

SUMMARY
Rapid and selective erasures of certain types of memories in the brain would be desirable under certain clinical circumstances. By employing an inducible and reversible chemical-genetic technique, we find that transient αCaMKII overexpression at the time of recall impairs the retrieval of both newly formed one-hour object recognition memory and fear memories, as well as 1-month-old fear memories. Systematic analyses suggest that excessive αCaMKII activity-induced recall deficits are not caused by disrupting the retrieval access to the stored information but are, rather, due to the active erasure of the stored memories. Further experiments show that the recall-induced erasure of fear memories is highly restricted to the memory being retrieved while leaving other memories intact. Therefore, our study reveals a molecular genetic paradigm through which a given memory, such as new or old fear memory, can be rapidly and specifically erased in a controlled and inducible manner in the brain.
INTRODUCTION
Memory retrieval is believed to be a rapid reconstructive process involving a recapitulation of the learned information (Sara, 2000; Hasselmo and Eichenbaum, 2005; Kensinger and Schacter, 2005). Pioneering studies have reported that memory retrieval can be enhanced by the activation of β-adrenergic receptors (Sara and Devauges, 1988; Devauges and Sara, 1991). It has been long wished that, under certain circumstances, some memories (e.g., traumatic war memories and unwanted fear or painful memories) can be selectively erased while leaving other memories intact (Sutherland and Bryant, 2005). Such controlled erasure of a specific set of memories in the brain is likely to be quite distinct from the natural and often slow forgetting process and may require an unnatural and rapid manipulation that can reverse the molecular and synaptic process involved in the formation of memories in the neural circuits (Bear and Malenka, 1994).
It is generally agreed that memory can be separated into four distinct temporal stages: namely, acquisition, consolidation, storage, and retrieval. It has been demonstrated that memory acquisition requires the activation of the NMDA receptor (Tsien et al., 1996; Tang et al., 1999; Cao et al., 2007; Rampon et al., 2000) and its downstream signaling molecules, including the CaMKII (Ouyang et al., 1997; Silva et al., 1992; Elgersma et al., 2002; Giese et al., 1998; Mayford et al., 1995; Silva et al., 1992), BDNF, integrins, and tPA (Chan et al., 2003; Hempstead and Lu, 2004; Pang et al., 2004). A series of inducible genetic knockout experiments also suggests that the memory consolidation requires the postlearning reactivations of the NMDA receptor to overcome the gradual draft in synaptic efficacy resulting from the routine metabolic turnovers of synaptic proteins (Shimizu et al., 2000; Wittenberg and Tsien, 2002; Cui et al., 2005). Moreover, inducible NR1 gene knockout in the mouse cortex during the storage phase has been shown to impair the storage of remote memories (Cui et al., 2004). Furthermore, various experiments have reported that reconsolidation of recently retrieved memories seems to require protein synthesis (Nader et al., 2000; Biedenkapp and Rudy, 2004; Alberini, 2005; Torras-Garcia et al., 2005; Rudy et al., 2006; Sara, 2000) and synaptic protein degradation (Lee et al., 2008). Finally, memory retrieval can be also influenced by activation of β-adrenergic receptors (Sara and Devauges, 1988; Devauges and Sara, 1991; Murchison et al., 2004; Sara, 2000) or application of protein kinase M zeta inhibitor (Shema et al., 2007). Therefore, the emerging view is that memory traces are highly dynamic at the various stages of the memory process.
Here, we describe the application of our recently developed chemical genetic strategy that can combine the molecular and tissue/region specificity of genetics with the high time-resolution of typical pharmacological inhibitions (Wang et al., 2003; Cho et al., 2007). This method is based on the principle of convergent protein engineering and organic chemical synthesis of its sensitized inhibitor for generating highly specific novel inhibition interface (Liu et al., 1999; Bishop et al., 2000), and we have subsequently introduced this sensitized protein/inhibitor system into the genetically modified mice (Wang et al., 2003). By using a genetically sensitized inhibitor (NM-PP1), we have rapidly manipulated the enzymatic activity of transgenically expressed αCaMKII exclusively in the retrieval process. We describe the consequence of the chemical genetic manipulation of transgenic αCaMKII activity on the retrieval of one-hour, short-term fear memories and novel object recognition memory, as well as one-month, long-term fear memories.
