

Abstract
The ultrafast excited-state dynamics underlying the receptor state photorecovery is resolved in the M100A mutant of the photoactive yellow protein (PYP) from Halorhodospira halophila. The M100A PYP mutant, with its distinctly slower photocycle than wt PYP, allows isolation of the pB signaling state for study of the photodynamics of the protonated chromophore cis-p-coumaric acid. Transient absorption signals indicate a subpicosecond excited-state proton-transfer reaction in the pB state that results in chromophore deprotonation prior to the cis–trans isomerization required in the photorecovery dynamics of the pG state. Two terminal photoproducts are observed, a blue-absorbing species presumed to be deprotonated trans-p-coumaric acid and an ultraviolet-absorbing protonated photoproduct. These two photoproducts are hypothesized to originate from an equilibrium of open and closed folded forms of the signaling state, I2 and I2’.
Photochromic photoreceptor proteins can be switched between different receptor states in response to different colors of applied light. While most photoreceptors progress (often as photocycles) through a series of metastable conformations with different absorption spectra,1 photochromic proteins exhibit light-triggered recovery pathways that “short-circuit” the thermal kinetics and accelerate the recovery of the dark-adapted receptor state by orders of magnitude. For example, plant phytochromes switch between red-absorbing (Pr) and far-red-absorbing (Pfr) states by photoisomerization of a bound bilin chromophore.1 While thermal reversion of the Pfr state occurs on a time scale of hours,2 absorption of a far-red photon recovers the Pr state within milliseconds.3 In slow-cycling photoreceptors, photoreversion is relevant to in vivo activity because each state has a comparable probability of absorbing a photon.4 Knowledge of the molecular mechanisms of photochromism, which requires characterization of the primary photochemical events, is therefore important for constructing a microscopic understanding of the complete activity of photochromic proteins.
PYP is a small, water-soluble photoreceptor first found in the extremophilic bacterium Halorhodospira halophila (Hh), where it is presumed to be responsible for blue-light phototactic activity.5 PYP has become a model system for characterization of the chromophore photochemistry responsible for initiating signal transduction. PYP domains are characterized by a p-coumaric acid (pCA) chromophore that is covalently bound to a conserved PAS (Per-Arnt-Sim) domain structure.6 In all known PYPs, the dark-adapted state (pG) is thought to contain the trans isomer of the pCA chromophore, which is stabilized by hydrogen bonds with several highly conserved residues within the binding pocket.6-9 The forward photocycle (Scheme 1) is initiated by a trans–cis isomerization on a picoseconds time scale, followed (in most PYPs) by much slower ground-state deprotonation to form the light-adapted signaling state (pB).
Scheme 1.
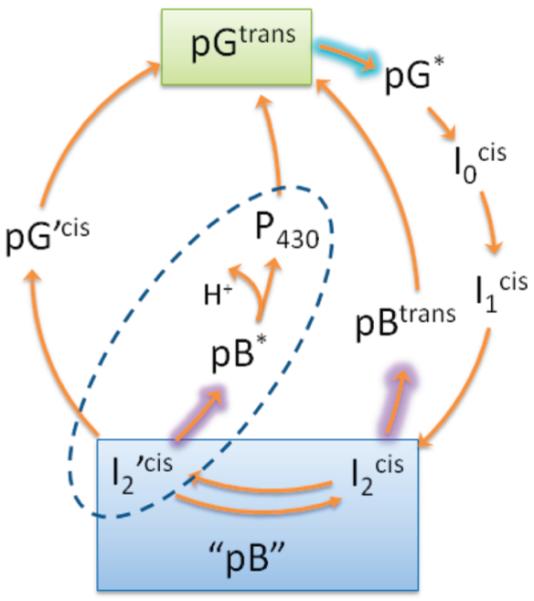
PYP Photocycle Indicating the Dark-Adapted State (pG), the Light-Adapted State (pB), and Photointermediatesa
aThe isomerization state of the p-coumaric acid chromophore is denoted. In the pH range of 3–10, the pB intermediates have a protonated chromophore, while pG, I0, and I1 intermediates are deprotonated.10-12 The dashed ellipse shows the photoreversal pathway investigated in this paper, while the pBtrans pathway is based on previous work.13
Photochromism in PYP was first demonstrated in wild-type Hh PYP14 and later in the M100A mutant of Hh PYP.5 While the dark recovery in M100A PYP has a half-life of 5.5 min, 365 nm irradiation accelerates pG recovery by 6 orders of magnitude.5 A four-order of magnitude photoactivated increase in pG recovery was reported in wild-type Hh PYP.12,13 In both cases, the mechanism of photorecovery was presumed to be photoinduced cis–trans isomerization of the pCA chromophore, in large part because thermal cis–trans isomerization was long considered to be the rate-limiting step in nonphotoactivated pG recovery.5,13,15,16 However, recent studies on pH dependence and kinetic isotope effects in the dark recovery dynamics of PYP have suggested that deprotonation of pCA, not isomerization, is likely the rate-limiting step for pG recovery.10,17 Determining the mechanism of pG photorecovery has been further complicated by the observation of several pB intermediates existing in a pH- and salt-dependent equilibrium, which exhibit different photorecovery kinetics (Scheme 1).11-13,17,18 Essentially no information is available for the excited-state dynamics underlying the photoreversal phenomenon.
