group and others on the mammalian ADARs point to dimerization that is independent of RNA binding (42, 43), other studies in mammalian cells and Drosophila indicate that dsRNA binding is required (40, 44). Furthermore, some studies on human ADAR2 suggest that the protein exists only as a monomeric form (32, 45). These discrepancies may result from a difference in species and techniques used. In summary, it remains to be established whether formation of the dimer complex is mediated by an amino acid interface made through protein-protein interactions of two monomers or via binding of the two subunits to a dsRNA substrate. In this present study, we have addressed the question by creating mutations for ADAR1 and ADAR2 within the dsRBDs that result in the total loss of all binding for long and short dsRNA. These dsRNA binding-deficient ADARs nevertheless dimerize identically to their wild type counterparts, revealing that ADAR dimerization is not mediated by dsRNA. Furthermore, our data indicate that two monomers with functional dsRBDs are required by a dimer for dsRNA binding and A → I editing activities.
EXPERIMENTAL PROCEDURES
Plasmid Construction
Wild type plasmids pBac-F-ADAR1, pBac-H-ADAR1, pBac-F-ADAR2, and pBac-H-ADAR2, corresponding to the human genes (42), were used as the starting basis for plasmid construction. The full-length ADAR1 (p150) and the ADAR2a splicing isoform (42) were used in this study. Mutations were created in each of the three dsRBDs of ADAR1 and in each of the two dsRBDs of ADAR2 separately by PCR and then combined through cloning (Fig. 1A). This introduced mutations in the lysines of the KKXXK motif to generate EAXXA at these amino acid locations (Fig. 1B). The mutations in ADAR1 that were introduced at dsRBD1 were K554E, K555A, and K558A; at dsRBD2, K665E, K666A, and K669A; and at dsRBD3, K777E, K778A, and K781A. Mutations created in the first dsRBD of ADAR2 included K127E, K128A, and K131A, and those in the second dsRBD consisted of K281E, K282A, and K285A. PCR mutagenesis using the QuikChange kit (Stratagene) and oligonucleotides creating specific nucleotide changes for silent DNA restriction sites and amino acid mutations are in bold, with the newly created silent endonuclease sites underlined. The mutagenic oligonucleotides for ADAR1 used in this study are A1-EAA1s (+BglI), 5′-GCTGAAGCTGGAAGCGAGGCCGTGGCGGCGCAGGATGCAGCTATGAAAGC-3′; A1-EAA2s (+ApaLI), 5′-CCAGTGTGAGTGCACCCAGCGAGGCAGTGGCAGCGCAGATGGCCGCAGAGG- 3′ and A1-EAA3s (+BglI), 5′-CGTCTGCGCACACAGCGAGGCCCAAGGGGCGCAGGAAGCAGCAGATGC- 3′. The ADAR2 mutant oligonucleotides are A2-EAA1s (+SphI), 5′-GGCTCTGGTCCCACAGAGGCAAAGGCAGC GGCTCTGGTCCCACAGAGGCAAAGGCAGCACTGCATGCTGCTGAGAAGG- 3′ and A2-EAA2s (+SacII), 5′-GGCTCGGGGAGAAACGAGGCGCTTGCCGCGGCCCGGGCTGCGC- 3′.
FIGURE 1. ADAR double-stranded RNA binding domains and the KKXXK motif.
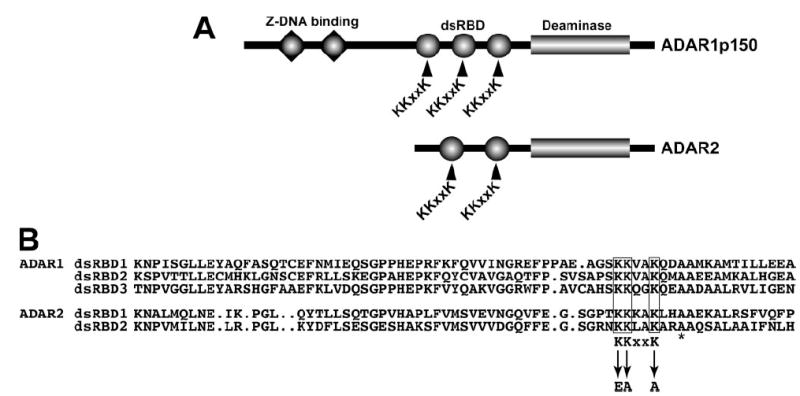
A, domain structure of ADAR1 and ADAR2 indicating Z-DNA binding domains (triangles), dsRBD (circles), and the C-terminal deaminase domain (rectangle). The KKXXK motif in each dsRBD is indicated. B, sequence alignment of the ADAR1 (top) and ADAR2 (bottom) dsRBD displaying the homology around the KKXXK motif and the mutated lysines to EAXXA (termed EAA). The asterisk denotes an alanine mutated in other studies.