RESULTS
Rapid Manipulation of αCaMKII Activity in the Transgenic Mouse Forebrain
αCaMKII has significant homology to its CaMKII isoforms such as β, γ, and δCaMKII, as well as other members of CaMK superfamily such as CaMKI, CaMKIII, CaMKIV, CaMKV, and CaMK kinase (Hanson and Schulman, 1992). Thus, the structural conservancy of the catalytic ATP-binding sites among various kinases has created intrinsic difficulty in obtaining truly specific inhibitors (Hanks et al., 1988; Hunter, 1994). We turned this structural constraint to our advantage by creating a novel bump-and-hole interface inside of the ATP-binding pocket in αCaMKII to contain a hidden cavity (αCaMKII-F89G) accessible only to a rationally designed bulky inhibitor that is too large to fit into the natural ATP-binding pocket of unmodified kinases (Liu et al., 1999; Bishop et al., 2000). As previously described, we then overexpressed this αCaMKII-F89G transgene using the forebrain-specific CaMKII promoter (Figure S1A available online). The activity of this “enlarged” αCaMKII-F89G can be selectively inhibited by a rationally designed, genetically sensitized small-molecule inhibitor, NM-PP1 (Bishop et al., 2000; Wang et al., 2003; Cho et al., 2007). We again confirmed the forebrain specificity of the transgene expression in our transgenic offspring using a probe recognizing a unique SV40 intron sequence (Figure S1B). Our western blots estimated the relative amount of transgenic αCaMKII protein to be ~36% of the endogenous αCaMKII level. This overexpression in the protein amount resulted in ~200% increase in the total CaMKII activity in the transgenic brain, presumably due to intrasubunit autophosphorylation of neighboring subunits in the holoenzyme (Ouyang et al., 1997).
We also confirmed the acute inhibition of αCaMKII-F89G activity by intraperitoneal injection of NM-PP1 (5 μM). The CaMKII activity assay showed that a single i.p. injection of NM-PP1 can produce a significant suppression of CaMKII-F89G activity in the forebrain regions within 10 min (Figure S1C). This inhibition is readily reversible by a postinjection of 40 min as demonstrated both by pharmacokinetics and an enzyme activity assay in Tg-F89G mice. This time course fits well with the measured pharmacokinetics of [3H]-labeled NM-PP1 in the brain. Thus, a single i.p. injection of NM-PP1 into freely behaving transgenic mice can significantly suppress the αCaMKII-F89G’s enzymatic activity and maintained this suppression level 10–40 min after injection. This pharmacokinetic time course served as a guide to our subsequent behavioral experiments.
Excessive CaMKII Activity Impairs Retrievals of Newly Formed Memories
In considering the view that short-term memory traces are represented in a more labile state than long-term memory traces (Gerard, 1949; Squire, 1987) and that the storage sites for short-term memories and long-term memories may differ as a result of consolidation (Zola-Morgan and Squire, 1990; Frankland and Bontempi, 2005), we are interested in investigating the question of whether the retrievals of newly formed memory and old memory share some common convergence that can be subjected to similar molecular manipulation. Thus, we examined the functional consequence of chemical-genetic manipulation of CaMKII on the memory using short-term duration. We used three different memory tests, namely one-hour novel object recognition test (Clark et al., 2000; Rampon et al., 2000), contextual fear conditioning, and cued fear conditioning (Phillips and LeDoux, 1992; Kim and Fanselow, 1992; Davis et al., 2003). All behavioral experiments were performed on the littermate wild-type and transgenic mice and were conducted by two experimenters blind to the genotype of each mouse. Both wild-type and transgenic mice received the same NM-PP1 injection protocols.
We first tested the performance of the transgenic mice in one-hour memory tests. We found that the transgenic mice were profoundly impaired in the novel object recognition memory in comparison to the wild-type mice (Figure 1A). The impairment in one-hour memory was further observed in the contextual fear conditioning test (Figure 1B) and the cued fear conditioning test (Figure 1C). It is important to note that the amount of immediate freezing after training is similar, indicating that transgenic mice have a normal freezing behavioral expression. Thus, those results suggest that the elevated CaMKII in the forebrain impaired the memory process.