In this Letter, the M100A mutant of Hh PYP was used to resolve the pB photoreversal dynamics on the ultrafast (100 fs to 3 ns) time scale. Although M100A has similar steady-state pG absorption spectrum as wt PYP (Figure 1), it has a distinctly slower dark recovery of the pG state (42 min vs 1 s).5,19 The long recovery time allowed generation of a photoequilibrium under 440 nm light that is nearly 100% in the pB state. Continuous irradiation with 440 nm light (which the pB state does not absorb) excludes any interfering pG signals that would plague the faster-cycling wt PYP.
Figure 1.
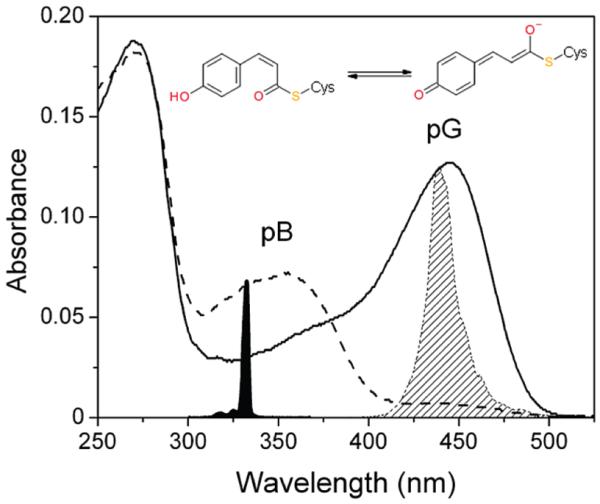
Steady-state absorption spectra of the pG (solid curve) and the pB (dashed curve) state of the M100A mutant of PYP are shown with the corresponding structures of the pCA chromophore. The slight shoulder at 360 nm in the pG spectrum is due to partial photoconversion of the sample during spectrophotometry. The spectra of the 330 nm femtosecond laser pulse (filled) and 440 nm LED (diagonal hatched) are indicated.
The measured femtosecond transient absorption signals of the pB state in M100A PYP (Figure 2) demonstrate rich dynamics. The kinetics varies strongly with probe wavelength. The early transient spectra include an excited-state absorption band from 360 to 425 nm with a negative stimulated emission band between 425 and 600 nm (Figure 2a). Excited-state absorption largely obscures the bleach of the ultraviolet absorption of pB. The excited-state signals decay within 150 ps, after which the transient spectrum plateaus to a broad 0.5 mOD absorption between 430 and 740 nm with a negative band at 377 nm (Figure 2a, inset). This spectrum does not change from 200 ns to the ~3.3 ns time range of the experiment.
Figure 2.

Transient absorption data from M100A mutant. The sample was excited at 332 nm with 270 nJ. (a) Transient spectra (100 fs–100 ps) show evolution of the stimulated emission in the region of 450–600 nm. Inset: Spectral evolution between 200 ps and 3 ns. (b) Kinetic traces with fits from global analysis. Black lines correspond to fits from global analysis. (c) Evolution associated difference spectra (EADS) coexisting with a fourth static spectrum (designated ∞) estimated by means of global analysis.
Global analysis of the wavelength-dependent transient data was used to characterize spectral changes associated with differing time scales.20 The data were fit to a set of first-order rate equations representing a simple model of the underlying dynamics (Supporting Information). The global fit results in evolution associated difference spectra (EADS), which are composite difference spectra that describe a kinetics feature generally linked to an excited-state decay process but which are not necessarily the difference spectra of populations of two pure molecular states (i.e., species associated difference spectra or SADS).20 The M100A data are well described with three EADS decaying sequentially with exponential time constants of 1.9, 15, and 120 ps, which coexist with a fourth static spectrum (designated ∞) (Figure 2c). The three transient EADS all exhibit stimulated emission components, indicating multiexponential excited-state kinetics. The similarity of the EADS suggests the possibility of either continuous evolution of a single-species spectrum (e.g., solvation) or evolution between multiple species with similar spectral properties (e.g., cis to trans isomerization). The processes accompanying these time constants are interpreted by examining the spectral characteristics of the EADS.
