

Abstract
Synthetic inhibitors of protein-protein interactions are being discovered despite the inherent challenge in targeting large contact surfaces with small molecules. An analysis of available examples identifies common features of complexes that make them tractable for small molecules. We deduced that relative disposition and energetic contributions of “hot spot” residues provide a predictive scale for the potential of protein-protein interactions to be inhibited by small molecules. Based on this model, we analyzed the full set of helical protein interfaces in the Protein Data Bank to identify those that are potentially suitable candidates for synthetic ligands.
Most modern pharmaceuticals are small molecules that target molecular pockets in enzymes or protein receptors but in general they fail to achieve sufficient specificity and affinity to target extended, and often flat, interfaces common to protein-protein interactions (PPI). However, successful examples of small molecule PPI inhibitors are emerging.(1) Analysis suggests that although protein interfaces are large, often a small subset of the residues contributes significantly to the free energy of binding.(2, 3) Small molecules that reproduce the functionality of these hot spot residues have the potential to inhibit the relevant interfaces.
Alanine scanning mutagenesis offers a powerful approach for identifying hot spot residues.(4) For example, in the well studied p53/HDM2 interaction, three residues (F19, W23 and L26) from a helix in the p53 activation domain reside in a deep hydrophobic groove (Figure 1, panel a).(5) Mutation of any of these residues to alanine leads to a significant (> 2 kcal/mol) decrease in the stability of the resulting complex.(6) Similar alanine scanning results are obtained with pro-apoptotic partners of the anti-apoptotic protein Bcl-xL (Figure 1, panel b).(7) The complex between transcription factor p53 and its regulator HDM2 is inhibited by nutlins (Figure 1, panel c),(8, 9) and there are highly potent small molecule antagonists, including ABT-737 and A-385358, of the interactions between Bcl-xL and BH3 domains (Figure 1, panel d).(10, 11) We conjectured that these interactions can be inhibited with nanomolar affinity by small molecules because the critical residues lie within a small radius of each other on one of the partner proteins, allowing their arrangement on a low molecular weight scaffold. For instance, the two chlorobenzene groups in nutlin-3 span 6 Å (Figure 1, panel e), and occupy the binding pockets of the key tryptophan and leucine residues from the p53 helix.(8) Similarly A-385358 targets same key pockets on Bcl-xL as the helical BH3 domains.(12) Using these two examples of successfully inhibited protein-protein interactions as a guide, we surveyed the Protein Data Bank (PDB)(13) to identify protein-protein interactions as likely targets for small molecule inhibitors. While a number of studies have focused on predicting the physicochemical properties of small molecule protein-protein interaction inhibitors,(14–17) we sought to develop a method to gauge the "inhibitability" of protein complexes.
Figure 1.

(a) The p53/HDM2 interaction (PDB code: 1YCR). A helix in the p53 activation domain resides in a deep hydrophobic groove. (b) The pro-apoptotic protein partner Bak bound to the anti-apoptotic protein Bcl-xL (PDB code: 1BXL). (c) Nutlin-3 binds to HDM2 in the same hydrophobic groove occupied by the p53 helix (PDB code: 1rv1). (d) ABT-785358 targets Bcl-xL at the site of its pro-apoptotic binding partners (PDB code: 2o22) (e) The structures of nutlin-3 and A-385358.
Here we focus on protein complexes that feature α-helices at the interfaces. α-Helices constitute the largest class of protein secondary structure and mediate many protein interactions.(18, 19) Helices located within the protein core are vital for the overall stability of protein tertiary structure, whereas exposed α-helices on protein surfaces constitute central bioactive regions for the recognition of numerous proteins, DNAs, and RNAs. Helix mimetics have emerged as a highly effective class of PPI inhibitors.(20–26) A catalog of targetable helical interfaces should significantly enhance the utility of these helix mimetics.
We began by identifying the full set of α-helical interfaces in the PDB (Figure 2). The PDB (version 08/04/2009) was queried for structures containing more than one protein entity (Supporting Information).(18) This query extracted 9,339 complexes. We clustered these complexes according to sequence similarity of all protein chains in each complex using the CD-HIT(27) sequence alignment program at a 95% similarity threshold. This yielded a dataset of 4,143 unique protein complexes. For each <4 Å resolution structure, we extracted potential chain partners belonging to separate molecules as specified in the PDB file. Identification of secondary structure, interfacial residues, and hot spot residues was accomplished using the Rosetta suite of programs.(28–30) Rosetta determines secondary structure by calculating the φ and ϕ angles of the protein backbone. We define a helical segment as one that contains at least four contiguous residues with φ and ϕ angles characteristic of an α- or the closely related 310-helix (Supporting Information).(18) An interfacial residue is defined as a residue that has at least one atom within a 5 Å radius of an atom belonging to a binding partner in the protein complex. Hot spot residues were predicted using a computational alanine scan.(29, 30) Hot spot residues were defined as residues that upon mutation to alanine are predicted to decrease the binding energy by a threshold value ΔΔGbind ≥ 1.0 kcal/mol, as measured in Rosetta energy units. Our method identified 2,561 PDB entries possessing helix interfaces in protein-protein (HIPP) interactions and suggests that roughly 62% of the protein complexes in the PDB feature helical interfaces.