ADAR Expression, Purification, and Detection
The resulting expression constructs pBac-F-ADAR1(EAA1,2,3), pBac-HADAR1(EAA1,2,3), pBac-F-ADAR2(EAA1,2), and pBac-HADAR2(EAA1,2) contain a FLAG or His6 tag, respectively. Briefly, Sf9 insect cells were individually infected or co-infected with two ADAR recombinant baculoviruses (42). The extracted proteins were either purified with an anti-FLAG M2 monoclonal antibody (mAb)-agarose gel (Sigma) column or, for the His6-containing proteins, with a TALON metal resin (BD Biosciences) column. Proteins expressed as a co-infection of FLAG and His6 epitope-tagged ADARs were sequentially purified with both affinity columns, beginning in either direction. If necessary, the final eluted proteins were concentrated using Centriplus spin columns (Millipore). The yield of eluted proteins was determined by Western blotting analysis. The primary monoclonal antibodies used in this study are anti-FLAG M2 (Sigma), anti-His6 (BD Biosciences), mAb15.8.6 (anti-ADAR1), and mAb1.3.1 (anti-ADAR2) (42).
Functional Assays
Filter binding assays were carried out in triplicate using the recombinant ADAR proteins and a synthetic long c-Myc dsRNA (583 base pairs) labeled with [α-32P]ATP (29). The amount of dsRNA substrate ranged from 0.1 to 6.4 nm. For the base modification assay of the A → I editing activity, the recombinant protein was tested also with the c-Myc dsRNA (38). The electrophoretic mobility shift assay was conducted using a 19-base-pair enhanced green fluorescent protein short interfering RNA as the short dsRNA substrate, as described previously (18).
Size Exclusion Column Chromatography
Purified protein (1 ug) was injected into a 500- μl loop and applied to a Superose 12 10/300 GL gel filtration column (GE Healthcare) (42). Fractions of 0.5 ml were collected at a flow rate of 0.35 ml/min using a fast protein liquid chromatography system. The molecular weight of ADAR proteins was ascertained by comparison to known molecular mass standards obtained from Sigma. The column was calibrated with blue dextran 2000 (void), thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), albumin (67 kDa), ovalbumin (43 kDa), and chymotrypsinogen A (25 kDa). The peak position of each protein standard was determined by optical absorption at 280 nm. The peak position for ADAR1 and ADAR2 was confirmed by Western blotting analysis of the fractions.
RESULTS
Mutating the KKXXK Motif Lysines in the dsRBDs Abolishes Binding to Long dsRNA
The co-structures of a dsRBD protein bound to dsRNA revealed the importance of several lysines in a KKXXK motif for making crucial contacts across the major groove of dsRNA (36, 37). Our goal was to generate point mutations for ADAR1 and ADAR2 in the KKXXK motif of each dsRBD to create a protein that would no longer bind dsRNA (Fig. 1A) and then investigate its dimerization aspects. For the Staufen protein, conversion of these KKXXK amino acid residues to EAXXA results in a dramatic loss of dsRNA binding in vivo (37). These mutations have been shown not to alter the protein folding as assayed by various structural methods (37). To this end, we created substitutions in ADAR1 and ADAR2 for each KKXXK motif converting it to EAXXA. The KKXXK mutation to EAXXA will be referred to as “EAA” throughout the text for the glutamate, alanine, and alanine substitution introduced (Fig. 1B).
Our new FLAG-tagged ADAR (EAA) mutants were purified, and Western blotting analysis confirmed production of the full-length proteins. ADAR1 (EAA) and ADAR2 (EAA) were examined for their ability to bind long dsRNA by using a 585-base-pair c-Myc dsRNA (29). As anticipated, these dsRBD mutant ADARs were deficient in dsRNA binding as compared with the wild type forms of the protein (Fig. 2A). This demonstrated the importance of the KKXXK motif in dsRNA binding for ADAR. As expected, our ADAR1 (EAA) and ADAR2 (EAA) mutants that abolished binding to dsRNA completely lacked RNA editing activity as tested on a long synthetic c-Myc dsRNA substrate (Fig. 3).
FIGURE 2. ADAR dsRBD mutation abolishes binding to long and short dsRNA.
