

Abstract
Pyrazoles are important azole heteroarenes frequently found in pharmaceuticals and protein ligands and there has been a growing interest in new synthetic methods for their preparation. We report the first catalytic intermolecular C-H arylation of pyrazoles, namely SEM-protected pyrazoles and N-alkylpyrazoles, which lays the foundation for a new approach to the synthesis of complex arylated pyrazoles, where new arene rings are directly attached to predetermined positions of the heteroarene nucleus. Through a systematic search we identified a palladium-pivalate catalytic system as the most effective protocol and mapped the reactivity of all three C-H bonds of the pyrazole (C-5 > C-4 ≫ C-3). To circumvent the low reactivity of C-3 position, we developed a “SEM switch”, which transposes the SEM-protecting group from one nitrogen to the other in one step, and in the process transforms the unreactive C-3 position to the reactive C-5 position. The SEM switch thus enables sequential arylation of C-5 and C-3 position, providing rapid access to protected or free 3,4,5-triarylpyrazoles (C-4 arene ring is readily introduced by bromination and Suzuki coupling). Furthermore, N-alkylation of SEM-protected pyrazoles allows for regioselective introduction of the amine substituent, addressing the low regioselectivity of N-alkylation of pyrazoles lacking sufficient steric bias. Thus, the catalytic C-H arylation combined with the protecting group transposition and N-alkylation provides a rapid route to fully substituted pyrazoles with complete regiocontrol of all substituents. The particular strength of this strategy is the ability to commence the synthesis from either the parent pyrazole or practically any pyrazole intermediate.
Introduction
Azole heteroarenes are structural units frequently found in biologically active compounds, including natural products, protein ligands, and pharmaceuticals.1 These structures represent indispensable components of the modern medicinal chemistry repertoire.2 Pyrazoles, while rarely found in nature, serve as important core structures of many pharmaceuticals and pharmaceutical leads with a wide range of biological activities (e.g., cholesterol lowering,3 anti-inflammatory,4 anti-cancer,5 anti-depressant and anti-psychotic agents6). As a result, there is a continuing interest in the development of versatile methods to access highly substituted pyrazoles. Traditionally, pyrazoles have been synthesized via de novo condensation reactions of 1,3-dicarbonyls and hydrazines or by cycloadditions of diazoalkanes and alkynes.7 Although recent efforts greatly expanded the generality of de novo approaches, each method has its scope and efficiency limitations.8
We propose a conceptually different approach to the assembly of complex arylated pyrazoles based on direct C-H bond arylation (Figure 1). Following this logic on a strategic level, new aromatic rings would be directly attached to the existing pyrazole core at desirable positions (“topologically obvious synthesis”),9 which suggests a rapid entry into functionalized heterocycles and an approach enabling efficient synthesis of series of pyrazole analogs and regioisomers.
Figure 1.
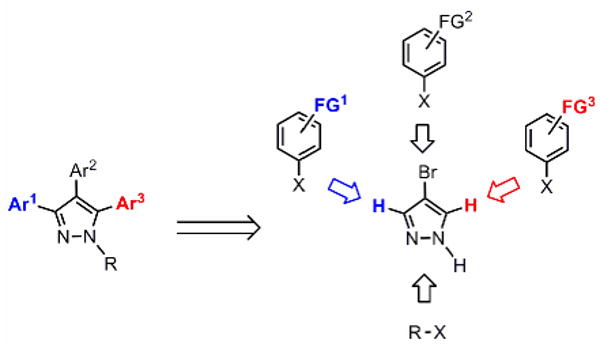
A new approach to synthesis of complex pyrazoles via direct C-H arylation
In order to realize this goal, we developed the first catalytic method for intermolecular C-arylation of pyrazoles and explored its regioselectivity trends. On the basis of these results, a general approach was devised for the synthesis of mono-, di- and tri-arylpyrazoles (Scheme 1). The first aryl group can be installed regioselectively in the 4-position of pyrazole by bromination and Suzuki coupling; other substituents can also be introduced at this position due to its high nucleophilicity. The first direct C-H arylation takes place with good selectivity at the 5-position, providing a concise approach to unsymmetrical diarylpyrazoles. The subsequent arylation is however inefficient, owing to the low reactivity of the 3-position. To address this problem, we have developed a simple one-step protocol to shift the SEM-group from one nitrogen to the other, which transforms the unreactive position 3 to the reactive position 5, and enables the second C-H arylation to proceed in an efficient manner, affording a short route to protected and free triarylpyrazoles (Scheme 1).10 Furthermore, regioselective introduction of a nitrogen substituent becomes possible by the N-alkylation of unsymmetrical SEM-pyrazoles (Scheme 1). Thus, this strategy allows for rapid assembly of fully substituted pyrazoles with complete regio-control of all C- and N-substituents. Complex pyrazoles with different number and position of aryl rings can be readily synthesized from common precursors by choosing the desirable haloarene donor and the order of the arylation and alkylation reactions.