A prominent feature in all of the EADS is that the stimulated emission band occurs at significantly lower energy than other PYP samples containing a protonated cis-pCA chromophore.21-23 For example, the fast-cycling PYP from Rhodobacer sphaeroides, which contains a thermal equilibrium of pG and pB states, exhibits two emission bands; fluorescence excitation spectra have indicated that a fluorescence band peaking at 440 nm originates from the pB state with protonated cis-pCA, while a second fluorescence band at 500 nm was ascribed to the pG state with deprotonated trans-pCA.21 Acid denaturation studies in wt Hh PYP have also shown that emission from the protonated forms of both isomers of pCA peak between 420 and 430 nm.22,23 PYP denatured with guanidinium chloride at high pH, and containing anionic pCA, has emission peaking at 500 nm.24 Red-shifted emission from the anionic form compared to the neutral form is also observed in isolated pCA and similar chromophores in solution.25,26 The stimulate demission in the 1.9 ps EADS extends to 600 nm, making this emission spectrum comparable to deprotonated trans-pCA in the pG state.27 Because the 1.9 ps EADS forms near-instantaneously after excitation (within the instrument response), the excitedstate population in M100A is, at least partially, deprotonated within ~300 fs through an excited-state proton-transfer (ESPT) reaction.
Analysis of the 15 ps EADS provides further evidence of rapid ESPT in M100A. In Figure 3, the EADS from the M100A pB state data are compared to SADS obtained from wt PYP pG state data (Supporting Information).27 The difference of these difference spectra (EADSpB – SADSpG) closely resembles the difference spectrum of the static ground-state absorption spectra, pG – pB. This comparison indicates that the transient spectra obtained from Hh PYP in the pG and pB states differ only in their ground-state bleach components, implying that the excited-state absorption spectrum and stimulated emission from pG* and pB* are nearly identical. Such an observation is remarkable considering that the chromophores in these two states initially differ in geometry, protonation, and local protein structure. While previous studies have suggested that distinguishing between isomers could be difficult because the cis-and trans-pCA ground-state absorption spectra differ more in extinction coefficient than peak wavelength,10,22,23 the 50–150 nm blue shift in ground-state absorption and emission due to protonation of pCA is unmistakable. The nearly indistinguishable excited-state signals in the pB state of M100A PYP (after 335 nm excitation) and the pG state of wt PYP (after 440 nm excitation) are strong evidence that the pCA chromophore is fully deprotonated and relaxed in the excited state in the 15 ps EADS. The decay of the partially deprotonated 1.9 ps EADS puts an upper limit on the time scale of formation of the 15 ps EADS, but the 1.9 ps time constant is likely related to structural relaxation in the excited state.
Figure 3.
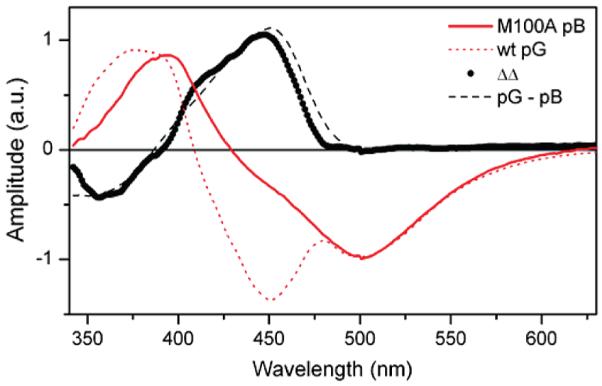
Comparison of 15 ps EADS from the pB state of M100A (red, solid) with SADS obtained from photoexcitation of the pG state in wild-type PYP (red, dotted). The difference spectrum ΔΔ (black, circles) indicates that the pB and pG states have identical picosecond emission spectra, suggesting that the predominate fluorescent excited-state species in photoexcited pB has a deprotonated chromophore. It resembles the static difference spectrum of pG and pB (black, dashed).
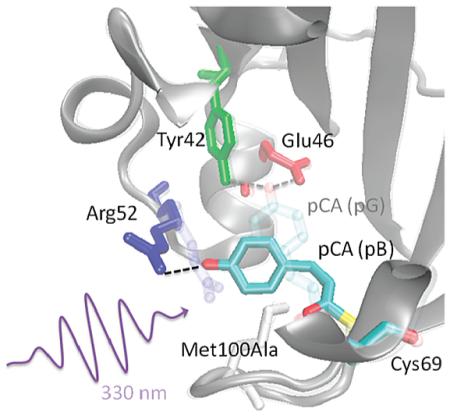
Subpicosecond ESPT indicates that the proton-transfer reaction has a low barrier in the excited state, suggesting a large photoinduced change in pKa or a highly favorable hydrogen acceptor–donor geometry. For instance, an ESPT reaction occurring within 50 fs was recently reported28 in the S65T/H148D mutant of green fluorescent protein (GFP). The GFP chromophore is theoretically predicted to be a strong photoacid,29 with the large decrease in the pKa of the hydroxyl group upon photoexcitation facilitating the proton-transfer kinetics. Furthermore, the hydrogen donor and acceptor groups are in unusually close proximity in this mutant, with the phenol hydroxyl group of the GFP chromophore forming a 2.4 Å hydrogen bond with D148. In wt GFP, solvent-mediated ESPT from the chromophore to Glu222 (4.55 Å) occurs on a slower, but still rapid, time scale of 2–3 ps.30 The PYP chromophore pCA is also predicted to be a strong photoacid based on the large difference in transition energies between neutral and ionized states.29 Additionally, Stark spectra