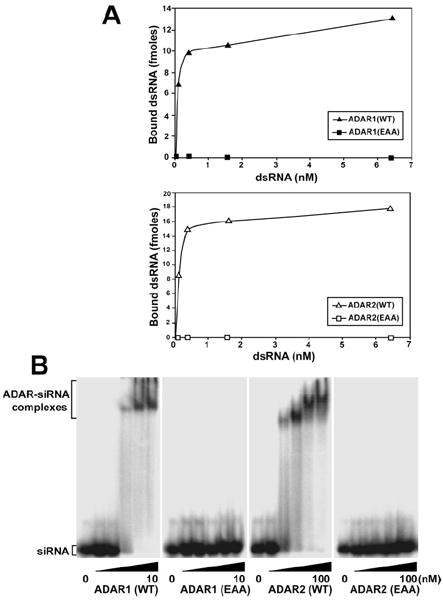
A, binding of recombinant proteins to c-Myc dsRNA (583 base pairs) was analyzed by a nitrocellulose filter binding assay. 10 ng of purified ADAR1 (WT and EAA) (top panel) and ADAR2 (WT and EAA) (bottom panel) protein were incubated at 30 °C for 5 min in triplicate with various concentrations of dsRNA substrate. B, binding of recombinant ADAR1 (WT and EAA) and ADAR2 (WT and EAA) proteins to short dsRNA was examined by electrophoretic mobility shift assay. Binding of ADAR1 proteins up to 50-fold over Kd values for wild type (10 nm) and ADAR2 proteins, up to 10-fold over Kd values for wild type (100 nm) to 10 pm 32P-labeled 19 base pairs enhanced green fluorescent protein short interfering RNA was analyzed on a native 4.5% polyacrylamide gel.
FIGURE 3. dsRBD mutant ADARs completely lack the A → I modification activity.
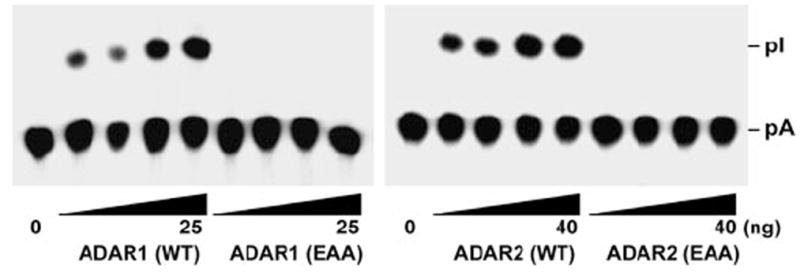
The A → I conversion of c-Myc dsRNA (20 fmol) was monitored with increasing amounts of purified recombinant ADAR1 (WT and EAA) (right panel, 3.13, 6.25, 12.5, and 25 ng) and ADAR2 (WT and EAA) (left panel, 5, 10, 20, and 40 ng) proteins. Following incubation for 1 h at 37 °C, the reaction products were deproteinized, digested with P1 nuclease, and analyzed by thin layer chromatography. pA, 5′-AMP; pI, 5′-IMP.
dsRBD Mutant ADARs Are Unable to Bind Short dsRNA
One outstanding question on ADAR dimerization was whether this association was truly RNA binding-independent. Previously, RNases were utilized to show that dimerization was RNA binding-independent in vitro (42) and subsequently in vivo (43). However, uncertainties still remained with the RNase treatment as to whether short dsRNA duplexes could have been protected by the ADAR protein and consequently lead to the two monomers becoming associated. We therefore needed to determine that the ADAR (EAA) mutants could not bind these types of substrate. Short dsRNA duplexes, such as short interfering RNAs, are bound with very high affinity by ADAR (18). Electrophoretic mobility shift analysis using a 19-base-pair short interfering RNA duplex shows that the wild type proteins are able to bind this ligand, whereas ADAR1 (EAA) and ADAR2 (EAA) mutants are incapable of binding (Fig. 2B). These results rule out the possibility of short dsRNA duplexes bringing the two subunits together for dimer formation.