Scheme 1.
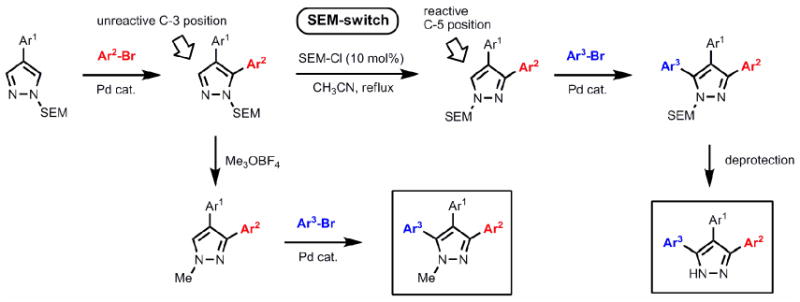
Sequential C-arylation enabled by SEM group switch provides a rapid access to triarylpyrazoles with complete control of regioselectivity. The SEM group also allows for regioselective N-alkylation.
Results
Development of C-H Arylation Protocol for SEM-protected Pyrazoles
We have previously reported catalytic methods for direct C-arylation of indoles, pyrroles, and imidazoles using palladium11 and rhodium12 catalysts and carboxylate bases. However, these protocols were inefficient for arylation of pyrazoles, calling for the development of new conditions.13,14,15 We rationalized the low reactivity of pyrazoles in terms of relatively high Lewis basicity of these compounds and the ability to deactivate the catalyst. We chose to examine SEM-protected pyrazoles as the substrates [SEM = 2-(trimethylsilyl)ethoxymethyl] due to the stability of this protecting group under the catalytic arylation conditions11c,16 as well as the ability of SEM group to be transposed from one nitrogen to another and enabling sequential arylations as outlined above.
Systematic examination of the reaction parameters (metal catalyst, ligand, base, and solvent) for the coupling of SEM-pyrazole and bromobenzene led to a robust method which uses the following conditions: 5 mol % Pd(OAc)2, 7.5 mol % P(n-Bu)Ad2, and 3 equivalents KOPiv, in DMA as the solvent and heating at 140 °C (Figure 2). Once again, an unbiased search for catalytic C-H arylation conditions identified a carboxylate salt as the best base, consistent with the previous results in our group that demonstrated the importance of carboxylate base in the metallation step of heteroarenes.11,12 Furthermore, these conditions are very similar to those developed independently by Fagnou and colleagues for arylation of benzene aromatics.17 Following Fagnou's lead, potassium pivalate can be replaced with potassium carbonate and a sub-stoichiometric amount of pivalic acid, resulting in a more practical procedure; both versions of this protocol (PivOK or PivOH/K2CO3) afford comparable yields of arylated SEM-pyrazoles.
Figure 2.
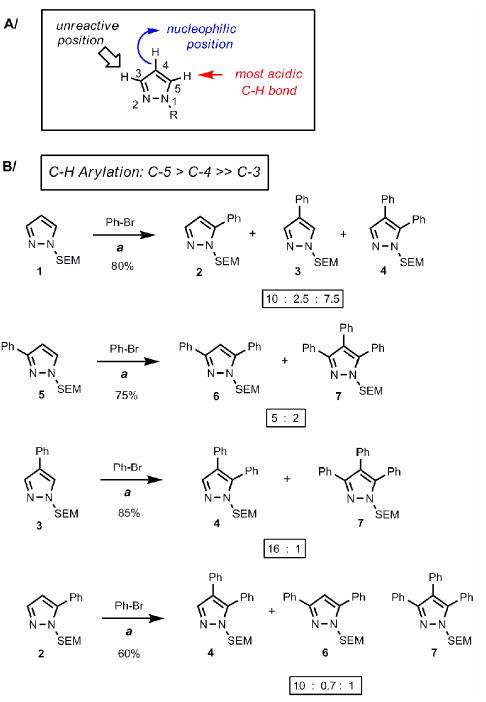
A/ General reactivity properties of pyrazole. B/ Reactivity profile of pyrazoles toward palladium-catalyzed C-H arylation. The C-5 position exhibits the highest reactivity. (a) Reaction conditions: Pyrazole, PhBr (1.5 equiv), Pd(OAc)2 (5 mol %), P(n-Bu)Ad2 (7.5 mol %), K2CO3 (3 equiv), HOPiv (25 mol %), 2.5 M DMA, 140 °C for 12 hours. Isolated yields are shown except for substrate 1, where the substrate conversion and the product ratio was determined by 1H NMR of the crude mixture. All product ratios were confirmed by 1H NMR of crude mixtures.