of the pB state of M100A have shown that pCA undergoes a large change in static dipole moment (|Δμ| = 19 D) upon excitation,15 resulting in a decrease in charge density on the hydroxyl oxygen that would acidify this group in the excited state. A pKa* value substantially lower than the pKa of trans-pCA (~9) would promote ESPT to a nearby proton acceptor. Structural models based on time-resolved X-ray crystallography on wt PYP indicate that Arg52 forms a hydrogen bond with the hydroxyl group of cis-pCA in the pB state (PDB: 1TS0)31 and is a potential proton acceptor. On the other hand, solution structures of the pB state, based on the N-terminal truncate (Δ25 PYP), argue a high degree of disorder in residues 42–56 (PDB: 1XFQ)32 that would make specific hydrogen bonds between pCA and the protein unlikely. Kinetic isotope effect studies and additional mutant studies will be necessary to determine the identity of the proton acceptor in M100A PYP.26
We have interpreted the spectral evolution between the 1.9 and 15 ps EADS as structural relaxation of the chromophore, but we note that the spectroscopic data do not conclusively support complete isomerization in the excited state. Recently, Martínez and co-workers argued that acid–base chemistry and photoisomerization dynamics are coupled in the GFP chromophore, which shares the same phenolate monomethine motif as pCA.33 In the GFP simulations, as in many other theoretical and experimental studies,34 photoisomerization proceeds through conical intersections with the ground electronic state and is therefore necessarily coupled to a loss of excited-state population. In the present M100A data, the persistence of stimulated emission (an excited-state signal) in the 15 and 120 ps EADS clearly indicates that the excited electronic state is not strongly quenched with the 1.9 ps time constant. Therefore, we do not believe that chromophore deprotonation and isomerization are coupled in the case of M100A PYP. Cis–trans isomerization more likely occurs after deprotonation, possibly associated with the slower 15 and 120 ps excited-state decay constants. Notably, these time scales are at least a factor of 3 longer than those observed in the forward trans–cis reaction. The slower reverse photoisomerization likely reflects the role of the binding pocket in stabilizing isomerization in the folded protein, particularly by lowering an excited-state activation barrier.
Although evidence of isomerization in the ultrafast experiments is indirect, long-lived photoproducts are observed in the “terminal” (> 300 ps) transient spectrum (Figure 2c). Because the red absorption band in the static EADS was consistent with a solvated electron from a two-photon ionization process, observed in previous ultrafast studies on the pG state27 and isolated chromophores in solution,35-37 the intensity dependence of the 300 ps transient spectrum was measured (Figure 4a). The shape of the spectrum is relatively flat, with distinct bands at 430 and 640 nm. When the power-dependent spectra are normalized at the red band, the 430 nm peak exhibits different power dependence than the red band, indicating that the visible region of the 300 ps spectrum consists of contributions from two photoproducts with different power dependence. Global analysis of the data using a spectral model resolves two spectra, (1) a600 nm band with a quadratic power dependence (Supporting Information) expected for two-photon photoionization27 and (2) a band with a peak at 430 nm representing a population with linear power dependence at low pulse energies. The 430 nm band is ascribed to an unidentified photocycle intermediate, P430, in which the red-shifted spectrum suggests that the species has a deprotonated pCA chromophore (e.g., in pG). The kinetics of P430 formation is obscured by overlapping photoionization and excited-state signals; however, generation of a deprotonated photoproduct is consistent with the observed ESPT reaction discussed above.
Figure 4.
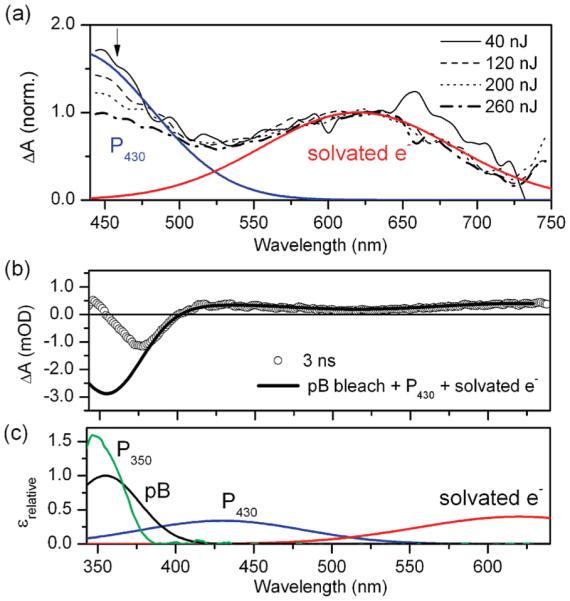
(a) Power dependence study of the 300 ps transient absorption spectrum of M100A, showing spectra normalized at 640 nm. Excitation energies varied from 40 to 260 nJ. SVD analysis indicated two species with difference power dependence assigned to solvated electron and a primary photoproduct P430. (b) Comparison of the terminal spectrum (3 ns, circles) with the pB bleach, P430, and the solvated electron contributions indicates absorption of an UV photoproduct. (c) Absorption spectra of species present at ~10 ns, pB, P430, solvated electron, and the deduced UV photoproduct P350.
