

Abstract
Apoptin, a chicken anemia virus-derived protein, selectively induces apoptosis in transformed but not in normal cells, thus making it a promising candidate as a novel anticancer therapeutic. The mechanism of apoptin-induced apoptosis is largely unknown. Here, we report that contrary to previous assumptions, Bcl-2 and Bcl-xL inhibit apoptin-induced cell death in several tumor cell lines. In contrast, deficiency of Bax conferred resistance, whereas Bax expression sensitized cells to apoptin-induced death. Cell death induction by apoptin was associated with cytochrome c release from mitochondria as well as with caspase-3 and -7 activation. Benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone, a broad spectrum caspase inhibitor, was highly protective against apoptin-induced cell death. Apoptosis induced by apoptin required Apaf-1, as immortalized Apaf-1-deficient fibroblasts as well as tumor cells devoid of Apaf-1were strongly protected. Thus, our data indicate that apoptin-induced apoptosis is not only Bcl-2- and caspase dependent, but also engages an Apaf-1apoptosome-mediated mitochondrial death pathway.
Keywords: Apaf-1, apoptin, apoptosis, caspases, Bcl-2, Bcl-xL
Introduction
Apoptosis is essential for ontogenesis, tissue homeostasis and proper function of the immune system (Los et al., 1999; Zheng and Flavell, 2000). Viruses have developed various mechanisms that either prevent apoptotic elimination of infected cells or facilitate apoptosis in target cells, thus weakening an antiviral response (Everett and McFadden, 2001; Thomson, 2001). Chicken anemia virus (CAV) induces apoptosis in infected tissues of the avian host resulting in depletion of thymocytes and erythroblastoid cells in bone marrow (Adair, 2000). Infection with CAV causes a disease in young chickens that is characterized by a generalized lymphoid atrophy, severe anemia and increased mortality. The death of infected cells is caused by apoptin (VP3), a small 14 kDa virally encoded and proline-rich protein, which has no homologous cellular counterparts (Noteborn et al., 1994).
One of the most remarkable features of apoptin is its unique ability to cause apoptosis rather selectively in tumor cells, but not in primary, nontransformed cells (Danen-Van Oorschot et al., 1997). The cellular localization appears to be crucial for apoptin’s selective toxicity in transformed cells. In most primary cells apoptin remains in the cytoplasm, whereas in transformed cells it migrates into the nucleus and kills the cell. Nuclear translocation occurs upon phosphorylation of apoptin at Thr-108 by a still unknown kinase (Rohn et al., 2002). The transformation-dependent toxicity of apoptin makes it an interesting lead molecule for the development of novel anticancer strategies.
The mechanism of apoptin-induced apoptosis is largely unknown. It has been proposed that cell death caused by apoptin is independent of the p53 and Bcl-2 status of the cell (Zhuang et al., 1995; Danen-Van Oorschot et al., 1999; Schoop et al., 2004) that largely determine the susceptibility of cells towards radio-and chemotherapy. Colocalization of apoptin with the death regulators Bcl-10 and FADD in cytoplasmic structures, called death effector filaments, has been recently reported (Guelen et al., 2004), but the significance of this finding remains to be determined. Apoptin-induced death is resistant to overexpression of the cowpox virus caspase inhibitor CrmA, which inhibits proinflammatory caspases as well the initiator caspase-8 and -9. However, overexpression of the broad-spectrum baculoviral caspase inhibitor p35 was reported to delay apoptin-mediated cell death (Danen-van Oorschot et al., 2000). Furthermore, downstream caspases including caspase-3 become activated upon apoptin expression in transformed cells (Danen-van Oorschot et al., 2000). Cytochrome c release from mitochondria has also been observed upon death induction by apoptin, and it is not inhibited by p35 coexpression.
The release of cytochrome c and other proapoptotic mitochondrial proteins is essentially controlled by Bcl-2 family proteins which comprise pro- and antiapoptotic homologues (Gross et al., 1999; Antonsson and Martinou, 2000; Daniel et al., 2003). The antiapoptotic family members (e.g. Bcl-2, Bcl-xL) share three to four Bcl-2 homology (BH) regions and localize to cytoplasmic sides of intracellular membranes (Bouillet and Strasser, 2002). Proapoptotic Bcl-2 proteins, such as Bax and Bak, have two or three BH regions and can bind to antiapoptotic Bcl-2 proteins (Suzuki et al., 2000). Other proapoptotic Bcl-2 proteins (e.g. Bad, Bid, Bim) share only the short BH3 region (Bouillet and Strasser, 2002). The mechanism of apoptosis regulation by Bcl-2 family members is not fully understood. It is widely believed that Bcl-2 functions to preserve mitochondrial membrane integrity and to prevent the release of cytochrome c from mitochondria (Strasser et al., 2000; Reed and Green, 2002). BH3-only proteins appear to sense stimuli that cause cellular stress and initiate the death cascade. Bax and Bak are essential for cell killing governed by BH3-only proteins and antagonized by Bcl-2 (Antonsson and Martinou, 2000; Marsden and Strasser, 2003). Overexpression of Bcl-2 provides a survival advantage for cancer cells and it is associated with increased frequency of lymphoma development in a murine model (McDonnell et al., 1989). Loss of proapoptotic Bax function seems to be important for the pathogenesis of colorectal cancers (Rampino et al., 1997). However, clinical studies on the predictive value of Bcl-2 family protein expression only partially support the in vitro observations (Hamilton and Piccart, 2000).
The attractiveness of apoptin as a lead compound for the development of anticancer therapies has prompted us to study its molecular mechanism of action. Contrary to previous reports (Zhuang et al., 1995; Danen-Van Oorschot et al., 1999; Schoop et al., 2004), we show here that Bcl-2 and Bcl-xL inhibit apoptin-induced death under various conditions. On the other hand, the Bax status of the cell markedly affected the resistance towards apoptin. Cell death induced by apoptin was associated with cytochrome c release from mitochondria, activation of caspase-3, -7, and sensitivity to a broad-spectrum caspase inhibitor. Furthermore, cells lacking Apaf-1, a crucial molecule in the mitochondrial death pathway, were resistant against apoptin. In conclusion, our data clearly indicate that apoptin induced death engages a caspase dependent mitochondrial pathway and is controlled by pro- and antiapoptotic Bcl-2 family members.