Regioselectivity of C-Arylation of SEM-Protected Pyrazoles
The next key question centered on establishing the regioselectivity of the catalytic C-H arylation method (Figure 2). The parent SEM-pyrazole 1 when submitted to the catalytic conditions gave a mixture of products, which indicated the higher reactivity of the 5-position relative to the 4-position, and very low reactivity of the 3-position (Figure 2B). In addition to the monoarylated products, the bis-arylation product 4 was also formed in a significant amount. This trend was confirmed by examining the reaction of 1-SEM-3-phenylpyrazole 5, which gave a 5:2 ratio of products 6 and 7. In contrast, compound 3 was arylated at the 5-position with high selectivity to provide compound 4 in 80% yield; the regioisomer stemming from arylation at the 3-position was not detected, and only a small amount of the bis-arylation product 7 was formed (16:1 ratio, 4:7). Arylation of compound 2 was also selective, taking place at the 4-position to afford product 4 as the major product; however, the yield was lower compared to arylation of the 5-position. These results demonstrate that the arylation is inefficient at the 3-position, which is consistent with the low reactivity of this site (to both electrophiles and strong bases), while arylation can be achieved at both C-4 and C-5 positions with preference for the latter.
These reactivity trends laid the foundation for the general synthetic strategy shown in Scheme 1. The information provided by these reactions is sufficient for planning regioselective elaboration of either the parent pyrazole or substituted pyrazoles.
Rationale for the Observed Regioselectivity
Although this paper focuses on the method and strategy development, we provide some context for the potential rationale for the observed selectivity preferences. It is well established that the 4-postion of pyrazole is most nucleophilic and readily undergoes electrophilic substitution while the 5-position carries the most acidic C-H bond which can selectively be deprotonated by strong bases (e.g. lithiation).18 The previous results suggest that both the palladium-acetate11b and the rhodium-pivalate12 catalytic systems developed in our laboratory, as well as related systems developed by others, proceed via an electrophilic-like mechanism,19 in the context of indoles, pyrroles and imidazoles, where the metal acts as an electrophile, breaking the aromaticity, and the carboxylate ligand as the base, removing the proton. On the other hand, Fagnou's laboratory generated compelling evidence which shows that the palladium-pivalate system, in the context of benzene arenes, selectively targets acidic C-H bonds via a “σ-deprotonation mechanism” where the metal-carboxylate moiety directly engages the C-H bond.17,20 The C-5 selectivity may thus be explained by the preference of the catalytic system for the acidic C-H bonds; however, an alternative rationale akin to the one proposed for C-2 arylations of indoles involving an electrophilic-like mechanism with the migration of palladium to the position adjacent to the nitrogen, is also reasonable.11b Importantly, the ability to accommodate both electron-deficient benzenes and electron-rich heteroarenes suggests substantial plasticity of the palladium-pivalate catalytic system, which may also account for the ability to arylate both C-5 and C-4 positions of the pyrazole system. The phosphine ligand modulates the reactivity of this catalytic system; in the context of pyrazoles, strong σ-donor phosphines also protect the catalyst against the inhibition by the basic sp2 nitrogen of the substrate. Detailed mechanistic studies to answer these mechanistic questions are under way in our laboratories and will be reported in the future.
Arene Donor Substrate Scope
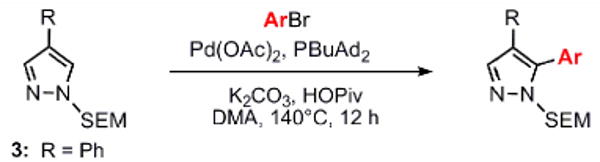
Compound 3 was used to examine the reaction scope of the arylation method; it was synthesized in short order by bromination of free (NH)-pyrazole at the 4-position,21 N-alkylation with SEM-Cl and the Suzuki reaction with phenylboronic acid (see Supporting Information). The arylation reactions may be performed on the benchtop under argon, and the scope of the reaction is quite broad, tolerant of a wide variety of functional groups on the bromoarene donor, including ketone, ester, nitro, dimethylamino, and pyridyl groups (Table 1). Electron-deficient and electron-neutral bromoarenes perform best in the reaction, whereas bromoarenes with electron-donating substituents in the para-position or steric bulk in the ortho-position give lower yields of desired products.
Table 1.