The populations extracted from the SVD analysis were also used to decompose the broad-band terminal EADS from the global analysis of the transient data (Figure 2c). The negative signal at 377 nm is attributed to the ground-state bleach of pB. Since the absorption peak of the pB state is located at 355 nm, a photoproduct absorption with maximum absorption at around 350 nm is deduced (Figure 4b). This unidentified photoproduct, P350, peaks in the UV, suggesting that the chromophore is protonated. Moreover, its spectrum is very similar to a protonated trans-pCA species (pBtrans) observed in wild-type Hh PYP following 355 nm laser excitation of a steady-state mixture of pB and pG populations.13
Protonated and deprotonated photoreversal pathways are not necessarily conflicting observations; rather, two photointermediates could potentially arise from conformational heterogeneity. It is well-documented in wt PYP that the pB state consists of two states I2’ and I2 in a pH-dependent equilibrium (Scheme 1); the pK ≈ 5.9–6.4 is thought to be associated with protonation of Glu46.10,18,38 Far-UV circular dichroism measurements indicate that these states have different degrees of helical secondary structure,38 while transient absorption measurements show that the states follow different photorecovery kinetics.18 For the M100A mutant, dark recovery is known to be pH-dependent, with pK values of 6.5 and 10.39 At 7.4 pH, the conditions of our M100A experiment, the photoequilibrium mixture is 80–90% I2’, where the protein is in a partially unfolded conformation called the “open folded” signaling state.10 In contrast, previous photoreversal studies on wt PYP (in which pBtrans was observed) were conducted at pH 5.6,13 where 80–90% of the signaling state is I2, which has a folded protein structure. The difference in folding states would have a significant effect on the pKa of pCA and neighboring residues and could affect chromophore photochemistry. Additional pH dependence studies will elucidate this question.
Unfortunately, our data do not conclusively rule out the possibility that P350 is a photoionization product. Two-photon ionization in wild-type PYP,27 as well as solutions of isolated pCA35 and chromophore analogues,37,40 has been observed to form a radical species with absorption peaks near 370 nm.35,37 Radical formation should follow a quadratic power dependence like the solvated electron band at 600 nm; however, poor signal-to-noise in the UV region of the data prohibited measurement of a power dependence of P350. The simple kinetics model presented in this Letter is insufficient for separating P350 kinetics from excited-state absorption kinetics.
In conclusion, the photodynamics of the pB state in the M100A mutant of PYP was studied by femtosecond transient absorption. Spectral and kinetic analysis indicated that in the initial photoreaction, the chromophore undergoes a rapid subpicosecond ESPT with chromophore isomerization occurring on 15 and 120 ps time scales. A long-lived deprotonated photoproduct absorbing at 430 nm and an UV-absorbing product in which the chromophore remains protonated were observed. Future studies will further explore the nature of the ESPT during the photorecovery of PYP with deuterium kinetic isotope effect and pH dependence studies.
EXPERIMENTAL SECTION
Expression, Reconstitution, and Purification of PYP
Histidine-tagged M100A apoPYP from H. halophila was overexpressed in E. coli (BL21 DE3) and extracted with 8 M urea, as described previously41 with modifications. The apoPYP was diluted two-fold using a 10 mM Tris-HCl buffer at pH 7.5 buffer and was reconstituted by the addition of an excess of p-coumaric acid anhydride. After removal of the urea by dialysis against a 10 mM Tris-HCl buffer at pH 7.5, the protein was purified by chromatography on Ni-NTA resin (using 200 mM imidazol as the eluent) and DEAE-sepharose CL6B (using 100 mM NaCl for elution) until a purity index of 0.6 was achieved.
Ultrafast Spectroscopy
Upon blue-light irradiation of PYP-M100A, the pG population with an absorption spectrum peaking at 444 nm shifts to the pB population with a blue-shifted 355 nm peak.17 In the absence of light, the pG state thermally recovers with a 42 min time constant it the dark in PYP-M100A. To maintain an excess of pB state during the ultrafast transient absorption measurements and prevent signal contamination from recovered pG, the sample was continuously irradiated with CW 430 nm light from a high-power LED (720 mW). The PYP-M100A sample was exposed for 5 min to the LED prior to collecting data to ensure that the measured dynamics originated from photoexcitation of the pB state without contamination from the pB’ state.42 The protein sample was buffered with 10 mM Tris-HCl buffer at pH 7.4. The absorption spectra of the samples were measured before and after data collection, with no significant changes observed. The data were collected at room temperature.
Femtosecond transient absorption spectra were collected with an ultrafast spectrometer.43 Pump pulses at 332 nm were generated by sum frequency generation of the laser fundamental (800 nm, 40 fs, 270 μJ) and 537 nm pulses (50 fs, 1.9 μJ) generated by a noncollinear optical parametric amplifier, overlapped with an external angle of ~1.6° in a 1 mm BBO crystal. After passing through a UG-11 transmission filter, the pump pulses were 300 nJ, the bandwidth was 5 nm (Full Width at Half Maximum), and the pulse duration was ~300 fs. White-light probe pulses were generated by focusing part of the fundamental into a CaF2 window, with magic angle polarization. The sample was inserted into a 1 mm quartz cell, which was translated by a motorized actuator. Transmitted probe pulses were focused into a spectrograph and the signals recorded with a homemade silicon diode array detector system.