Results
Apoptin expression is not toxic for primary cells
A unique feature of apoptin is its selective toxicity for transformed but not primary cells. In transformed cells apoptin localizes in the nucleus, whereas in primary nontransformed cells it remains largely in the cytoplasm (Danen-Van Oorschot et al., 1997). To investigate the mechanism of apoptin action, its cDNA was cloned into the pEGFP-C1 vector. The control and green fluorescent protein (GFP)-apoptin vectors were transiently transfected into primary human umbilical vein endothelial cells (HUVECs) and human breast carcinoma MCF-7 cells (Figure 1). The control GFP protein was evenly expressed in the HUVECs, and no signs of apoptosis could be observed (Figure 1a). GFP-apoptin at early time points post-transfection was, similarly to the control, found in the cytoplasm and in the nucleus, however, with a higher nuclear concentration (Figure 1b). After 48 h, in many HUVECs the expression pattern of GFP-apoptin changed to dot-like structures that were predominantly localized in the nucleus. However, even in cells with nuclear apoptin no signs of cell death could be observed 4 days after transfection (Figure 1c). In MCF-7 cells, like in HUVECs, the control GFP protein showed an even distribution in the cells (Figure 1d), whereas GFP-apoptin revealed an exclusive localization in the nucleus (Figure 1e, f). At later time points, MCF-7 cells expressing apoptin showed morphological changes of apoptosis, such as chromatin condensation and cell detachment (data not shown, see Figure 2).
Figure 1.

Cellular localization and toxicity of GFP-apoptin in primary endothelial and MCF-7 cells. HUVECs (a–c) and MCF-7 cells (d–f) were transiently transfected with the pEGFP-C1 control vector (a, d) or with a pEGFP-apoptin construct (b, c, e, f). The pictures were taken 24 h (a, b, d, e, f) and 96 h (c) post-transfection. (f) DAPI staining of cells shown in (e).
Figure 2.

Apoptin-mediated cell death is caspase dependent. (a) Wild-type caspase-3-deficient MCF-7 carcinoma cells and MCF-7 cells expressing caspase-3 (MCF-7/C3) were transiently transfected either with pEGFP-C1 alone or with a pEGFP-C1-apoptin construct and then either left untreated or incubated with 30 μM zVAD-fmk. Photographs were taken 24 h post-transfection. (b) At the indicated time points the GFP-apoptin transfected MCF-7 and MCF-7/C3 cells were examined microscopically and scored for apoptosis. The percentage of live (flat with intact nuclei) and apoptotic cells (round cells with condensed chromatin) was determined for each time point. At least 300 green cells per sample were independently analysed by two scientists. The mean values of three independent experiments are given with s.d. (c) MCF-7/C3 cells transiently expressing DsRed or DsRed-apoptin in the presence or absence of 30 μM zVAD-fmk were lysed 24 h post-transfection, incubated with the fluorogenic caspase substrate DEVD-AMC and assayed for caspase activity in a fluorimeter. Caspase activity is given in arbitrary units (AU). Red fluorescent protein instead of GFP was used for tracing apoptin in order to avoid interference of protein fluorescence with the flourogenic caspase assay. (d) MCF-7 and MCF-7/C3 cells transiently expressing GFP or GFP-apoptin in the presence or absence of 30 μM zVAD-fmk were lysed 24 and 48 h post-transfection and subjected to SDS–PAGE. Cleavage of caspase-3 and PARP-1 was detected by Western blot analysis. The full-length forms and proteolytic fragments of the respective proteins are indicated.
Apoptin-mediated cell death is caspase dependent
To investigate whether caspase inhibitors could block apoptin-induced apoptosis, caspase-3-deficient MCF-7 cells and MCF-7 cells stably overexpressing caspase-3 (MCF-7/C3) were transiently transfected with the pEGFP-apoptin construct or the vector control in the presence or absence of 30 μM of the broad-range caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk). At 24 h post-transfection cells were examined microscopically for protein expression and the effects of zVAD-fmk. Both, MCF-7 and MCF-7/C3 cells were highly sensitive to apoptin (Figure 2a). In contrast, cells transfected with apoptin vector in the presence of zVAD-fmk showed no signs of cell death.
To investigate the effect of caspase inhibition in detail, a time course study was performed. MCF-7 and MCF-7/C3 cells were transfected with GFP-apoptin and either left untreated or supplemented with zVAD-fmk. The percentage of apoptotic cells among apoptin-transfected cells was determined morphologically 24, 48 and 72 h post-transfection. The caspase inhibitor protected both cell lines up to 72 h post-transfection against apoptin-induced apoptosis (Figure 2b). In MCF-7/C3 cells this protection was less efficient. About 43% of the transfected MCF-7/C3 cells died within 72 h, in comparison to only 24% of MCF-7 cells that had died at this time point in the presence of zVAD-fmk. The difference in protection efficiency might be caused by the short half-life time of the inhibitor (Lamkan. et al., 2002) in combination with the overexpression of caspase-3 in MCF-7/C3 cells.
The finding that zVAD-fmk protected MCF-7 cells suggested caspases as essential mediators of apoptin-induced apoptosis. To further verify this assumption, enzymatic caspase assays were performed. MCF-7/C3 cells were transfected with the pDsRed-C1-apoptin construct or with pDsRed-C1 control vector in the presence or absence of zVAD-fmk. After 24 h cell extracts were prepared and incubated with the fluorogenic caspase-3 substrate Ac-DEVD-7-aminomethyl-4-coumarin (AMC). A three- to four fold increase of caspase activity could be observed in DsRed-apoptin transfected cells as compared to cells transfected with the vector control. zVAD-fmk efficiently inhibited induction of caspase-3 activity (Figure 2c).
Caspase activation was also confirmed by Western blot analysis. Overexpression of apoptin in MCF-7/C3 cells caused a marked activation of caspase-3, as visualized by the decreased level of its zymogen and the appearance of the 17-kDa active subunit of caspase-3 (Figure 2d). Furthermore, cleavage of the caspase-3 substrate PARP-1 correlated with the activation of caspase-3 upon apoptin expression (Figure 2d). zVAD-fmk inhibited apoptin-induced activation of caspase-3 as well as PARP-1 cleavage. A slight but clearly visible cleavage of PARP-1 was also evident in caspase-3-deficient MCF-7 cells.