Arylation Substrate Scope
 | ||||
|---|---|---|---|---|
| Entry | R | ArBr | Product | Isolated Yield, % |
| 1 | Ph | 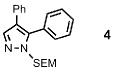 |
80% (5% bis) | |
| 2 | Ph | 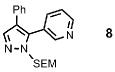 |
65% (15% bis) | |
| 3 | Ph |  |
 |
82% |
| 4 | Ph | 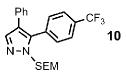 |
74% (13% bis) | |
| 5 | Ph | 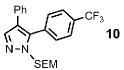 |
58% | |
| 6 | Ph | 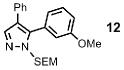 |
76% | |
| 7 | Ph | 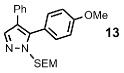 |
35% | |
| 8 | Ph | 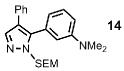 |
74% | |
| 9 | Ph | 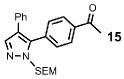 |
58% | |
| 10 | o-tol | 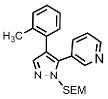 |
79% | |
| 11 | 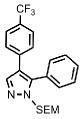 |
69% (8% bis) | ||
| 12 | COOEt | 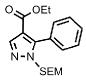 |
75% | |
Reaction conditions: Pyrazole, ArBr (1.5 equiv), Pd(OAc)2 (5 mol %), P(n-Bu)Ad2 (7.5 mol %), K2CO3 (3 equiv), HOPiv (25 mol %), 2.5 M DMA, 140 °C for 12 hours. Isolated yields are an average of at least two separate reactions.
Pyrazole Substrate Scope
Substitution on the phenyl ring in the 4-position of the pyrazole is also tolerated as illustrated by the ortho-tolyl- and para-trifluoromethylphenyl-substrates, which give the corresponding products 16 and 17, respectively, in good yields (Table 1, entry 10 and 11). Furthermore, the method is compatible with substituents other than aryl groups as demonstrated by efficient arylation of the substrate containing electron-withdrawing ester functionality directly attached to the pyrazole nucleus in the 4-position, affording product 18 in 75% isolated yield (Table 1, entry 12). Thus, this method allows for synthesis of 4,5-diaryl-SEM-pyrazoles in one step from readily available 4-aryl-SEM-pyrazoles.
SEM-group Transposition (SEM-group Switch)
After the installment of the second arene ring, the subsequent arylation would have to take place at the 3-position which bears the last available C-H bond. However, as shown is Figure 2, the arylation of this site is not feasible due to its low reactivity; no reaction or very low yields of the desired product were obtained. To solve this problem, we considered switching the SEM group from one nitrogen atom to another, which would transform the unreactive position 3 to the reactive position 5 (Scheme 2).
Scheme 2.
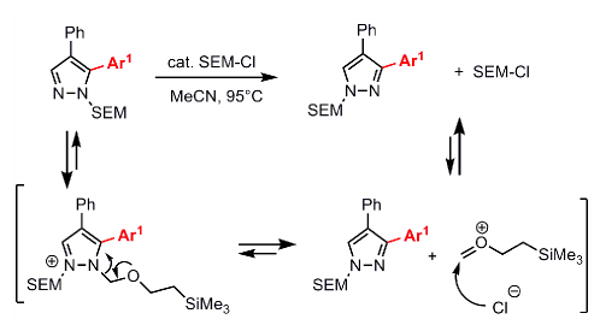
SEM group transposition (SEM switch)
It has been reported that a mixture of unsymmetrical N-protected imidazoles could be equilibrated by heating in the presence of alkylating agents, leading to formation of the thermodynamic product.22 Applying this approach to SEM-pyrazoles, we were able to achieve the SEM switch by heating the starting material with 10 mol % of SEM-Cl in acetonitrile. Alkylation of the pyrazole nitrogen lone pair by SEM-Cl forms a pyrazolium salt, which enables the equilibration of the two regioisomers, ultimately producing the thermodynamically favored, less hindered product (Scheme 2). The SEM-switch gives an average of ∼90% conversion as demonstrated with four different substrates (Table 2). The two isomers were frequently difficult to separate via flash chromatography; in such cases, reverse-phase HPLC was used to separate and purify these compounds. However, when triarylpyrazoles are desirable, separating the two regioisomers is not necessary, as the “unswitched material” may readily be removed after the subsequent arylation. The SEM-group switch transforms in one step an unreactive compound to a reactive substrate, avoiding the need for deprotection and reprotection, and enables the two sequential C-H arylations.
Table 2.
SEM switch scope
Determined by 1H NMR.
Isolated yield (flash chromatography).
Isolated yield (HPLC).
The Second C-Arylation and Preparation of 3,4,5-Triarylpyrazoles
Scheme 3 illustrates the brevity of the sequential arylation scheme for preparation of 3,4,5-triarylpyrazoles. Compound 8, available in one arylation step from starting material 3, was submitted to the SEM-group transposition to provide compound 20. The second arylation with 4-bromo-trifluoromethylbenzene proceeded in high yield under the standard catalytic conditions to give SEM-protected triarylpyrazole 23, which after deprotection furnished triarylpyrazole 24. Thus, all three arene rings are introduced to the pyrazole nucleus with complete regiochemical control; two of the rings are attached directly via C-H bond functionalization.