Dimerization of dsRNA Binding-defective ADARs
Having demonstrated the incapability of ADAR (EAA) mutants for the binding to long and short dsRNA, we investigated their dimerization by sequential co-purification of differentially epitope-tagged ADAR proteins as employed previously (42). Briefly, His-tagged versions of ADAR1 (EAA) and ADAR2 (EAA) were produced to differentiate it from the FLAG-tagged partner. Co-infection of two separate baculoviruses in insect cells resulted in co-expression of FLAG (denoted as F)- and His (denoted as H)-tagged subunits of ADAR1 or ADAR2. First, the ADAR recombinant proteins were purified by affinity chromatography on the M2 anti-FLAG mAb column and then on the TALON metal resin column for the His6 tag purification. Each chromatography step was monitored by Western blot analysis using the anti-FLAG- or anti-His6-specific antibody, which indicated that an F/H dimeric complex was retrieved from the second column (Fig. 4, lanes 9–12). Both ADAR1 (EAA) and ADAR2 (EAA) were purified as oligomeric complexes containing both FLAG- and His-tagged protein (Fig. 4, lanes 4 and 10 for ADAR1 and lanes 6 and 12 for ADAR2). The reversed purification sequence was also performed to confirm the results of our co-purification scheme (data not shown). These dsRBD mutant ADARs that lack dsRNA binding but are capable of homodimerization just like their wild type counterparts suggest that ADAR dimerization is independent of its binding to dsRNA.
FIGURE 4. Homodimerization of ADAR is independent of dsRNA binding.

The ADAR1 (WT and EAA) and ADAR2 (WT and EAA) proteinswere co-expressed in Sf9 cells with its differentially tagged partner. These recombinant proteins were sequentially purified on an anti-FLAGmAb affinity column (left panel) and then on a second TALON affinity column (right panel). Western blotting analysis specific for the first purification using the anti-FLAG M2 mAb indicates the F-tagged protein eluted (left panel) and the anti-His6 mAb reveals the H-tagged protein that is retrieved (right panel), confirming the interaction. Single purified recombinant F-ADAR2 and H-ADAR2 expressed with one tag was included to show the specificity of the two mAbs used forWestern blotting analysis (lanes 1, 2, 7, and 8). These F/H-tagged ADAR1 and ADAR2 oligomeric complexes were alsoidentified with the reciprocal mAb toconfirm the presence of the other monomer subunit partner (not shown). Approximately 10 ng of each purified protein was loaded onto the SDS-polyacrylamide gels.
In our dimerization model, we hypothesized that mostly all ADAR protein is in an oligomeric form. This would make the major three combinations of monomer subunit for dimer formation to be F/F, F/H, and H/H in a relative ratio of 1:2:1 but would not necessarily exclude the possibility that a small fraction of monomeric protein exists. We quantitated each step of the affinity column chromatography by Western blot and calibration with known ADAR amounts that were used as loading controls. From the material that bound on the first affinity column (FLAG M2 Ab), 80–90% of the FLAG-tagged subunit was eluted from this resin, whereas a substantial amount of the His-tagged subunit was washed away; however, 30–45% still remained (Table 1, see column titled “FLAG column elution”). Upon collecting, pooling, and loading the complete FLAG eluate (containing F/F- and F/H-tagged dimers) onto the TALON column for His6 purification, we then quantitated again each tagged subunit. The His-tagged protein completely bound and eluted from the TALON resin, whereas the FLAG protein was partially washed away (Table 1, see column titled “His column elution”). Presumably, the protein that was displaced off of the second column represents F/F homodimers of ADAR1 and ADAR2, respectively. We anticipated, according to our expected statistical distribution on dimerization, that a significant amount of 2× purified ADAR proteins should be retrieved, and indeed 30–45% of the subunits were recovered (Table 1, see column titled “Total protein recovered”). The level of recovery further establishes that the oligomer represents the major form of the complex and illustrates that this technique is not selectively enriching a rare form of the complex. More importantly, this demonstrates that the ADAR dsRBD mutants behave identically to their wild type counterparts and that dimerization is not a function of binding dsRNA. Furthermore, these co-purification experiments also illustrate that the introduced EAA substitutions are not disrupting ADAR protein structure as indicated by its protein-protein interactions, identical to the wild type, and are therefore intact.
TABLE 1. Percentage of protein yield from sequential co-purification.