Antiapoptotic Bcl-2 family members prevent apoptin-induced cell death
It has been postulated that Bcl-2 expression accelerates apoptin-induced killing instead of inhibiting it (Noteborn et al., 1998; Danen-Van Oorschot et al., 1999). In this case, apoptin would be a very promising killer molecule to treat cancers that overexpress Bcl-2, which of ten hinders radio- and chemotherapy. Bcl-2, Bcl-xL and other antiapoptotic members of this protein family are efficient blockers of apoptosis mediated by the mitochondrial pathway (Antonsson and Martinou, 2000). Therefore, we examined the role of Bcl-2 and Bcl-xL in apoptin-induced killing of transformed cells. We compared the sensitivity towards apoptin-induced death between the parental MCF-7 cell line and its derivatives stably overexpressing Bcl-2 or Bcl-xL. Transient expression of GFP-apoptin in MCF-7 cells induced apoptosis within 48 h, whereas MCF-7/Bcl-2 and MCF-7/Bcl-xL cells remained viable and appeared morphologically healthy for at least 72 h post-transfection (Figure 3a). DNA staining with DAPI showed evenly distributed chromatin in the nuclei of Bcl-2 or Bcl-xL overexpressing cells, in contrast to condensed chromatin in MCF-7 cells that expressed apoptin. To further examine the protective effect of Bcl-2 and Bcl-xL we performed a kinetic study. GFP-apoptin was transiently expressed in MCF-7, MCF-7/C3, MCF-7/Bcl-2 and MCF-7/Bcl-xL cells, and the percentage of dead cells among the total green fluorescent cells was determined by microscopy at the indicated time points (Figure 3b). Up to 70–80% of MCF-7 parental cells or MCF-7/C3 cells died within 48 h after transfection with apoptin, whereas MCF-7 cells overexpressing Bcl-2 or Bcl-xL were resistant. At later time points Bcl-xL seemed to be less protective than Bcl-2. Overexpression of both, Bcl-2 and Bcl-xL conferred resistance in MCF-7 cells for up to 96 h post-transfection with GFP-apoptin (data not shown).
Figure 3.
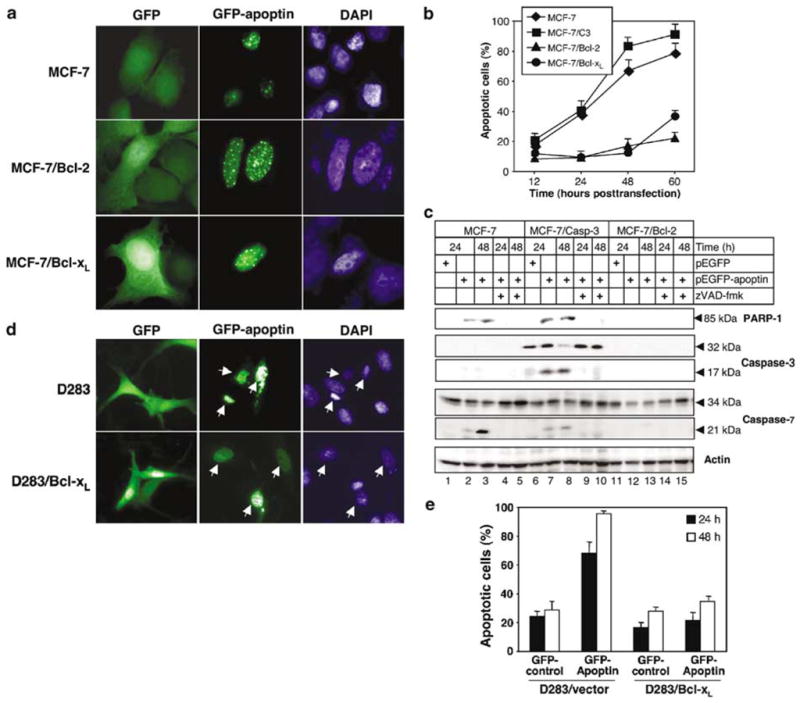
Antiapoptotic Bcl-2 family members prevent apoptin-induced cell death. (a) MCF-7 cells, MCF-7/Bcl-2 and MCF-7/Bcl-xL cells were transfected with GFP alone or the GFP-apoptin construct. The cells were fixed and stained with DAPI 48 h post-transfection. (b) MCF-7, MCF-7/C3, MCF-7/Bcl-2, and MCF-7/Bcl-xL cells transiently transfected with GFP-apoptin were scored microscopically by two independent researchers, and the percentage of dead cells among the transfected cells was calculated. The mean values of three independent experiments are given with s.d. (c) MCF-7, MCF-7/C3, MCF-7/Bcl-2 were transiently transfected with GFP-apoptin or with the GFP vector in the presence of absence of 30 μM zVAD-fmk. Cell lysates were prepared 24 and 48 h post-transfection followed by SDS–PAGE. Cleavage of caspase-3, -7, and PARP-1 was detected by Western blot analysis. Actin served as a control for equal protein loading. (d) D283 vector control and D283/Bcl-xL cells were transfected with GFP or the GFP-apoptin expression vector. At 24 h post-transfection the cells were stained with DAPI and analysed by fluorescence microscopy. Arrows mark corresponding cells in the GFP and DAPI fluorescence channels. (e) Quantification of apoptosis induction in D283 vector control and D283/Bcl-xL cells transiently transfected with GFP-apoptin. Cell death was assessed microscopically and independently by two scientists at the indicated time points, and the percentage of dead cells among the transfected cells was calculated. The mean values of three independent experiments are given with s.d.
Since MCF-7 cells overexpressing Bcl-2 were not killed by apoptin, we have next investigated whether this effect correlated with the inhibition of effector caspases. As detected by Western blot analysis, caspase-3 was activated in MCF-7/C3 cells after transfection with apoptin, followed by the cleavage of cellular substrates, such as PARP-1 (Figure 3c). In addition, activation of caspase-7, another important executioner caspase, could be demonstrated in MCF-7C3 cells and the parental cell line upon expression of GFP-apoptin (Figure 3c). The cleavage of PARP-1 in MCF-7 cells provided further evidence for caspase-7 activation. In contrast, in MCF-7/Bcl-2 cells neither caspase-7 activation nor PARP-1 cleavage were observed upon GFP-apoptin expression (Figure 3c).