Supplementary Material
ACKNOWLEDGMENT
This work was supported (D.S.L.) with a Career Development Award (CDA0016/2007-C) from the Human Frontiers Science Program Organization and support from NIH Grant GM063805 and OCAST Grant HR07-135S and from startup funds provided by Oklahoma State University (W.D.H.).
Footnotes
SUPPORTING INFORMATION AVAILABLE Details of global analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
SECTION Biophysical Chemistry
REFERENCES
- (1).Briggs WR, Spudich JL. Handbook of Photosensory Receptors. Wiley-VCH; New York: 2005. [Google Scholar]
- (2).Briggs WR, Rice HV. Phytochrome — Chemical and Physical Properties and Mechanism of Action. Annu. Rev. Plant Physiol. 1972;23:293. [Google Scholar]
- (3).Chen EF, Lapko VN, Lewis JW, Song PS, Kliger DS. Mechanism of Native Oat Phytochrome Photoreversion: A Time-Resolved Absorption Investigation. Biochemistry. 1996;35:843–850. doi: 10.1021/bi952115z. [DOI] [PubMed] [Google Scholar]
- (4).Kennis JT, van Stokkum IH, Crosson S, Gauden M, Moffat K, van Grondelle R. The LOV2 Domain of Phototropin: A Reversible Photochromic Switch. J. Am. Chem. Soc. 2004;126:4512–4513. doi: 10.1021/ja031840r. [DOI] [PubMed] [Google Scholar]
- (5).Devanathan S, Genick UK, Canestrelli IL, Meyer TE, Cusanovich MA, Getzoff ED, Tollin G. New Insights into the Photocycle of Ectothiorhodospira halophila Photoactive Yellow Protein: Photorecovery of the Long-Lived Photobleached Intermediate in the Met100Ala Mutant. Biochemistry. 1998;37:11563–11568. doi: 10.1021/bi9803776. [DOI] [PubMed] [Google Scholar]
- (6).Hellingwerf K, Hendriks J, Gensch T. Photoactive Yellow Protein, A New Type of Photoreceptor Protein: Will This “Yellow Lab” Bring Us Where We Want to Go? J. Phys. Chem. A. 2003;107:1082–1094. [Google Scholar]
- (7).Kyndt JA, Fitch JC, Meyer TE, Cusanovich MA. Thermochromatium tepidum Photoactive Yellow Protein/Bacteriophytochrome/Diguanylate Cyclase: Characterization of The PYP Domain. Biochemistry. 2005;44:4755–4764. doi: 10.1021/bi047373n. [DOI] [PubMed] [Google Scholar]
- (8).Haker A, Hendriks J, Gensch T, Hellingwerf K, Crielaard W. Isolation, Reconstitution and Functional Characterisation of the Rhodobacter sphaeroides Photoactive Yellow Protein. FEBS Lett. 2000;486:52–56. doi: 10.1016/s0014-5793(00)02242-0. [DOI] [PubMed] [Google Scholar]
- (9).Kumauchi M, Hara MT, Stalcup P, Xie AH, Hoff WD. Identification of Six New Photoactive Yellow Proteins — Diversity and Structure-Function Relationships in a Bacterial Blue Light Photoreceptor. Photochem. Photobiol. 2008;84:956–969. doi: 10.1111/j.1751-1097.2008.00335.x. [DOI] [PubMed] [Google Scholar]
- (10).Hendriks J, Hellingwerf KJ. pH Dependence of the Photoactive Yellow Protein Photocycle Recovery Reaction Reveals a New Late Photocycle Intermediate with a Deprotonated Chromophore. J. Biol. Chem. 2009;284:5277–5288. doi: 10.1074/jbc.M805904200. [DOI] [PubMed] [Google Scholar]
- (11).Joshi CP, Borucki B, Otto H, Meyer TE, Cusanovich MA, Heyn MP. Photocycle and Photoreversal of Photoactive Yellow Protein at Alkaline pH: Kinetics, Intermediates, And Equilibria. Biochemistry. 2006;45:7057–7068. doi: 10.1021/bi0517335. [DOI] [PubMed] [Google Scholar]
- (12).Joshi CP, Borucki B, Otto H, Meyer TE, Cusanovich MA, Heyn MP. Photoreversal Kinetics of the I1 and I2 Intermediates in the Photocycle of Photoactive Yellow Protein by Double Flash Experiments with Variable Time Delay. Biochemistry. 2005;44:656–665. doi: 10.1021/bi0481141. [DOI] [PubMed] [Google Scholar]
- (13).Hendriks J, van Stokkum IH, Crielaard W, Hellingwerf KJ. Kinetics of and Intermediates in a Photocycle Branching Reaction of the Photoactive Yellow Protein from Ectothiorhodospira halophila. FEBS Lett. 1999;458:252–256. doi: 10.1016/s0014-5793(99)01136-9. [DOI] [PubMed] [Google Scholar]
- (14).Miller A, Leigeber H, Hoff WD, Hellingwerf KJ. A Light-Dependent Branching-Reaction in the Photocycle of the Yellow Protein from Ectothiorhodospira-halophila. Biochim. Biophys. Acta. 1993;1141:190–196. [Google Scholar]