Figure 2.

Evaluation of structures from the Protein Data Bank to identify and assess helical interfaces in protein–protein (HIPP) interactions. The helical interfaces were segregated based on binding interfaces and computational alanine scanning mutagenesis analysis. *ΔΔGavg ≥ 2 kcal/mol; ** ΔΔGavg = 1–2 kcal/mol.
After analyzing the energetic contributions and spatial arrangement of the hot spot residues in the helix of p53 in the p53/HDM2 interaction and the BH3 helix of Bak in the Bcl-xL/Bak interaction we found common features that may provide insight into the reason HDM2 and Bcl-xL are tractable targets for inhibition by small molecules. For both complexes the calculated average ΔΔGbind of the hot spot residues in the helix of the protein partner (p53 in the p53/HDM2 complex and Bak in the Bcl-xL/Bak complex) is greater than 2 kcal/mol and the radius between the hot spot residues in the helix of the protein partners was on the order of 7 Å. The HIPP dataset was analyzed to screen for protein receptors that possessed these same features. Using these criteria we placed the HIPP interactions into three broad categories: (1) receptors which contain a cleft for helix binding (Figure 3, panel a), as in the p53/HDM2 complex, where at least two nearby residues contribute strongly to binding; (2) extended interfaces that require multiple contacts from two to five turn helices featuring two or more residues that contribute strongly to binding (Figure 3, panel b); and (3) receptors with clefts and extended interfaces characterized by weaker interactions.(31) We defined hot spot residues as strong or weak contributors based on the change in free energy (ΔΔGavg) when these residues on a given helix are mutated to alanine, with a ΔΔGavg cutoff of 2 kcal/mol. Receptors with clefts are targeted by helices with two or more hot spot residues within a 7 Å radius, while the extended interfaces category features a distribution of hot spot residues over a larger distance of 7–30 Å (Figure 3). Category 2 consists of interfaces where the helical segment spans 20 residues or roughly five helical turns; longer sequences were placed in category 3. We then calculated the proportion of hot spots residing on the helix versus the rest of the chain to determine if a simple mimic of an interfacial helix can inhibit a large interface. This analysis is complicated by the fact that a number of the complexes in the PDB do not have full-length proteins, and in fact a number of the relevant complexes only consist of a truncated helical domain.
Figure 3.

Helical Interfaces: we have divided helical protein-protein interactions between those that feature clefts for binding (a) and those with extended interfaces (b). The p53/MDM2 (PDB code: 1YCR) (a) and cyclin-dependent kinase6/D-type viral cyclin (PDB code: 1G3N) (b) complexes are representative examples of binding cleft and extended interfaces, respectively. The distance between flanking hot spot residues in the helix of the protein partner of a binding cleft target spans a radius of 7 Å or less (a) and greater than 7 Å but less than 30 Å for an extended interface target.
This procedure provided 159 complexes that we predict are targets for small molecules and another 252 interfaces that may be inhibited by helix mimetics. The remainder of complexes that did not meet the criteria of the first two categories but were not eliminated from consideration for other reasons (Supporting Information) feature weaker interactions between the candidate helix and the protein receptor. Table 1 illustrates examples of interfaces with binding clefts and extended interfaces. Full lists of complexes that fall in each of the three aforementioned categories, along with the sequence information for each PDB entry, are included in the Supporting Information along with a list of the redundant HIPP interactions. It is interesting to note that proteins from the same family do not necessarily fall into the same category. For example, when Bcl-xL is cocrystallized with the Bak peptide this HIPP interaction is classified as a binding cleft target. On the other hand, the closely related Bad peptide, when cocrystalized with the same Bcl-xL protein, is classified as an extended HIPP interaction. These results reflect the distinct binding profiles observed with Bcl-2 family proteins and BH3 domains.(32)
Table 1.