Since the protective effect of Bcl-2 or Bcl-xL against apoptin-induced death could have been cell type specific, we further tested the inhibitory activity of Bcl-xL in the human medulloblastoma D283 cell line and its derivative overexpressing Bcl-xL (D283/Bcl-xL). About 70% of GFP-apoptin-expressing D283 cells died within 24 h post-transfection, and almost 100% of apoptin-transfected cells were dead within 48 h (Figure 3d and e). In contrast, only about 35% of the D283/Bcl-xL apoptin-expressing cells underwent apoptosis 48 h post-transfection (Figure 3e). Most of the Bcl-xL over-expressing cells remained normal and only cells that expressed high levels of GFP-apoptin died, but still cell death occurred much slower than in control cells.
The above experiments were performed using transfected cells stably overexpressing Bcl-2 or Bcl-xL. Next, we examined the apoptins’ toxicity in cell lines expressing elevated amounts of Bcl-2 either spontaneously or after stimulation with physiological stimuli. For example, Bcl-2 expression in MCF-7 cells can be increased by the stimulation of estrogen receptors (Somai et al., 2003). As shown in Figure 4a, β-estradiol increased Bcl-2 expression in different MCF-7 cell lines. Upregulation of Bcl-2 expression by β-estradiol decreased the sensitivity of both MCF-7/C3 and MCF-7/Bcl-2 cells towards apoptosis induced by a cell permeable, recombinant Tat-apoptin protein. Furthermore, we investigated apoptins’ toxicity in the three cell lines KG-1, K562 and DOHH-2 that were previously described by Noteborn and colleagues to spontaneously express elevated amounts of Bcl-2 (Zhuang et al., 1995). As shown in Figure 4b, KG-1 and K562 cells expressed much higher levels of Bcl-2 as compared to DOHH-2 cells. Both KG-1 and K562 cells were also much less sensitive towards apoptin-induced cell death in comparison to the DOHH-2 cell line, in which apoptin potently reduced cell survival. Unlike in transformed cells, Tat-apoptin was unable to induce apoptosis in HUVEC. Less than 10% of endothelial cell death occurred following treatment with Tat-apoptin for 96 h, which was, however, also observed with Tat-GFP and therefore not attributable to apoptin itself (data not shown).
Figure 4.

Induced and spontaneous Bcl-2 expression inhibits apoptin-triggered cell death in different cell lines. (a) The expression of Bcl-2 was upregulated in MCF-7 breast cancer cells and its derivatives stably transfected with either caspase-3 (MCF-7/C3) or Bcl-2 (MCF-7/Bcl-2) by cell stimulation with 10 nM β-estradiol for 2 days. Cytotoxicity of cell-permeable Tat-apoptin (1 μM) or a Tat-GFP control was evaluated in MCF-7/C3 and MCF-7/Bcl-2 cells before and after Bcl-2 upregulation using the MTT assay. (b) Three unrelated cell lines (KG-1, DOHH-2, and K562) expressing variable amounts of Bcl-2 were treated with 1 μM recombinant Tat-apoptin or Tat-GFP for 4 days. Cell viability was then measured by the MTT assay. The status of Bcl-2 and procaspase-3 expression was examined by Western blot analysis. The level of cell death in both experiments was calculated from internal reference samples incorporated into each assay plate. Mean values of three independent experiments are shown. The results obtained by the MTT assay were confirmed by microscopic assessment of cell death as described in Materials and method.
Bax promotes apoptin-mediated cell death
The experiments described above demonstrate an inhibitory role of antiapoptotic Bcl-2 family members towards apoptin-induced death. To investigate whether apoptin-induced death is facilitated by the proapoptotic Bcl-2 family member Bax, we used the Bax-deficient prostate carcinoma cell line DU145 and the corresponding Bax-transfected derivative (DU145/Bax). Upon transfection with GFP-apoptin, Bax-negative, mock-transfected DU145 cells remained viable and morphologically healthy for the duration of the experiment (48 h). In contrast, Bax-expressing cells showed morphological changes typical for apoptosis (Figure 5a). To examine the Bax dependence of apoptin-induced kill, time course experiments were performed. The percentage of dead cells among the transfected ones was determined by fluorescent microscopy (Figure 5b). Most (~70%) of the cells that expressed Bax were killed by apoptin already within 36 h post-transfection. 60 h post-transfection ~80% of DU145/Bax cells were dead, compared to only ~25% of cell death observed in the parental cell line. The majority of DU145 cells revealed no morphological changes even after 96 h.
Figure 5.

Bax promotes apoptin-mediated death. (a) Bax-deficient, wild-type mock transfected DU145 prostate carcinoma cells and Bax-expressing DU145 cells were transfected with the GFP-vector alone or with GFP–apoptin. The cells were fixed and stained with DAPI 48 h post-transfection. Arrows indicate apoptotic cells expressing GFP-apoptin. (b) DU145 and DU145/Bax cell lines transiently transfected with GFP-apoptin were examined microscopically and the percentage of dead cells among transfected cells was assessed. The mean values of three independent experiments are given with s.d.
Apoptin triggers the apoptosome pathway
Bcl-2 family proteins modulate the intrinsic apoptosome-dependent death pathway. However, Bcl-2 might also have a broader role, because Bcl-2 overexpression can regulate caspase activation independently of the Apaf-1/caspase-9 pathway (Bitzer et al., 2002; Marsden et al., 2002). Furthermore, it has been described that Bcl-2 prevents the mitochondrial release of other proapoptotic factors, including AIF and endonuclease G, which might be involved in caspase-independent cell death (Haraguchi et al., 2000). To further explore the importance of the apoptosome pathway for death execution by apoptin, we investigated the requirement of the key players of this pathway, Apaf-1 and cytochrome c. The apoptin-triggered cytochrome c release from mitochondria was directly observed in MCF-7 cells stably expressing a cytochrome c–GFP fusion protein (Figure 6a). To examine the importance of Apaf-1, two approaches were applied. First, we employed Apaf-1 siRNA to suppress the levels of Apaf-1 in MCF-7/C3 cells. Western blot analysis confirmed that this approach strongly reduced Apaf-1 expression (Figure 6b). Inhibition of Apaf-1 expression by RNA interference almost completely blocked the cell death induced by apoptin as well by the chemotherapeutic drug etoposide (Figure 6c). The requirement of Apaf-1 for apoptin-induced apoptosis was confirmed in Apaf-1(−/−) fibroblasts transformed by the SV40 large T-antigen. Apaf-1(−/−) fibroblasts were completely resistant to apoptin, whereas about 70% of the immortalized wild-type fibroblasts were killed by apoptin (Figure 6d). Nontransformed fibroblasts were resistant to apoptin, again demonstrating the specific toxicity of apoptin for transformed cells (data not shown).