Percentages are calculated from quantitative Western blot analysis using anti-FLAG and anti-His6 mAbs
| FLAG column elution | His column elution | Total protein recovered | |
|---|---|---|---|
| % | % | % | |
| F-ADAR1 (WT) | 87 | 47 | 41 |
| H-ADAR1 (WT) | 30 | 100 | 30 |
| F-ADAR1 (EAA) | 81 | 52 | 42 |
| H-ADAR1 (EAA) | 32 | 100 | 32 |
| F-ADAR2 (WT) | 89 | 41 | 36 |
| H-ADAR2 (WT) | 45 | 100 | 45 |
| F-ADAR2 (EAA) | 90 | 33 | 30 |
| H-ADAR2 (EAA) | 40 | 100 | 40 |
Analysis of ADAR Mutant Oligomeric Complexes by Size Fractionation Chromatography
To further confirm dimerization of our ADAR (EAA) mutants, we subjected the proteins through size exclusion chromatography on a Superose 12 gel filtration column. Previously, this method confirmed that the ADAR1 and ADAR2 proteins eluted as homodimers with an apparent molecular mass of ~300 kDa for ADAR1 (monomer is 150 kDa) and~180 kDa for ADAR2 (monomer is 90 kDa) (42). Based on standard size markers, the oligomeric form of ADAR1 (EAA) fractionated to an estimated peak of ~300 kDa (Fig. 5A), and the ADAR2 (EAA) peak position was ~180 kDa (Fig. 5B). The EAA mutant forms of ADAR1 and ADAR2 migrated at similar fractions to wild type, displaying comparable elution profiles indicative of RNA-independent dimerization. Interestingly, no distinctive free monomer peak was detected for all ADAR proteins tested. Our results suggest that these proteins form a stable dimer and that the subunits are not bridged by RNA binding. For the elution pattern of ADAR1 (WT), we did observe a large molecular mass aggregate (Fig. 5A, Fraction 18) that was sometimes seen in our previous studies (42) and could have been due to some large ADAR1 oligomeric complex bound to some unknown dsRNAs derived from host Sf9 cells. In addition, we tested a full-length ADAR2 that was purified from a yeast-expressing recombinant system that was previously used for domain analysis and crystallography of a C-terminal deaminase domain fragment (32, 45). This yeast-derived human ADAR2 was reported to be monomeric, as determined by analytical ultracentrifugation (32, 45). In our hands, the yeast-derived ADAR2 migrated at the same dimeric position along with our baculovirus Sf9 insect cell-purified human ADAR2 (Fig. 5B).
FIGURE 5. Analysis of dsRNA binding mutant ADAR oligomeric complexes by size exclusion chromatography.
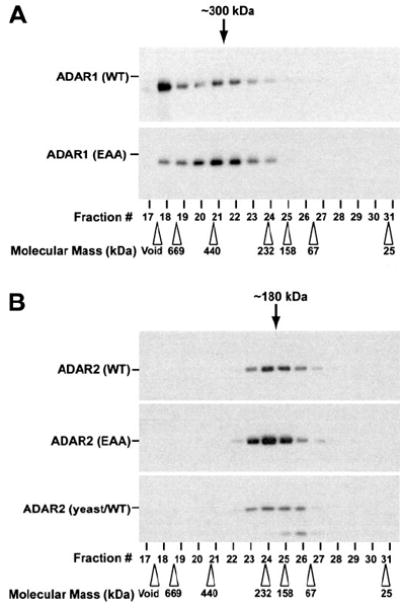
Recombinant purified ADAR1 (WT and EAA) (A) and ADAR2 (WT and EAA) (B) proteins as well as a yeast-derived wild type ADAR2-purified protein (B, lower panel) were fractionated by Superose 12 gel filtration column chromatography and analyzed by Western blotting using specific mAb for ADAR1 (A) or ADAR2 (B). The positions of molecular 32 size marker protein used as the calibration standards are indicated by open arrowheads. The estimated dimer size for fractions of ADAR1 (~300 kDa) and ADAR2 (~180 kDa) are indicated by black arrows. Running the samples on a denaturing SDS-polyacrylamide gel allowed confirmation of the monomeric size for ADAR1 (150 kDa) and ADAR2 (90 kDa).
A dsRBD Mutant Monomer Inhibits the Activities of Its Wild Type Partner
To obtain more insights into the possible functional interaction between the two monomers, we next examined whether the EAA mutant could act in a dominant negative fashion over wild type. A chimeric dimer of F/H-ADAR2 (EAA/WT) was produced and tested side by side with the completely wild type version F/H-ADAR2 (WT/WT). An additional chimeric dimer F/H-ADAR2 (E396A/WT) was also tested. The glutamate residue substituted to alanine in F/H-ADAR2 (E396A/WT) is essential for formation of the catalytic center of ADAR2 (32). Furthermore, chimeras of F/H-ADAR2 (E396A/WT) exhibit half the activity of a full wild type (WT/WT) dimer (42). The equivalent glutamate to alanine mutation in ADAR1 was shown to abolish editing activity, while retaining dsRNA binding affinity identical to that of wild type (22, 38, 42). We compared the editing activity of the chimeras and found that the editing activity was totally inhibited in the presence of the dsRBD mutant monomer for the F/H-ADAR2 (EAA/WT) protein (Fig. 6A). Similar results were obtained with the F/H-ADAR1 (EAA/WT) chimeric dimer (data not shown). Interestingly, the preformed mutant homodimer of ADAR2 (EAA/EAA) had no inhibitory effects on the editing activity of the wild type homodimer ADAR2 (WT/WT) up to an 8-fold ratio examined (data not shown). As expected, the F/H-ADAR2 (E396A/WT) displayed 50% editing activity as compared with the wild type (Fig. 6A). This suggested that the EAA dsRNA mutant monomer has a dominant effect over the activity of the wild type monomer provided that the two monomers are complexed together as a single dimeric molecule. To determine whether this was an effect based on the dsRNA binding properties of the F/H-ADAR2 (EAA/WT) chimera, we performed dsRNA binding assays. Interestingly, the F/H-ADAR2 (EAA/WT) chimera completely lacked all dsRNA binding, whereas the F/H-ADAR2 (E396A/WT) chimera displayed normal levels of binding (Fig. 6B). The F/H-ADAR1 (EAA/WT) chimeric dimer also lacked all dsRNA binding (data not shown). The total loss of dsRNA binding and editing activities observed with the (EAA/WT) chimeric ADAR dimers indicated that intact dsRBDs from each monomer subunit were required to recognize the dsRNA substrate and for editing activity to occur.