Scheme 3.
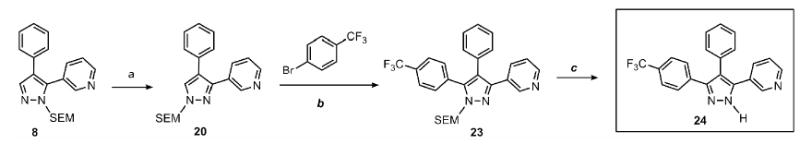
Synthesis of triarylpyrazoles via sequential C-arylation
Conditions: (a) Pyrazole, SEMCl (10 mol %), MeCN, 95°C, 24 hr, 84% yield. (b) Pyrazole, ArBr (1.5 equiv), Pd(OAc)2 (5 mol %), P(n-Bu)Ad2 (7.5 mol %), K2CO3 (3 equiv), HOPiv (25 mol %), 2.5 M DMA, 140°C, 12 hours; 77% yield. (c) 3N HCl, EtOH, reflux, 3 h, 75% yield. In DMSO-d6, compound 24 exists as a mixture of tautomers. Yields are an average of at least two separate isolated yields.
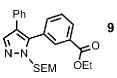
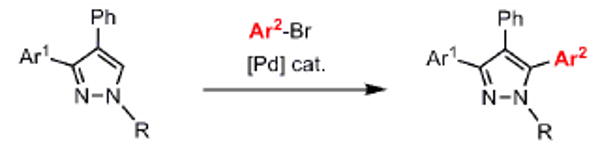
The scope of the arylation catalytic method was further examined in the context of 1-SEM-3,4-diarylpyrazole substrates (Table 3). Good to excellent yields of SEM-protected triarylpyrazoles were obtained with a variety of functional groups on both the pyrazole substrate and the bromoarene donor.
Table 3.
C-arylation of diarylpyrazoles. Substrate scope.
 | ||
|---|---|---|
| Entry | Product | Isolated Yield |
| 1 | 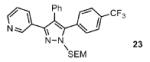 |
77% |
| 2 |  |
88% |
| 3 | 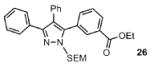 |
64% |
| 4 | 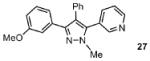 |
70% |
| 5 | 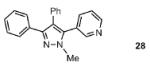 |
67% |
| 6 | 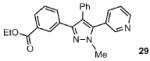 |
53% |
| 7 | 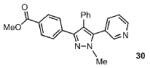 |
54% |
| 8 | 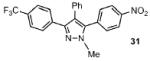 |
43% |
Reaction conditions: Pyrazole, ArBr (1.5 equiv), Pd(OAc)2 (5 mol %), P(n-Bu)Ad2 (7.5 mol %), K2CO3 (3 equiv), HOPiv (25 mol %), 2.5 M DMA, 140 °C, 12 hours. Yields are an average of at least two separate isolated yields.
Deprotection of SEM-pyrazoles
Free (NH)-pyrazoles are readily accessible by the deprotection of the SEM group, which is accomplished by the action of hydrochloric acid in ethanol.23 To confirm the generality of this protocol in the context of complex pyrazoles, triarylpyrazoles 23 and 25 (Table 3) as well as diarylpyrazoles 4, 8, and 10 (Table 1) were deprotected to afford high yields of the corresponding free pyrazoles (Supporting Information, section VI). It is well known that free pyrazoles exist as a mixture of tautomers.24,25 While the signal for the NH proton is broad in CDCl3, the two peaks are resolved in DMSO-d6 (δ 13-14.5 ppm); in the case of compound 24, both tautomers are present in a 56:44 ratio (Supporting Information).
Regioselective N-Alkylation/C-Arylation Sequence Enabled by the SEM Group
The sequential arylation mediated by the SEM-group switch, described above, provides a rapid access to protected and free triarylpyrazoles and offers an attractive alternative to the de novo approaches. However, when N-alkylated products are desirable, the N-alkylation of free N-H pyrazoles that lack a sufficient steric bias – such as the triarylpyrazole 24 (Scheme 3) – suffers from lack of regioselectivity, giving a mixture of two regioisomers. To address this issue, we considered the idea of using the SEM group to selectively introduce the alkyl group at the desired nitrogen of the pyrazole. We tested a number of methylating reagents (dimethyl sulfate, methyl iodide, and trimethyloxonium tetrafluoroborate), out of which Me3O-BF4 gave best results, yielding N-methylated pyrazolium salts at room temperature in dry dichloromethane. For example, compound 12 was methylated and deprotected to produce a single regioisomer of the alkylated pyrazole, namely the compound 33 (Scheme 4). This two-step procedure not only selectively alkylates the nitrogen of choice, but also serves to transform the unreactive 3-position of the pyrazole to the reactive 5-position. The N-methylpyrazole 33 was then arylated with 3-bromopyridine to afford complex pyrazole 27. Thus, the regioselective N-alkylation is incorporated into the sequential arylation scheme to afford the fully substituted pyrazole as a single isomer.