Predicted helical protein-protein interactions as targets for synthetic ligands.
| PDB code, interface chains; candidate helixa | ΔΔGavg/helix (kcal/mol)b | Helix contributionc | Hot spot residue helix positiond | Chain name/Functione |
|---|---|---|---|---|
| Examples of Interfaces with Binding Clefts | ||||
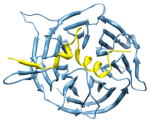 2i3s, c d; d |
2.8 | 55% | i | c: Cell cycle arrest protein |
| i + 1 | d: Checkpoint serine/threonine-protein kinase | |||
| i + 3 | Function: cell cycle | |||
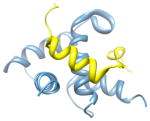 22ka6, a b; b |
2.6 | 54% | i | a: CREB-binding protein |
|
i + 1 i + 3 |
b: Signal transducer and activator of transcription 1-α/β | |||
| i + 4 | Function: transcription and gene regulation | |||
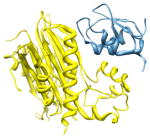 2pop, c d; c |
2.3 | 100% | i | c: Mitogen-activated protein kinase kinase kinase 7- interacting protein 1 |
| i + 1 | d: Baculoviral IAP repeat-containing protein | |||
| i + 4 | Function: signaling | |||
| Examples of Extended Interfaces | ||||
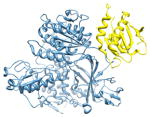 2nup, b c; c |
2.5 | 72% | i | b: Protein transport protein Sec24A |
|
i + 4 i + 5 |
c: Vesicle-trafficking protein SEC22b | |||
| i + 8 | Function: protein transport | |||
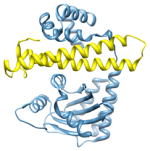 2q0o, a c; c |
2.4 | 66% | i | a: Probable transcriptional activator protein traR |
|
i + 10 i + 13 |
c: Probable transcriptional repressor tram | |||
| i + 14 | Function: transcription and gene regulation | |||
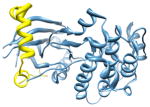 2vgo, a d; d |
2.5 | 24% | a: Serine/threonine-protein kinase 12-A | |
| i | d: Inner centromere protein A | |||
| i + 8 | Function: enzyme (transferase) | |||
Chains in the complex featuring a helix at the interface; candidate helix to be mimicked is part of the indicated chain.
ΔΔGavg/helix is derived from Rosetta computational alanine mutagenesis studies and indicates the average free energy penalty for mutating two or more key residues at the interface to alanine.
Helix contribution refers to the proportion of key contact residues positioned on the candidate helix as compared to the chain (see text for a detailed explanation).
Relative positioning of the hot spot residues on a helix.
Description of chains featuring the helical interface and cellular function.
We propose that receptors with binding clefts are candidates for high-throughput screening efforts with small molecule libraries currently available to chemical biologists.(1, 33) It is expected that several of these targets would not be of interest for inhibitor design owing to their biological function and other criteria.(15) The overall numbers of targets in each category are expected to increase as the PDB is further populated with helical protein-protein interactions. The aim of this inquiry is to devise an algorithm to gauge the “inhibitability” of protein-protein interactions. Existing examples of potent small molecules disrupting protein-protein interfaces as predicted in Category 1 are listed in Supporting Information, Table S1.(14, 15) Our analysis suggests that stabilized helices, and other structured oligomers, are potentially better candidates for targeting extended interfaces (Category 2);(21, 22) although these helix mimetics can also effectively modulate Category 1 interactions.(24, 34, 35) It is likely that direct mimics of helices from Category 3 interfaces, where the hot spot residues do not contribute strongly, will not target the cognate protein receptor with high affinity; although utilization of non-natural residues or use of covalent crosslinks with protein receptor could overcome the inherent weak affinities at these interfaces.
We sorted the helical interactions in the HIPP dataset according to function as defined in the PDB (Supplementary Information, Figure S2). Although some HIPP interactions could fall into more than one functional group, we limited each HIPP interaction to one group. Helical interfaces are involved in a broad range of functions from enzymatic activity to gene regulation. Interfaces with binding clefts are involved in a comparable range of functions as extended interfaces. We anticipate that the methods and results described here will guide development of next generation protein-protein interaction inhibitors.
Supplementary Material
Acknowledgments
This work was financially supported by the National Institutes of Health (GM073943). A.L.J. thanks New York University for the Dean’s Dissertation Fellowship.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet.