FIGURE 6. Dominant negative effects of one dsRBD mutant monomer paired with a wild type partner.

A, the A → I base modification assay for F/H-ADAR2 (WT/WT), F/H-ADAR2 (EAA/WT), and F/H-ADAR2 (E396A/WT) was carried out with 10 ng of protein for 30 min at 37 °C with 20 fmol of c-Myc dsRNA. B, dsRNA binding assays similar to Fig. 2 were performed with 10 ng of F/H-ADAR2 (WT/WT), F/H-ADAR2 (EAA/WT), F/H-ADAR2 (E396A/WT), and 6.4 nm c-Myc dsRNA. The experiments were normalized to F/H-ADAR2 (WT/WT), and all experiments were done in triplicate.
DISCUSSION
Some conflicting data have been reported previously regarding ADAR RNA-independent dimerization and its RNA binding function, because these two processes almost overlap in the N-terminal dsRBDs (40, 43, 44). These differing conclusions obtained might be due to the dsRBD mutation employed. A mutant substituted for a highly conserved alanine to glutamate (A/E) three amino acids downstream of the KKXXK motif (see Fig. 1B, asterisk) used in other investigations (40, 44) may not have been the best choice for analysis. Structural studies indicate that this conserved alanine is in a α-helix and required for proper folding of the dsRBD as a hydrophobic residue, whereas the lysines are on a surface-exposed loop that interacts across the major groove of a dsRNA molecule (36, 37). Thus, this mutation is likely to disrupt the dsRBD structure (36, 37). The A/E mutation may appear to lose RNA binding together with dimerization, causing one to conclude that RNA-dependent dimerization resulted, but this may just be a result of destabilization of the entire domain structure (40, 44).
By utilizing the A/E mutation, it has been shown by bioluminescence resonance energy transfer analysis of mammalian ADAR2 that dimerization is dependent on RNA binding (44). Furthermore, a two-hybrid study performed on rat ADAR2 demonstrated that the protein could not dimerize by utilizing the same A/E mutation, similar to the dADAR studies performed in the past (40, 44). In contrast, the recent fluorescence resonance energy transfer analysis indicated RNA-independent dimerization of mammalian ADAR2 in vivo by use of RNases (43). This group re-evaluated their earlier model that dimerization was dependent on RNA binding, which was based on cross-linking analysis (46). The authors speculate that, in light of their newer data, these cross-links may represent an RNA-dependent rearrangement upon binding RNA and not indicative of dimerization itself (43).
In this study, we have addressed the question of whether dimerization of ADAR1 and ADAR2 is mediated by protein-protein interactions or through their binding to dsRNA. Using new ADAR1 and ADAR2 mutants defective in their dsRBDs, we have shown that the proteins could no longer bind dsRNA but were still able to dimerize independent of its substrate. For this analysis, we chose to make mutations in a KKXXK motif that was located within the dsRBD (33, 36, 37). Initially, we created mutations in the first lysine of each KKXXK motif, but this K/A substitution still retained the ability to bind and edit dsRNA (data not shown). We then chose to make a more severe mutation in the dsRBDs by substituting the KKXXK motif lysines with EAA.
The EAA mutations created in ADAR1 and ADAR2 completely abolished their binding to both a long entirely complementary dsRNA as well as a short dsRNA duplex. As expected, ADAR1 (EAA) and ADAR2 (EAA) did not have any enzymatic activity, confirming that the presence of functional dsRBDs is essential for A → I editing activity. By sequential purification of differentially tagged monomers and size exclusion chromatography, we have shown that dsRBD mutant ADAR1 and ADAR2 form homodimers which are independent of binding dsRNA.