Figure 6.

Requirement of Apaf-1 for apoptin-induced apoptosis. (a) Apoptin induces cytochrome c release from transformed cells. MCF-7/GFP-cytochrome c cell line was transiently transfected with pDsRed-C1 vector or with pDsRed-C1-apoptin. At 24 h post-transfection the cells were fixed with 3% formaldehyde, stained with DAPI and evaluated under a microscope. The pictures show representative cells. (b) Apaf-1 expression was suppressed by siRNA in MCF-7/caspase-3 cells as indicated. At 24 h the cells were transfected with GFP-apoptin or treated with 50 μM etoposide. After culture for further 48 h, the cells were lysed and subjected to immunoblotting with anti-Apaf-1 (upper panel) or anticaspase-3 antibodies (lower panel). Arrows indicate the position of the full-length proteins and processed fragment of caspase-3. (c) MCF-7/caspase-3 cells were transfected as in (b) and then stained with DAPI to reveal chromatin structure. Cells with condensed chromatin were scored as apoptotic. (d) To substantiate the effect of Apaf-1 deficiency on apoptin-induced apoptosis, immortalized Apaf-1 knockout mouse fibroblasts were used for transfection with GFP and GFP-apoptin vector. After 48 h the cells were fixed and stained with DAPI. The number of apoptotic cells among the transfected cells was assessed as described above.
Discussion
Tumor cell specificity is an important prerequisite for successful cancer therapy (Fischer and Schulze-Osthoff, 2005). Apoptin can induce apoptosis in cell lines derived form a great variety of human tumors, for example, hepatoma, lymphoma, cholangiocarcinoma, melanoma, breast and lung tumor, and colon carcinoma. In contrast, apoptin does not induce apoptosis in normal, nontransformed cells such as fibroblasts, keratinocytes or smooth muscle cells (Oro and Jans, 2004). The manner by which apoptin is able to distinguish between tumor and normal cells remains to be elucidated. Surprisingly, it has been reported that Bcl-2 facilitates and accelerates rather than inhibits apoptosis (Zhuang et al., 1995; Noteborn et al., 1998; Danen-Van Oorschot et al., 1999; Schoop et al., 2004), suggesting that apoptin-induced apoptosis does not feed into the classical apoptotic pathway. These appealing features, which would make apoptin an interesting candidate for novel antitumor strategies, prompted us to investigate the molecular mechanisms of apoptin-induced apoptosis in more detail.
The subcellular localization of apoptin has been suggested to be crucial for the induction of apoptosis. In all cells undergoing apoptin-induced apoptosis, the protein was detected in the nucleus, while in primary cells apoptin is preferentially localized in the cytoplasm (Danen-Van Oorschot et al., 1997). Recently, apoptin was described to be phosphorylated specifically at Thr-108 in tumor and transformed cells (Rohn et al., 2002). This modification was shown to be important for the nuclear localization and cell killing ability of apoptin. However, nuclear localization of apoptin might be important, but does not seem sufficient for inducing apoptosis. Even in normal cells such as HUVECs, we found that apoptin could be translocated to the nucleus without inducing apoptosis. Consistent with these data, fusion of a heterologous nuclear localization sequence to apoptin forces it into the nucleus, but does not induce apoptosis in normal cells (Danen-Van Oorschot et al., 2003). It was also shown that a C-terminally truncated apoptin construct, in which the phosphorylation site was deleted, was still able to translocate to the nucleus, although with lower efficiency, and induce apoptosis (Guelen et al., 2004; Tavassolli et al., 2005). Thus, the interaction with other molecules or additional modifications of apoptin that are not present in nontransformed cells may be required for apoptin’s toxicity.
Recently, it was proposed that the nuclear translocation of apoptin is not specific for transformed cells but correlates with the cellular concentration of apoptin (Wadia et al., 2004). Indeed, GFP-apoptin is able to enter nuclei of primary cells, but still the nuclear transfer of GFP-apoptin appears to be much more efficient in transformed cells (Wadia et al., 2004; and Figure 1 of this paper). This phenomenon was so far observed only by using apoptin fused to GFP. Whether nonmodified apoptin can enter nuclei of primary cells remains to be determined. Nevertheless, using a GFP-apoptin transfection system in isogenic cell pairs (transformed and nontransformed phenotypes), it has been shown recently that the C-terminus of apoptin (aa 74–121) contains a nuclear localization signal whose activity is highly specific for transformed cells (Poon et al., 2005).
Although caspase-independent forms of apoptosis have been described, activation of caspases plays a central role in most apoptotic pathways (Schulze-Osthoff et al., 1998). Activation of caspases is achieved via two principal signaling pathways, namely the extrinsic and intrinsic death pathways, both of which depend on the inducible formation of large multiprotein complexes (Schwerk and Schulze-Osthoff, 2005). Upon formation of the death-inducing signaling complex in the extrinsic death receptor pathway or the apoptosome in the intrinsic mitochondrial pathway, initiator caspases are autoproteolytically processed resulting in the activation of downstream caspases and cleavage of numerous death substrates (Stroh and Schulze-Osthoff, 1998; Fischer et al., 2003). It has been proposed that apoptin-induced cell death occurs independently of the caspase cascade (Noteborn et al., 1998). To investigate this hypothesis, we studied the role of caspases in apoptosis induced by apoptin using the caspase-3-deficient MCF-7 cell line, its retransfected derivative MCF-7/C3, and by employing pharmacological caspase inhibition. The caspase inhibitor zVAD-fmk almost completely inhibited cell death caused by apoptin, indicating that caspases are the essential executors of apoptin-induced death. The inhibitory effect of zVAD-fmk on apoptin-induced apoptosis was not only observed in MCF-7 cells, but also in other tumor cell lines, such as Jurkat T-cell leukemia, HeLa cervix carcinoma, and D283 medulloblastoma cells (data not shown). Moreover, employing fluorogenic substrate assays and Western blot analysis, detection of activated caspase-3 and -7 fully confirmed these results. A significant increase in DEVDase activity upon apoptin expression as well as the proteolytic processing of caspase-3 and -7 were observed. The caspase inhibitor zVAD-fmk blocked caspase activity as well as the occurrence of active subunits of caspase-3, -7 and PARP-1 cleavage. Since both, the MCF-7 parental cell line and MCF-7/C3 caspase-3-expressing cells were killed, caspase-3 may be dispensable for death induction by apoptin. This is most likely explained by the fact that effector caspases such as caspase-6 and -7 can substitute for caspase-3 in apoptin-induced apoptosis.