Scheme 4.
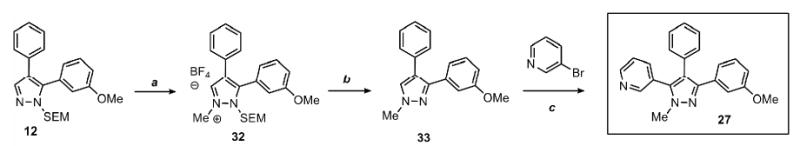
Sequential C-arylation and N-methylation provides a rapid access to 1-methyl-3,4,5-triarylpyrazoles with complete regioselectivity control
Conditions: (a) Pyrazole, Me3O-BF4 (1.2 equiv), CH2Cl2, RT, 1 hr. (b) 3N HCl, EtOH, reflux, 1 h; 70% yield over 2 steps. (c) Pyrazole, ArBr (1.5 equiv), Pd(OAc)2 (5 mol %), P(n-Bu)Ad2 (7.5 mol %), K2CO3 (3 equiv), HOPiv (25 mol %), 2.5 M DMA, 140 °C for 12 hours; 70% yield. Yields are an average of at least two separate isolated yields.
One limitation of this approach is that other nucleophilic functions present in the substrate would also undergo methylation; namely, pyridyl and dimethylamino groups are not compatible with the methylation reaction conditions. For anilines, this problem can be solved by introducing the nitrophenyl ring (Table 1 and Table 3), and reducing the nitro group after the alkylation step. Another solution is to conduct the alkylation earlier in the sequence and thus introduce the alkylation-prone groups by C-arylation after the N-alkylation step as illustrated in Scheme 4. The pyridine ring is attached in the last step of the sequence, combining the utility of the regioselective N-alkylation with direct C-H arylation.
Conclusions
Pyrazoles are an important class of heteroarenes frequently found in pharmaceuticals and protein ligands and there has been a growing interest in new synthetic methods for their preparation. We here present a strategically new approach based on the synthetic logic enabled by direct C-H bond functionalization, where new substituents are directly attached to predetermined positions of the heteroarene nucleus.9 This approach enables functionalization of all five pyrazole positions with complete regiocontrol and allows for the efficient synthesis of analog series and regioisomeric series from common precursors. The particular strength of this strategy is the ability to commence the synthesis from either the parent pyrazole or practically any pyrazole intermediate.
Owing to the intrinsic reactivity preferences of pyrazole, the sequential C-arylation of free N-H pyrazoles or N-alkylated pyrazoles is not possible and therefore we devised a strategy based on the SEM-protecting group and its transposition. The SEM group fulfills three major roles: first, it protects the pyrazole amine group and thus enables the C-arylation (free pyrazoles are not good substrates); second, it enables the regioselective sequential C-H arylation via the SEM group switch; and third, it allows for regioselective N-methylation which can be coupled to subsequent C-arylation. In sum, the catalytic C-H arylation combined with the protecting group transposition and N-alkylation, provides a rapid route to fully substituted pyrazoles with complete regiocontrol of all substituents.
Significant advances have recently been reported for the de novo synthesis of pyrazoles, both in terms of regiocontrol and scope.8 Also, major improvements have been achieved in the area of Suzuki coupling of pyrazoles; many of the problems associated with inefficient preparation of azolyl boronate esters and the low yielding coupling process have recently been addressed.10,26 Our approach is complementary to these methods and together they will greatly enable molecular designers to rapidly access a wide variety of complex pyrazole compounds. It is apparent that the sequential C-arylation approach will also be applicable to imidazoles and triazoles; these results will be described in separate publications in the near future.
Supplementary Material
Acknowledgments
We thank the National Institute of General Medical Sciences (NIGMS) for support of this work. We also thank Dr. Barry Touré (Novartis) for important intellectual contributions. Dr. Denis Gribkov is acknowledged for performing the preliminary experiments.
Footnotes
Supporting Information Available. Reaction conditions and compound characterization. This material is available on the World Wide Web at http://pubs.acs.org.