- (15).Premvardhan L, van der Horst MA, Hellingwerf KJ, van Grondelle R. How Light-Induced Charge Transfer Accelerates the Receptor-State Recovery of Photoactive Yellow Protein from Its Signaling State. Biophys. J. 2005;89:L64–L66. doi: 10.1529/biophysj.105.075275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Imamoto Y, Harigai M, Morimoto T, Kataoka M. Low-Temperature Spectroscopy of Met100Ala Mutant of Photoactive Yellow Protein. Photochem. Photobiol. 2008;84:970–976. doi: 10.1111/j.1751-1097.2008.00336.x. [DOI] [PubMed] [Google Scholar]
- (17).Hendriks J, van Stokkum IH, Hellingwerf KJ. Deuterium Isotope Effects in the Photocycle Transitions of the Photoactive Yellow Protein. Biophys. J. 2003;84:1180–1191. doi: 10.1016/S0006-3495(03)74932-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Borucki B, Joshi CP, Otto H, Cusanovich MA, Heyn MP. The Transient Accumulation of the Signaling State of Photoactive Yellow Protein Is Controlled by the External pH. Biophys. J. 2006;91:2991–3001. doi: 10.1529/biophysj.106.086645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kumauchi M, Hamada N, Sasaki J, Tokunaga F. A Role of Methionine 100 in Facilitating PYP(M)-Decay Process in the Photocycle of Photoactive Yellow Protein. J. Biochem. 2002;132:205–210. doi: 10.1093/oxfordjournals.jbchem.a003211. [DOI] [PubMed] [Google Scholar]
- (20).van Stokkum IHM, Larsen DS, van Grondelle R. Global and Target Analysis of Time-Resolved Spectra. Biochim. Biophys. Acta. 2004;1657:82–104. doi: 10.1016/j.bbabio.2004.04.011. [DOI] [PubMed] [Google Scholar]
- (21).Haker A, Hendriks J, van Stokkum IH, Heberle J, Hellingwerf KJ, Crielaard W, Gensch T. The Two Photocycles of Photoactive Yellow Protein from Rhodobacter sphaeroides. J. Biol. Chem. 2003;278:8442–8451. doi: 10.1074/jbc.M209343200. [DOI] [PubMed] [Google Scholar]
- (22).van Stokkum IH, Gobets B, Gensch T, Mourik F, Hellingwerf KJ, Grondelle R, Kennis JT. (Sub)-Picosecond Spectral Evolution of Fluorescence in Photoactive Proteins Studied with a Synchroscan Streak Camera System. Photochem. Photobiol. 2006;82:380–388. doi: 10.1562/2005-06-15-RA-572. [DOI] [PubMed] [Google Scholar]
- (23).Hoff WD, Croonquist PA, Lee BC. Folding and Signaling Share the Same Pathway in a Photoreceptor. Biophys. J. 2002;82:1452. doi: 10.1073/pnas.111153598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Changenet-Barret P, Espagne A, Plaza P, Hellingwerf KJ, Martin MM. Investigations of the Primary Events in a Bacterial Photoreceptor for Photomotility: Photoactive Yellow Protein (PYP) New J. Chem. 2005;29:527–534. [Google Scholar]
- (25).Espagne A, Paik DH, Changenet-Barret P, Martin MM, Zewail AH. Ultrafast Photoisomerization of Photoactive Yellow Protein Chromophore Analogues in Solution: Influence of the Protonation State. ChemPhysChem. 2006;7:1717–1726. doi: 10.1002/cphc.200600137. [DOI] [PubMed] [Google Scholar]
- (26).Vengris M, Larsen DS, van der Horst MA, Larsen OFA, Hellingwerf KJ, van Grondelle R. Ultrafast Dynamics of Isolated Model Photoactive Yellow Protein Chromophores: “Chemical Perturbation Theory” in the Laboratory. J. Phys. Chem. B. 2005;109:4197–4208. doi: 10.1021/jp045763d. [DOI] [PubMed] [Google Scholar]
- (27).Larsen DS, van Stokkum IH, Vengris M, van Der Horst MA, de Weerd FL, Hellingwerf KJ, van Grondelle R. Incoherent Manipulation of the Photoactive Yellow Protein Photocycle with Dispersed Pump–Dump–Probe Spectroscopy. Biophys. J. 2004;87:1858–1872. doi: 10.1529/biophysj.104.043794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kondo M, Heisler IA, Stoner-Ma D, Tonge PJ, Meech SR. Ultrafast Dynamics of Protein Proton Transfer on Short Hydrogen Bond Potential Energy Surfaces: S65T/H148D GFP. J. Am. Chem. Soc. 2009;132:1452–1453. doi: 10.1021/ja907690g. [DOI] [PubMed] [Google Scholar]