Pro- and antiapoptotic Bcl-2 family proteins are important modulators of the mitochondrial death pathway. Several reports by Noteborn and colleagues claimed that the antiapoptotic Bcl-2 molecule does not inhibit, but even facilitates apoptosis induction by apoptin (Noteborn et al., 1998; Danen-Van Oorschot et al., 1999; Schoop et al., 2004). However, in our experiments both Bcl-2 and Bcl-xL were highly protective against apoptin-triggered death, a finding that was confirmed with different, unrelated cell lines. Moreover, apoptin-induced apoptosis was strongly compromised in DU145 prostate carcinoma cells, which carry a frame-shift mutation in the Bax gene and do not express Bax protein. Consistent with these data, expression of Bax in DU145 cells strongly sensitized towards apoptin-induced apoptosis. Thus, these results demonstrate unequivocally that apoptin-induced apoptosis is controlled by Bcl-2 proteins. This notion is substantiated by our finding that apoptin also induced mitochondrial cytochrome c release.
It is widely believed that Bcl-2 family proteins regulate mitochondrial membrane pores that release cytochrome c and other apoptogenic factors. However, Bcl-2 proteins can also induce or suppress caspase-independent nonapoptotic cell death (Kane et al., 1995). Moreover, recent reports have shown that Bcl-2 can regulate activation of initiator caspases independently of the classical apoptosome, arguing that they operate by mechanisms other than by regulating caspase-activating proteins such as Apaf-1 (Marsden et al., 2002). We therefore compared the impact of loss of Apaf-1 in transformed fibroblasts from Apaf-1-deficient mice as well as by employing Apaf-1 RNA interference in MCF-7 cells. Both approaches clearly showed that Apaf-1 deficiency rendered cells resistant to GFP-apoptin. Similar results were obtained when Apaf-1-depleted or -deficient cells were treated with the recombinant Tat-apoptin (data not shown). Thus, in contrast to previous results by Noteborn and colleagues, these experiments clearly show that apoptin-mediated cell death is modulated by Bcl-2 proteins and ultimately feeds into the classical mitochondrial death pathway.
The mechanism of how apoptin triggers the mitochondrial pathway remains to be elucidated. Although apoptin presumably has to translocate to the nucleus, inhibitors of transcription or translation did not abrogate apoptosis induced by a bacterial cell-permeable Tat-apoptin fusion protein (data not shown). The tight association of apoptin with chromatin has suggested that apoptin could disturb chromatin assembly and trigger nuclease activation or an apoptotic DNA damage response (Zhang et al., 2003). Apoptin may also interfere with cell cycle progression, and since cancer cells frequently lack functional cell cycle check points, this may at least in part be responsible for the selectivity of apoptins’ toxicity (Teodoro et al., 2004). Furthermore, in some cells, apoptin localizes in the nucleoli (Guelen et al., 2004; and data not shown), in which transcription of ribosomal RNA and assembly of ribosomal subunits takes place. Induction of apoptosis by apoptin may therefore be due to interference with the ribosome synthesis process resulting in a shut-down of the cellular biosynthetic activities and final cell death. This assumption would be in line with our observation that apoptin can interact with different ribosomal proteins, as assessed by pull-down experiments with a bacterial GST-apoptin fusion protein (data not shown).
In conclusion, we show that, although apoptin is not cytotoxic for normal cells including HUVEC, its nuclear localization does not necessarily lead to cell death. Activation of caspases is required for apoptosis induction by apoptin. Contrary to previous reports, our findings provide compelling evidence that apoptin-mediated cell death is controlled by Bcl-2 proteins that regulate activation of the Apaf-1 apoptosome. Since apoptin induces apoptosis specifically in tumor cells, it might be an interesting lead molecule for cancer therapy development and useful to delineate the mechanisms of oncogenic transformation.
Materials and methods
Cells and reagents
DU145 prostate carcinoma cells were cultured in DMEM, MCF-7 breast carcinoma cells, the acute myeloid leukemia cells KG-1 and K562, immunoblastic B-cell lymphoma line DOHH-2 and D283 medulloblastoma cells in RPMI-1640, supplemented with 10% heat-inactivated fetal calf serum (FCS) and antibiotics (Gibco BRL, Eggenstein, Germany). Early passage HUVECs were obtained from the core facility of our department, and maintained in M199 medium containing 20% FCS, 2mM glutamine, antibiotics and 10 ng/ml basic fibroblast growth factor (Sigma, Deisenhofen, Germany). MCF-7 cells stably transfected with caspase-3 were a kind gift from RU Jänicke, and D283 cells stably expressing Bcl-xL (Poppe et al., 2001) and a vector control were provided by J Prehn. MCF-7 cells stably transfected with Bcl-2 or Bcl-xL (Jaattela et al., 1995) were obtained from M Jäättelä. MCF-7 expressing GFP-cytochrome c and DU145 cells stably transfected with Bax have been described (Gillissen et al., 2003). Primary Apaf1−/− fibroblasts (Cecconi et al., 1998) and the respective control cells were provided by F Cecconi and immortalized by retroviral transduction with a temperature-sensitive simian virus 40 large T antigen as described (Almazan and McKay, 1992). The broad-range caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (zVAD-fmk) was from Enzyme Systems (Dublin, CA, USA) and the fluorogenic caspase substrate N-acetyl-Asp-Glu-Val-Asp-aminomethylcoumarin (Ac-DEVD-AMC) from Biomol (Hamburg, Germany). MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and β-estradiol were from Sigma. Bacterial expression plasmids for the production of recombinant Tat-apoptin and Tat-GFP were obtained from Dr Tavassoli. Both proteins were expressed in bacteria as described (Guelen et al., 2004).
cDNAs, vectors, siRNA and transient transfections
Apoptin cDNA was inserted into the BamHI cloning site of pEGFP-C1 and pDsRed1-C1 vectors (Clontech, Palo Alto, CA, USA). The correct cloning was confirmed by restriction digestion and sequence analysis. Transfections were performed using lipofectamine reagent according to the manufacturer’s instructions (Gibco BRL). The broad-range caspase inhibitor zVAD-fmk was added to the cells at a concentration of 30 μM immediately after transfection and maintained during subsequent changes of medium. Sense and antisense oligonucleotides siRNA oligonucleotides corresponding to nucleotides 978–998 of Apaf-1 (AATTGGTGCACTTTTACGTGA (Lassus et al., 2002) were purchased from Qiagen (Hilden, Germany) and annealed to create the double-stranded siRNAs. Transfection of MCF-7/caspase-3 cells grown at 40% confluency was performed with TransMessenger™ reagent according to manufacturer’s instructions (Qiagen).