References
- 1.Pozharskii AF, Soldatenkov AT, Katritzky AR. Heterocycles in Life and Society. Wiley; Chichester, NY: 1997. [Google Scholar]
- 2.Elguero J. In: Comprehensive Heterocyclic Chemistry. Katrizky AR, Rees CW, Scriven EFV, editors. Vol. 5 Pergamon; Oxford: 1996. [Google Scholar]
- 3.Sliskovich DR, Roth BD, Wilson MW, Hoefle ML, Newton RS. J Med Chem. 1990;33:31–38. doi: 10.1021/jm00163a006. [DOI] [PubMed] [Google Scholar]
- 4.Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Roger RS, Rogier DJ, Yu SS, Anderson GD, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veenhuizen AW, Zhang YY, Isakson PC. J Med Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 5.Stauffer SR, Katzenellenbogen JA. J Comb Chem. 2000;2:318–329. doi: 10.1021/cc0000040. [DOI] [PubMed] [Google Scholar]
- 6.Moore KW, Bonner K, Jones EA, Emms F, Leeson PD, Marwood R, Patel S, Patel S, Rowley M, Thomas S, Carling RW. Bioorg Med Chem Lett. 1999;9:1285–1290. doi: 10.1016/s0960-894x(99)00169-9. [DOI] [PubMed] [Google Scholar]
- 7.Stanovnik B, Svete JD, editors. Science of Synthesis. Vol. 12. Stuttgart, NY: 2002. pp. 22–81.pp. 84–91. [Google Scholar]
- 8.(a) Aggarwal VK, De Vicente J, Bonnert RV. J Org Chem. 2003;68:5381–5383. doi: 10.1021/jo0268409. [DOI] [PubMed] [Google Scholar]; (b) Heller ST, Natarajan SR. Org Lett. 2006;8:2675–78. doi: 10.1021/ol060570p. [DOI] [PubMed] [Google Scholar]; (c) Deng X, Mani NS. Org Lett. 2008;10:1307–1310. doi: 10.1021/ol800200j. [DOI] [PubMed] [Google Scholar]
- 9.(a) Godula K, Sames D. Science. 2006;312:67–72. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]; (b) Kakiuchi F, Chatani N. Adv Synth Catal. 2003;345:1077–1101. [Google Scholar]
- 10.As recently reported, THP protecting group and its transposition has been used to achieve pyrazole lithiation and regioselective preparation of N-THP-pyrazolylboronate esters. McLaughlin M, Marcantonio K, Chen Cy, Davies IW. J Org Chem. 2008;73:4309–4312. doi: 10.1021/jo800321p.
- 11.(a) Lane BS, Sames D. Org Lett. 2004;6:2897–2900. doi: 10.1021/ol0490072. [DOI] [PubMed] [Google Scholar]; (b) Lane BS, Brown MA, Sames D. J Am Chem Soc. 2005;127:8050–8057. doi: 10.1021/ja043273t. [DOI] [PubMed] [Google Scholar]; (c) Touré BB, Lane BS, Sames D. Org Lett. 2006;8:1979–1982. doi: 10.1021/ol053021c. [DOI] [PubMed] [Google Scholar]; (d) Wang X, Gribkov DV, Sames D. J Org Chem. 2007;72:1476–1479. doi: 10.1021/jo061979v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Lane B, Sames D. J Am Chem Soc. 2005;127:4996–4997. doi: 10.1021/ja050279p. [DOI] [PubMed] [Google Scholar]
- 13.The development of catalytic methods for C-H arylation of arenes and heteroarenes is an active area of research. For recent reviews, see: Campeau L, Stuart DR, Fagnou K. Aldrichimica Acta. 2007;40:35–41.Satoh T, Miura M. Chem Lett. 2007;36:200–205.Seregin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173–1193. doi: 10.1039/b606984n.Alberico D, Scott SE, Lautens M. Chem Rev. 2007;107:174–238. doi: 10.1021/cr0509760.
- 14.Recent examples of azole C-arylation reported by others. Bowie AL, Hughes CC, Trauner D. Org Lett. 2005;7:5207–5209. doi: 10.1021/ol052033v.Deprez NR, Kalyani D, Krause A, Sanford MS. J Am Chem Soc. 2006;128:4972–4973. doi: 10.1021/ja060809x.Cerna I, Pohl R, Kleptarova B, Hocek M. Org Lett. 2006;8:5389–5392. doi: 10.1021/ol062324j.Lewis JC, Wu JY, Bergman RG, Ellman JA. Angew Chem Int Ed. 2006;45:1589–1591. doi: 10.1002/anie.200504289.Chuprakov S, Chernyak N, Dudnik AS, Gevorgyan V. Org Lett. 2007;9:2333–2336. doi: 10.1021/ol070697u.Chiong HA, Daugulis O. Org Lett. 2007;9:1449–51. doi: 10.1021/ol0702324.Bellina F, Cauteruccio S, Rossi R. J Org Chem. 2007;72:8543–8546. doi: 10.1021/jo701496p.Yang SD, Sun CL, Fang Z, Li BJ, Li YZ, Shi ZJ. Angew Chem Int Ed. 2008;47:1473–1476. doi: 10.1002/anie.200704619.Blaszykowski C, Aktoudianakis E, Alberico D, Bressy C, Hulcoop DG, Jafarpour F, Joushaghani A, Laleu B, Lautens M. J Org Chem. 2008;73:1888–1897. doi: 10.1021/jo702052b.