We also tested a recombinant yeast-derived full-length human ADAR2 similar to which the C-terminal crystal structure was solved and was shown to be monomeric in previous studies (32, 45). There may be some differences in a yeast expression system that does not have endogenous ADAR proteins, as opposed to a metazoan system that normally encodes for ADAR, perhaps for post-translational modifications. However, the full-length yeast-derived protein did fractionate at the dimeric size position by size fractionation chromatography alongside our insect-purified ADAR2, with no presence of a monomeric peak. We have no explanation as to why this full-length human ADAR2 protein has displayed monomeric properties in previous studies, but it may be due to the different buffer and/or more manipulation steps carried out to isolate the protein (45).
Previous studies have shown that having one monomer defective for the deaminase domain (E396A) disrupts the dimer function only by half (42). Taken together, these data indicate that a deaminase mutant chimeric dimer (E396A/WT) is able to bind dsRNA but that only one functional active site is formed and would therefore have partial activity. In contrast, A → I editing activity of the dsRNA binding mutant chimeric dimer (EAA/WT) is completely lost. This is because of the defective dsRBDs of one monomer and suggests that cooperative interactions of functional dsRBDs of both ADAR dimer subunits are required for dsRNA binding. Having one monomer in the dimer complex that is unable to bind the dsRNA excludes it from binding its substrate.
ADAR1 homozygous null mutations are embryonically lethal in mice, whereas heterozygotes are viable (47, 48). Dyschromatosis symmetrica hereditaria is a benign heterozygous human disease of a defective ADAR1 allele that causes aberrant skin pigmentation (9). Interestingly, more mutations are identified disproportionately in the deaminase domain than in the dsRBDs of ADAR1. It may perhaps be that mutations identified in the deaminase domain are less severe, because the chimeric dimers expected to form would still retain some editing activity. The likely distribution of monomer subunits in a dimer is 1:2:1 for (WT/WT), (Mut/WT), and (Mut/Mut), suggesting that a heterozygous deaminase mutation would not have as strong an effect due to its ability to maintain partial activity. In contrast, mutations are rarely found in the dsRBDs or KKXXK motif, because these alterations would have a more dominant effect when paired with a wild type partner, thus greatly reducing ADAR function. In this assumption, the reduced activity for ADAR could be as low as ¼ with only (WT/WT) dimers having editing activity, and this may be below a threshold for survival and possibly selected out naturally during development (47, 48). ADAR dimerization can be a potential source of modulation for RNA editing activity, and these ADAR (EAA) mutants may prove interesting for future studies in vivo.
Acknowledgments
We thank the Wistar Protein Expression Facility for recombinant protein productions. We also thank Drs. M. R. Macbeth and B. L. Bass for the generous gift of yeast-derived hADAR2 protein.
Supported by National Institutes of Health Postdoctoral Supplemental Award Grant R01 HL070045 and NCI National Institutes of Health Postdoctoral Training Grant T32 CA09171.
Footnotes
This work was partially supported by funds from the National Institutes of Health, the Juvenile Diabetes Research Foundation, and the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health (to K. N.).
The abbreviations used are: ADAR, adenosine deaminases acting on RNA; A → I, adenosine-to-inosine; dsRNA, double-stranded RNA; dsRBD, dsRNA binding domain; mAb, monoclonal antibody; WT, wild type.