Figure 1. CaMKII-F89G Tg Mice Exhibited Memory Deficit in Short-Term Memory Tests.

The black bar on the top column indicates the duration of CaMKII-F89G activity in one-hour short-term memory tests.
(A) The performance of Tg mice (n = 9) was greatly impaired in one-hour novel object recognition tests compared to WT mice (n = 10) (p < 0.01).
(B) Although no significant difference was observed in immediate freezing, the retention of one-hour contextual fear memory was significantly impaired in Tg mice (n = 9) compared to WT mice (n = 10) (p < 0.01).
(C) No significant difference was found in pretone freezing response in these mice; however, a significant difference in tone-elicited freezing response was observed between Tg mice (n = 9) and WT mice (n = 10) (p < 0.01).
(D) Transgenic mice (n = 10) with active CaMKII-F89G expression during learning and postlearning period (−5 to 55 min), but with CaMKII-F89G activity suppressed during recall period, showed normal memory retrieval in comparison to wild-type mice (n = 10) in one-hour novel object recognition recall test. NM-PP1 (16.6 ng/g) was given 15 min before recall.
(E) The Tg mice in which CaMKII-F89G activity was selectively inhibited during the recall stage also exhibited normal recall performance in one-hour contextual fear conditioning recall.
(F) The Tg mice recalled one-hour cued fear memory normally when CaMKII-F89G was suppressed at the time of recall. All values are mean ± SEM; **p < 0.01.
Because the observed memory impairments could be attributed to any one (or more) of the memory stages (e.g., acquisition, retention, or recall), we asked whether the elevated CaMKII in the forebrain affects learning and retention. Thus, we designed a “learning and retaining memory in the presence of elevated CaMKII” paradigm and used another group of transgenic and wild-type littermates. Under this paradigm, the identical training protocols were used, but mice were injected NM-PP1 15 min prior to the recall tests so that the transgenic mice would have the CaMKII-F89G activity suppressed in their brains. In other words, the transgenic mice were allowed to learn and maintain short-term memory in the presence of excessive CaMKII (endogenous CaMKII plus CaMKII-F89G) from learning stage and post-learning stage of 0 to 55 min. However, their CaMKII-F89G activities were temporarily suppressed during the time of recall. Interestingly, the transgenic mice exhibited completely normal performances in all three short-term memory retention tests in comparison to their wild-type littermates (Figures 1D–1F). These experiments suggest that the memory deficits observed in the transgenic mice reflect the impairment during retrieval.
To further examine whether the retrieval deficits in the transgenic mice were really due to the elevated CaMKII activity rather than the mere physical presence of the transgenic CaMKII-F89G protein itself, we then designed the “elevated CaMKII during recall” paradigm and conducted the experiments using a third group of wild-type littermates and transgenic mice in which NM-PP1 was injected twice over time. The first injection occurred 15 min prior to the training, and the second injection took place 10 min after training. As such, the learning and retention (0–55 min) occurred with the CaMKII-89G activity suppressed, but the memory recall took place with the elevated CaMKII level in the transgenic brains. Indeed, we found that these transgenic mice with the elevated αCaMKII-F89G activity at the time of recall showed significant retrieval deficits in the one-hour novel object recognition (Figure 2A), contextual fear memory (Figure 2B), and cued fear memory tests (Figure 2C). Therefore, our results using three different memory tests have demonstrated that the excessive amount of CaMKII activity at the time of recall impaired the retrieval of newly formed memories.
Figure 2. Transient Expression of CaMKII-F89G Activity Selectively during the Recall Stage Impaired Retrievals of One-Hour Memories.