References
- 1.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 2.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 3.Moreira IS, Fernandes PA, Ramos MJ. Hot spots--a review of the protein-protein interface determinant amino-acid residues. Proteins. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 5.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 6.Picksley SM, Vojtesek B, Sparks A, Lane DP. Immunochemical analysis of the interaction of p53 with MDM2;--fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene. 1994;9:2523–2529. [PubMed] [Google Scholar]
- 7.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Structure of Bcl-x(L)-Bak peptide complex: Recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 8.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 9.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 11.Shoemaker AR, Oleksijew A, Bauch J, Belli BA, Borre T, Bruncko M, Deckwirth T, Frost DJ, Jarvis K, Joseph MK, Marsh K, McClellan W, Nellans H, Ng S, Nimmer P, O'Connor JM, Oltersdorf T, Qing W, Shen W, Stavropoulos J, Tahir SK, Wang B, Warner R, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. A small-molecule inhibitor of Bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res. 2006;66:8731–8739. doi: 10.1158/0008-5472.CAN-06-0367. [DOI] [PubMed] [Google Scholar]
- 12.Bruncko M, Oost TK, Belli BA, Ding H, Joseph MK, Kunzer A, Martineau D, McClellan WJ, Mitten M, Ng SC, Nimmer PM, Oltersdorf T, Park CM, Petros AM, Shoemaker AR, Song X, Wang X, Wendt MD, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem. 2007;50:641–662. doi: 10.1021/jm061152t. [DOI] [PubMed] [Google Scholar]
- 13.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higueruelo AP, Schreyer A, Bickerton GR, Pitt WR, Groom CR, Blundell TL. Atomic interactions and profile of small molecules disrupting protein-protein interfaces: the TIMBAL database. Chem Biol Drug Des. 2009;74:457–467. doi: 10.1111/j.1747-0285.2009.00889.x. [DOI] [PubMed] [Google Scholar]
- 15.Bourgeas R, Basse MJ, Morelli X, Roche P. Atomic analysis of protein-protein interfaces with known inhibitors: the 2P2I database. PLoS One. 2010;5:e9598. doi: 10.1371/journal.pone.0009598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis FP, Sali A. The overlap of small molecule and protein binding sites within families of protein structures. PLoS Comput Biol. 2010;6:e1000668. doi: 10.1371/journal.pcbi.1000668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reynes C, Host H, Camproux AC, Laconde G, Leroux F, Mazars A, Deprez B, Fahraeus R, Villoutreix BO, Sperandio O. Designing focused chemical libraries enriched in protein-protein interaction inhibitors using machine-learning methods. PLoS Comput Biol. 2010;6:e1000695. doi: 10.1371/journal.pcbi.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jochim AL, Arora PS. Assessment of helical interfaces in protein-protein interactions. Mol BioSyst. 2009;5:924–926. doi: 10.1039/b903202a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones S, Thornton JM. Protein-Protein Interactions - A Review of Protein Dimer Structures. Prog Biophys Mol Bio. 1995;63:31–65. doi: 10.1016/0079-6107(94)00008-w. [DOI] [PubMed] [Google Scholar]
- 20.Henchey LK, Jochim AL, Arora PS. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation. Curr Opin Chem Biol. 2008;12:692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, Gellman SH. Structural and biological mimicry of protein surface recognition by alpha/beta-peptide foldamers. Proc Natl Acad Sci U S A. 2009;106:14751–14756. doi: 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis JM, Tsou LK, Hamilton AD. Synthetic non-peptide mimetics of alpha-helices. Chem Soc Rev. 2007;36:326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- 24.Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. Helical β-peptide inhibitors of the p53-hDM2 interaction. J Am Chem Soc. 2004;126:9468–9469. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]
- 25.Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. Inhibition of Hypoxia Inducible Factor 1–Transcription Coactivator Interaction by a Hydrogen Bond Surrogate Alpha-Helix. J Am Chem Soc. 2010;132:941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrison RS, Shepherd NE, Hoang HN, Ruiz-Gomez G, Hill TA, Driver RW, Desai VS, Young PR, Abbenante G, Fairlie DP. Downsizing human, bacterial, and viral proteins to short water-stable alpha helices that maintain biological potency. Proc Natl Acad Sci U S A. 2010;107:11686–11691. doi: 10.1073/pnas.1002498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W, Jaroszewski L, Godzik A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics. 2001;17:282–283. doi: 10.1093/bioinformatics/17.3.282. [DOI] [PubMed] [Google Scholar]
- 28.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kortemme T, Kim DE, Baker D. Computational alanine scanning of protein-protein interfaces. Sci STKE. 2004;2004:pl2. doi: 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- 31.Murray JK, Gellman SH. Targeting protein-protein interactions: Lessons from p53/MDM2. Biopolymers. 2007;88:657–686. doi: 10.1002/bip.20741. [DOI] [PubMed] [Google Scholar]
- 32.Fu X, Apgar JR, Keating AE. Modeling backbone flexibility to achieve sequence diversity: the design of novel alpha-helical ligands for Bcl-xL. J Mol Biol. 2007;371:1099–1117. doi: 10.1016/j.jmb.2007.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 34.Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. Inhibition of Hypoxia Inducible Factor 1–Transcription Coactivator Interaction by a Hydrogen Bond Surrogate Alpha-Helix. J Am Chem Soc. 2010;132:941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cummings CG, Hamilton AD. Disrupting protein-protein interactions with non-peptidic, small molecule alpha-helix mimetics. Curr Opin Chem Biol. 2010;14:341–346. doi: 10.1016/j.cbpa.2010.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.