References
- 1.Bass BL. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeCerbo J, Carmichael GG. Curr Opin Cell Biol. 2005;17:302–308. doi: 10.1016/j.ceb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Reenan RA. Trends Genet. 2001;17:53–56. doi: 10.1016/s0168-9525(00)02169-7. [DOI] [PubMed] [Google Scholar]
- 4.Toth AM, Zhang P, Das S, George CX, Samuel CE. Prog Nucleic Acid Res Mol Biol. 2006;81:369–434. doi: 10.1016/S0079-6603(06)81010-X. [DOI] [PubMed] [Google Scholar]
- 5.Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- 6.Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger JR, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH. Science. 1994;266:1709–1713. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- 7.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 8.Hoopengardner B, Bhalla T, Staber C, Reenan R. Science. 2003;301:832–836. doi: 10.1126/science.1086763. [DOI] [PubMed] [Google Scholar]
- 9.Maas S, Kawahara Y, Tamburro KM, Nishikura K. RNA Biol. 2006;3:1–9. doi: 10.4161/rna.3.1.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmauss C. Neuroscientist. 2003;9:237–242. doi: 10.1177/1073858403253669. [DOI] [PubMed] [Google Scholar]
- 11.Morse DP, Bass BL. Proc Natl Acad Sci U S A. 1999;96:6048–6053. doi: 10.1073/pnas.96.11.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, Olshansky M, Rechavi G, Jantsch MF. Nat Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 13.Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. Genome Res. 2004;14:1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Athanasiadis A, Rich A, Maas S. PLoS Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blow M, Futreal PA, Wooster R, Stratton MR. Genome Res. 2004;14:2379–2387. doi: 10.1101/gr.2951204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikura K. Nat Rev Mol Cell Biol. 2006;7:919–931. doi: 10.1038/nrm2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luciano DJ, Mirsky H, Vendetti NJ, Maas S. RNA. 2004;10:1174–1177. doi: 10.1261/rna.7350304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang W, Wang Q, Howell KL, Lee JT, Cho DS, Murray JM, Nishikura K. J Biol Chem. 2005;280:3946–3953. doi: 10.1074/jbc.M407876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blow MJ, Grocock RJ, van Dongen S, Enright AJ, Dicks E, Futreal PA, Wooster R, Stratton MR. Genome Biol. 2006;7:R27. doi: 10.1186/gb-2006-7-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Proc Natl Acad Sci U S A. 1994;91:11457–11461. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Connell MA, Krause S, Higuchi M, Hsuan JJ, Totty NF, Jenny A, Keller W. Mol Cell Biol. 1995;15:1389–1397. doi: 10.1128/mcb.15.3.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patterson JB, Samuel CE. Mol Cell Biol. 1995;15:5376–5388. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. Nature. 1996;379:460–464. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 26.Gerber A, O’Connell MA, Keller W. RNA. 1997;3:453–463. [PMC free article] [PubMed] [Google Scholar]
- 27.Lai F, Chen CX, Carter KC, Nishikura K. Mol Cell Biol. 1997;17:2413–2424. doi: 10.1128/mcb.17.5.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. J Biol Chem. 1996;271:31795–31798. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- 29.Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. RNA. 2000;6:755–767. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palladino MJ, Keegan LP, O’Connell MA, Reenan RA. Cell. 2000;102:437–449. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- 31.Hough RF, Lingam AT, Bass BL. Nucleic Acids Res. 1999;27:3424–3432. doi: 10.1093/nar/27.17.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stefl R, Xu M, Skrisovska L, Emeson RB, Allain FH. Structure. 2006;14:345–355. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 34.Xu M, Wells KS, Emeson RB. Mol Biol Cell. 2006;17:3211–3220. doi: 10.1091/mbc.E06-02-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hallegger M, Taschner A, Jantsch MF. RNA. 2006;12:1993–2004. doi: 10.1261/rna.125506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryter JM, Schultz SC. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramos A, Grunert S, Adams J, Micklem DR, Proctor MR, Freund S, Bycroft M, St. Johnston D, Varani G. EMBO J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai F, Drakas R, Nishikura K. J Biol Chem. 1995;270:17098–17105. doi: 10.1074/jbc.270.29.17098. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Samuel CE. J Virol. 1996;70:1961–1968. doi: 10.1128/jvi.70.3.1961-1968.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallo A, Keegan LP, Ring GM, O’Connell MA. EMBO J. 2003;22:3421–3430. doi: 10.1093/emboj/cdg327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sansam CL, Wells KS, Emeson RB. Proc Natl Acad Sci U S A. 2003;100:14018–14023. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho DS, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. J Biol Chem. 2003;278:17093–17102. doi: 10.1074/jbc.M213127200. [DOI] [PubMed] [Google Scholar]
- 43.Chilibeck KA, Wu T, Liang C, Schellenberg MJ, Gesner EM, Lynch JM, MacMillan AM. J Biol Chem. 2006;281:16530–16535. doi: 10.1074/jbc.M511831200. [DOI] [PubMed] [Google Scholar]
- 44.Poulsen H, Jorgensen R, Heding A, Nielsen FC, Bonven B, Egebjerg J. RNA. 2006;12:1350–1360. doi: 10.1261/rna.2314406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Macbeth MR, Lingam AT, Bass BL. RNA. 2004;10:1563–1571. doi: 10.1261/rna.7920904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaikaran DC, Collins CH, MacMillan AM. J Biol Chem. 2002;277:37624–37629. doi: 10.1074/jbc.M204126200. [DOI] [PubMed] [Google Scholar]
- 47.Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. J Biol Chem. 2004;279:4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- 48.Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. J Biol Chem. 2004;279:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]