Fluorescence microscopy
To monitor expression of fluorescent proteins, cells were grown on coverslips followed by transfection with the respective plasmids. The cells were washed twice with PBS and fixed with 3% formaldehyde in PBS for 20 min at room temperature. Cell nuclei were stained with DAPI for 15min. After extensive washing with PBS the glass slices were mounted with Hydromount™ (National Diagnostics, Atlanta). Fluorescence was detected using an Eclipse TE 300 inverted microscope and a ×40 oil immersion objective (Nikon, Düsseldorf, Germany) equipped with the appropriate filters (excitation: 490 nm, dichroic mirror: 505 nm, emission: 510 nm; or excitation: 510–560 nm, dichroic mirror: 575 nm, emission: 590 nm). Digital images were acquired with a SPOT-2 camera using Spot-2.2.1 software (Diagnostic Instruments, Sterling Heights, USA).
Cell death assessment and DEVDase assay
Cells were transfected with GFP-tagged apoptin (1 μg) expressed from the pEGFP-C1 vector. After the indicated time post-transfection cells were fixed with 3% formaldehyde in PBS, stained with DAPI and examined microscopically. For each time point at least 300 green cells were scored; the percentage of live (flat with intact nuclei) and apoptotic cells (round cells with condensed chromatin) was determined. Cytotoxicity of cell-permeable Tat-apoptin and a Tat-GFP control was measured using the MTT assay that was performed exactly as described (Ghavami et al., 2004). Fluorimetric assay of caspase activity (DEVDase assay) was performed as previously described (Essmann et al., 2003) using the fluorogenic caspase substrate Ac-DEVD-AMC. The release of aminomethylcoumarin was measured by fluorometry (Bio-Tek FL600; Bio-Tek Instruments, USA) using an excitation wavelength of 360 nm and an emission wavelength of 475 nm. The catalytic activities are given in arbitrary units.
Cell extracts and immunoblotting
Cleavage of caspase-3, -7 and poly(ADP-ribose)polymerase-1 (PARP-1) was detected by immunoblotting (Los et al., 2002). Briefly, 2 × 105 cells were transfected with pEGFP-C1 vector alone or with pEGFP-C1-apoptin expression vectors. The expression of GFP and GFP-apoptin was confirmed by fluorescence microscopy. Then, the cells were washed with cold PBS and lysed in 1% NP-40 buffer supplemented with protease inhibitors. Following SDS–PAGE and transfer, membranes were blocked for 1 h with 5% nonfat dry milk powder in TBS and then incubated overnight with the following primary antibodies: anti-caspase-3 recognizing the full-length protein (Transduction Laboratories, Heidelberg, Germany), antiactive caspase-3 (Cell Signaling Technology, Beverly, USA), anticaspase-7 (Alexis, Lausen, Switzerland), antiactive caspase-7 (Cell Signaling), anti-PARP-1 (Roche Diagnostics, Mannheim, Germany), anti-Apaf-1 (Pharmingen) and anti-Bcl-2 (Upstate). Following incubation for 1 h with the corresponding secondary antibodies and extensive washing with TBS, 0.02% Triton X-100 buffer, specific protein signals were visualized by enhanced chemiluminescent staining using ECL reagents (Amersham-Pharmacia Biotech).
Acknowledgments
This work was in part supported by grants from the ‘Deutsche Krebshilfe’ and the DFG. ML is supported by CIHR ‘Canada Research Chair’ and MHRC programs. SM acknowledges the generous support from CCMF. We are grateful to Drs J Prehn, RU Jänicke, F Prinz, F Cecconi, M Jäättela, LC Murphy for providing cell lines, expertise and reagents. We are in debt also to A Kemp and A Kania for help with experiments.
Abbreviations
- AMC
7-aminomethyl-4-coumarin
- CAV
chicken anemia virus
- GFP
green fluorescent protein
- HUVEC
human umbilical vein endothelial cell
- zVAD-fmk
benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone
References
- Adair BM. Dev Comp Immunol. 2000;24:247–255. doi: 10.1016/s0145-305x(99)00076-2. [DOI] [PubMed] [Google Scholar]
- Almazan G, McKay R. Brain Res. 1992;579:234–245. doi: 10.1016/0006-8993(92)90056-f. [DOI] [PubMed] [Google Scholar]
- Antonsson B, Martinou JC. Exp Cell Res. 2000;256:50–57. doi: 10.1006/excr.2000.4839. [DOI] [PubMed] [Google Scholar]
- Bitzer M, Armeanu S, Prinz F, Ungerechts G, Wybranietz W, Spiegel M, et al. J Biol Chem. 2002;277:29817–29824. doi: 10.1074/jbc.M111898200. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Strasser A. J Cell Sci. 2002;115:1567–1574. doi: 10.1242/jcs.115.8.1567. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez Bolado G, Meyer BI, Roth KA, Gruss P. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Danen-Van Oorschot AA, Fischer DF, Grimbergen JM, Klein B, Zhuang S, Falkenburg JH, et al. Proc Natl Acad Sci USA. 1997;94:5843–5847. doi: 10.1073/pnas.94.11.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danen-Van Oorschot AA, van der Eb AJ, Noteborn MH. Adv Exp Med Biol. 1999;457:245–249. doi: 10.1007/978-1-4615-4811-9_26. [DOI] [PubMed] [Google Scholar]
- Danen-van Oorschot AA, van der Eb AJ, Noteborn MH. J Virol. 2000;74:7072–7078. doi: 10.1128/jvi.74.15.7072-7078.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danen-Van Oorschot AA, Zhang YH, Leliveld SR, Rohn JL, Seelen MC, Bolk MW, et al. J Biol Chem. 2003;278:27729–27736. doi: 10.1074/jbc.M303114200. [DOI] [PubMed] [Google Scholar]
- Daniel PT, Schulze-Osthoff K, Belka C, Guner D. Essays Biochem. 2003;39:73–88. doi: 10.1042/bse0390073. [DOI] [PubMed] [Google Scholar]