- 15.Intramolecular C-arylation of pyrazoles have been reported via an alkylation/arylation sequence. Blaszykowski C, Aktoudianakis E, Bressy C, Alberico D, Lautens M. Org Lett. 2006;8:2043–2045. doi: 10.1021/ol060447y.
- 16.The SEM group is also a directing group for lithiation of azoles including pyrazoles. Edwards MP, Doherty AM, Ley SV, Organ HM. Tetrahedron. 1986;42:3723–729.Fugina N, Holzer W, Wasicky M. Heterocycles. 1992;34:303–314.Gerard AL, Bouillon A, Mahatsekake C, Collot V, Rault S. Tet Lett. 2006:4665–4669.Luo G, Chen L, Dubowchik G. J Org Chem. 2006;71:5392–5395. doi: 10.1021/jo060607j.
- 17.Lafrance M, Fagnou K. J Am Chem Soc. 2006;128:16496–16497. doi: 10.1021/ja067144j. [DOI] [PubMed] [Google Scholar]
- 18.(a) Stanovnik B, Svete JD. Science of Synthesis. Vol. 12. Stuttgart, NY: 2002. pp. 173–203. [Google Scholar]; (b) Gupta RR, Kumar M, Gupta V. Heterocyclic Chemistry. Springer; Berlin, NY: 1998. [Google Scholar]; (c) Behr LC, Fusco R, Jarboe CH. Pyrazoles, pyrazolines, pyrazolidines, indazoles, and condensed rings. In: Wiley RH, editor. The Chemistry of Heterocyclic Compounds. Vol. 22 Interscience Publishers; New York, NY: 1967. [Google Scholar]
- 19.Park CH, Ryabova V, Geregin IV, Sromek AW, Gevorgyan V. Org Lett. 2004;6:1159–1162. doi: 10.1021/ol049866q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The palladium-carbonate system also showed preference for acidic C-H bonds. Garcia-Cuadrado D, Braga AAC, Maseras F, Echavarren AM. J Am Chem Soc. 2006;128:1066–1067. doi: 10.1021/ja056165v. [DOI] [PubMed] [Google Scholar]
- 21.Zhao ZG, Wang ZX. Synth Comm. 2007;37:137–147. [Google Scholar]
- 22.He Y, Chen Y, Du H, Schmid LA, Lovely CJ. Tet Lett. 2004;45:5529–5532. [Google Scholar]
- 23.Luo G, Chen L, Dubowchik G. J Org Chem. 2006;71:5392–5395. doi: 10.1021/jo060607j. [DOI] [PubMed] [Google Scholar]
- 24.(a) Trofimenko S, Yap GPA, Jove FA, Claramunt RM, García MA, Santa Maria MD, Alkorta I, Elguero J. Tetrahedron. 2007;63:8104–8111. [Google Scholar]; (b) Vors JP, Gerbaud V, Gabas N, Canselier JP, Jagerovic N, Jimeno ML, Elguero J. Tetrahedron. 2003;59:555–560. [Google Scholar]; (c) Aguilar-Parrilla F, Cativiela C, Diaz de Villegas MD, Elguero J, Foces-Foces C, Laureiro JIG, Cano FH, Limbach HH, Smith JAS, Toiron C. J Chem Soc Perkin Trans 2. 1992:1737–1742. [Google Scholar]; (d) Habraken CL, Moore JA. J Org Chem. 1965;30:1892–1896. [Google Scholar]
- 25.For considerations regarding the binding of tautomers to protein targets, see: Chimenti F, Fioravanti R, Bolasco A, Manna F, Chimenti P, Secci D, Befani O, Turini P, Ortuso F, Alcaro S. J Med Chem. 2007;50:425–428. doi: 10.1021/jm060868l.
- 26.(a) Kudo N, Perseghini M, Fu GC. Angew Chem Int Ed. 2006;45:1282–1284. doi: 10.1002/anie.200503479. [DOI] [PubMed] [Google Scholar]; (b) Billingsley KL, Anderson KW, Buchwald SL. Angew Chem Int Ed. 2006;45:3484–3488. doi: 10.1002/anie.200600493. [DOI] [PubMed] [Google Scholar]; (c) Billingsley K, Buchwald SL. J Am Chem Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.