- (29).Jacquemin D, Perpete EA, Ciofini I, Adamo C. Fast and Reliable Theoretical Determination of pKa* for Photoacids. J. Phys. Chem. A. 2008;112:794–796. doi: 10.1021/jp7105814. [DOI] [PubMed] [Google Scholar]
- (30).Chattoraj M, King BA, Bublitz GU, Boxer SG. Ultrafast Excited State Dynamics in Green Fluorescent Protein: Multiple States and Proton Transfer. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8362–8367. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ihee H, Rajagopal S, Srajer V, Pahl R, Anderson S, Schmidt M, Schotte F, Anfinrud PA, Wulff M, Moffat K. Visualizing Reaction Pathways in Photoactive Yellow Protein from Nanoseconds to Seconds. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7145–7150. doi: 10.1073/pnas.0409035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bernard C, Houben K, Derix NM, Marks D, van der Horst MA, Hellingwerf KJ, Boelens R, Kaptein R, van Nuland NA. The solution Structure of a Transient Photoreceptor Intermediate: Delta25 Photoactive Yellow Protein. Structure. 2005;13:953–962. doi: 10.1016/j.str.2005.04.017. [DOI] [PubMed] [Google Scholar]
- (33).Olsen S, Lamothe K, Martinez TJ. Protonic Gating of Excited-State Twisting and Charge Localization in GFP Chromophores: A Mechanistic Hypothesis for Reversible Photoswitching. J. Am. Chem. Soc. 2010;132:1192. doi: 10.1021/ja907447k. [DOI] [PubMed] [Google Scholar]
- (34).Levine BG, Martinez TJ. Isomerization through Conical Intersections. Annu. Rev. Phys. Chem. 2007;58:613–634. doi: 10.1146/annurev.physchem.57.032905.104612. [DOI] [PubMed] [Google Scholar]
- (35).Changenet-Barret P, Plaza P, Martin MM. Primary Events in the Photoactive Yellow Protein Chromophore in Solution. Chem. Phys. Lett. 2001;336:439–444. [Google Scholar]
- (36).Jou FY, Freeman GR. Temperature and Isotope Effects on the Shape of the Optical-Absorption Spectrum of Solvated Electrons in Water. J. Phys. Chem. 1979;83:2383–2387. [Google Scholar]
- (37).Larsen DS, Vengris M, van Stokkum IHM, van der Horst MA, Cordfunke RA, Hellingwerf KJ, van Grondelle R. Initial PhotoInduced Dynamics of the Photoactive Yellow Protein Chromophore in Solution. Chem. Phys. Lett. 2003;369:563–569. [Google Scholar]
- (38).Shimizu N, Imamoto Y, Harigai M, Kamikubo H, Yamazaki Y, Kataoka M. pH-Dependent Equilibrium between Long Lived Near-UV Intermediates of Photoactive Yellow Protein. J. Biol. Chem. 2006;281:4318–4325. doi: 10.1074/jbc.M506403200. [DOI] [PubMed] [Google Scholar]
- (39).Kyndt JA, Savvides SN, Memmi S, Koh M, Fitch JC, Meyer TE, Heyn MP, Van Beeumen JJ, Cusanovich MA. Structural Role of Tyrosine 98 in Photoactive Yellow Protein: Effects on Fluorescence, Gateway, And Photocycle Recovery. Biochemistry. 2007;46:95–105. doi: 10.1021/bi061867y. [DOI] [PubMed] [Google Scholar]
- (40).Changenet-Barret P, Espagne A, Katsonis N, Charier S, Baudin JB, Jullien L, Plaza P, Martin MM. Excited-State Relaxation Dynamics of a PYP Chromophore Model in Solution: Influence of the Thioester Group. Chem. Phys. Lett. 2002;365:285–291. [Google Scholar]
- (41).Imamoto Y, Ito T, Kataoka M, Tokunaga F. Reconstitution Photoactive Yellow Protein from Apoprotein and p-Coumaric Acid-Derivatives. FEBS Lett. 1995;374:157–160. doi: 10.1016/0014-5793(95)01096-w. [DOI] [PubMed] [Google Scholar]
- (42).Cordfunke R, Kort R, Pierik A, Gobets B, Koomen GJ, Verhoeven JW, Hellingwerf KJ. Trans/cis (Z/E) Photoisomerization of the Chromophore of Photoactive Yellow Protein Is Not a Prerequisite for the Initiation of the Photocycle of This Photoreceptor Protein. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7396–7401. doi: 10.1073/pnas.95.13.7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Carroll EC, Compton OC, Madsen D, Osterloh FE, Larsen DS. Ultrafast Carrier Dynamics in Exfoliated and Functionalized Calcium Niobate Nanosheets in Water and Methanol. J. Phys. Chem. C. 2008;112:2394–2403. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.