- Essmann F, Bantel H, Totzke G, Engels IH, Sinha B, Schulze-Osthoff K, et al. Cell Death Differ. 2003;10:1260–1272. doi: 10.1038/sj.cdd.4401301. [DOI] [PubMed] [Google Scholar]
- Everett H, McFadden G. Virology. 2001;288:1–7. doi: 10.1006/viro.2001.1081. [DOI] [PubMed] [Google Scholar]
- Fischer U, Janicke RU, Schulze-Osthoff K. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Schulze-Osthoff K. Pharmacol Rev. 2005;57:187–215. doi: 10.1124/pr.57.2.6. [DOI] [PubMed] [Google Scholar]
- Ghavami S, Kerkhoff C, Los M, Hashemi M, Sorg C, Karami-Tehrani F. J Leukoc Biol. 2004;76:169–175. doi: 10.1189/jlb.0903435. [DOI] [PubMed] [Google Scholar]
- Gillissen B, Essmann F, Graupner V, Starck L, Radetzki S, Dorken B, et al. EMBO J. 2003;22:3580–3590. doi: 10.1093/emboj/cdg343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Guelen L, Paterson H, Gaken J, Meyers M, Farzaneh F, Tavassoli M. Oncogene. 2004;23:1153–1165. doi: 10.1038/sj.onc.1207224. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Piccart M. Ann Oncol. 2000;11:647–663. doi: 10.1023/a:1008390429428. [DOI] [PubMed] [Google Scholar]
- Haraguchi M, Torii S, Matsuzawa S, Xie Z, Kitada S, Krajewski S, et al. J Exp Med. 2000;191:1709–1720. doi: 10.1084/jem.191.10.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaattela M, Benedict M, Tewari M, Shayman JA, Dixit VM. Oncogene. 1995;10:2297–2305. [PubMed] [Google Scholar]
- Kane DJ, Ord T, Anton R, Bredesen DE. J Neurosci Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Declercq W, Depuydt B, Kalai M, Saelens X, Vandenabeele P. In: Caspases – Their Role in Cell Death and Cell Survival. Los M, Walczak H, editors. Kluwer Academic Press; New York: 2002. [Google Scholar]
- Lassus P, Opitz-Araya X, Lazebnik Y. Science. 2002;297:1352–1354. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A, et al. Mol Biol Cell. 2002;13:978–988. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los M, Wesselborg S, Schulze Osthoff K. Immunity. 1999;10:629–639. doi: 10.1016/s1074-7613(00)80062-x. [DOI] [PubMed] [Google Scholar]
- Marsden VS, O’Connor L, O’Reilly LA, Silke J, Metcalf D, Ekert PG, et al. Nature. 2002;419:634–637. doi: 10.1038/nature01101. [DOI] [PubMed] [Google Scholar]
- Marsden VS, Strasser A. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, et al. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- Noteborn MH, Todd D, Verschueren CA, de Gauw HW, Curran WL, Veldkamp S, et al. J Virol. 1994;68:346–351. doi: 10.1128/jvi.68.1.346-351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noteborn MH, Zhang YH, van der Eb AJ. Mutat Res. 1998;400:447–455. doi: 10.1016/s0027-5107(98)00016-5. [DOI] [PubMed] [Google Scholar]
- Oro C, Jans DA. Curr Drug Targets. 2004;5:179–190. doi: 10.2174/1389450043490631. [DOI] [PubMed] [Google Scholar]
- Poon IK, Oro C, Dias MM, Zhang JP, Jans DA. J Virol. 2005;79:1339–1341. doi: 10.1128/JVI.79.2.1339-1341.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe M, Reimertz C, Dussmann H, Krohn AJ, Luetjens CM, Bockelmann D, et al. J Neurosci. 2001;21:4551–4563. doi: 10.1523/JNEUROSCI.21-13-04551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- Reed JC, Green DR. Mol Cell. 2002;9:1–3. doi: 10.1016/s1097-2765(02)00437-9. [DOI] [PubMed] [Google Scholar]
- Rohn JL, Zhang YH, Aalbers RI, Otto N, Den Hertog J, Henriquez NV, et al. J Biol Chem. 2002;277:50820–50827. doi: 10.1074/jbc.M208557200. [DOI] [PubMed] [Google Scholar]
- Schoop RA, Kooistra K, Baatenburg De Jong RJ, Noteborn MH. Int J Cancer. 2004;109:38–42. doi: 10.1002/ijc.11675. [DOI] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Eur J Biochem. 1998;254:439–459. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- Schwerk C, Schulze-Osthoff K. Mol Cell. 2005;19:1–13. doi: 10.1016/j.molcel.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Somai S, Chaouat M, Jacob D, Perrot JY, Rostene W, Forgez P, et al. Int J Cancer. 2003;105:607–612. doi: 10.1002/ijc.11147. [DOI] [PubMed] [Google Scholar]
- Strasser A, O’Connor L, Dixit VM. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- Stroh C, Schulze-Osthoff K. Cell Death Differ. 1998;5:997–1000. doi: 10.1038/sj.cdd.4400451. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- Tavassoli M, Guelen L, Luxon BA, Gaken J. Apoptosis. 2005;10:717–724. doi: 10.1007/s10495-005-0930-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodoro JG, Heilman DW, Parker AE, Green MR. Genes Dev. 2004;18:1952–1957. doi: 10.1101/gad.1198404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson BJ. Int J Exp Pathol. 2001;82:65–76. doi: 10.1111/j.1365-2613.2001.iep0082-0065-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia JS, Wagner MV, Ezhevsky SA, Dowdy SF. J Virol. 2004;78:6077–6078. doi: 10.1128/JVI.78.11.6077-6078.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Leliveld SR, Kooistra K, Molenaar C, Rohn JL, Tanke HJ, et al. Exp Cell Res. 2003;289:36–46. doi: 10.1016/s0014-4827(03)00188-5. [DOI] [PubMed] [Google Scholar]
- Zheng TS, Flavell RA. Exp Cell Res. 2000;256:67–73. doi: 10.1006/excr.2000.4841. [DOI] [PubMed] [Google Scholar]
- Zhuang SM, Shvarts A, Jochemsen AG, van Oorschot AA, van der Eb AJ, Noteborn MH. Carcinogenesis. 1995;16:2939–2944. doi: 10.1093/carcin/16.12.2939. [DOI] [PubMed] [Google Scholar]