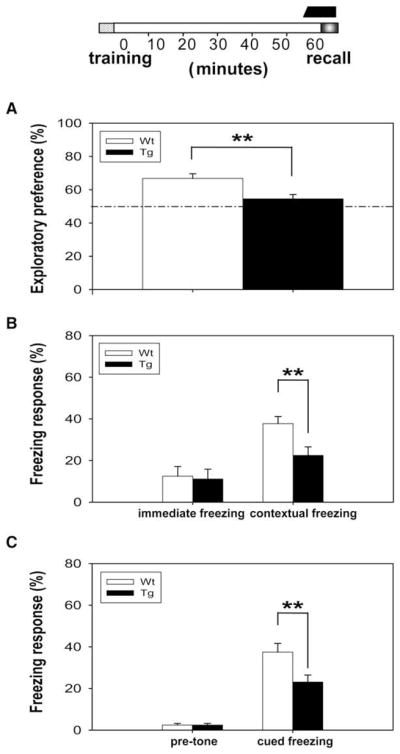
Memory retrieval deficits of Tg mice in one-hour novel object recognition recall test (A), one-hour contextual fear memory recall test (B), and one-hour cued fear memory recall test (C) (12 animals in each group per test; p < 0.01). Since a single i.p. injection can provide consistent and complete in vivo inhibition of αCaMKII-F89G activity between 10 and 40 min postinjection, we designed a two-injection protocol so that αCaMKII-F89G activity would be expressed by the time of recall. The black bar above the timeline on the top of Figures A–C represents the period in which CaMKII-F89G in Tg mice was expressed. The first NM-PP1 (16.6 ng/g) was given 15 min before training, while the second NM-PP1 was introduced by i.p. injection 10 min after training. All values are mean ± SEM; **p < 0.01.
Excessive CaMKII Activity Also Impairs Retrievals of Old Memories
We then examined the consequence of excessive CaMKII activity on the memory performance of the old memories with longer duration by using one-month contextual and cued fear conditioning paradigms (Phillips and LeDoux, 1992; Shimizu et al., 2000; Davis et al., 2003; Kim and Jung, 2006). Consistent with our short-term memory tests, we found that the transgenic mice exhibited normal immediate freezing but showed significant deficits in the one-month contextual fear memory (Figure 3A) and one-month cued fear memory tests (Figure 3B), indicating that expression of the CaMKII-F89G disrupts the long-term memory process.
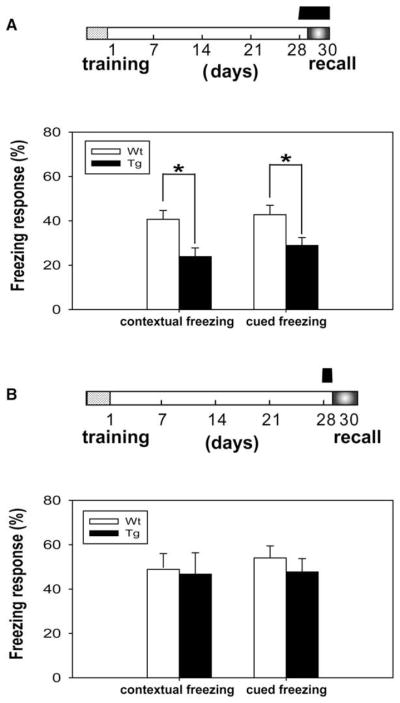
Figure 3. Expression of αCaMKII-F89G Activity in the Tg Mice Induced Memory Deficit in One-Month Long-Term Fear Memory Tests.

The black bar on top column indicates the duration in which the CaMKII-F89G activity is expressed.
(A) Contextual learning in Tg mice (n = 12) during initial training was comparable to WT mice (n = 11), as shown by the similar immediate freezing response. However, a significant difference was observed in the recall test of one-month contextual fear memory in Tg mice compared to WT mice (p < 0.01).
(B) Although no significant difference was found in pretone freezing response in these mice, a significant difference in tone-elicited freezing response during recall was observed between Tg mice (n = 12) and WT mice (n = 11) (p < 0.01). All values are mean ± SEM; **p < 0.01.
(C–D) Transgenic mice with temporal expression of CaMKII-F89G restricted to learning and postlearning consolidation period performed normally in long-term memory recall tests. The black bar on the top column indicates the duration in which the CaMKII-F89G activity is expressed. The CaMKII-F89G activity in the Tg mice was selectively inhibited during the recall stage. Tg mice (n = 9) that received an i.p. injection of NM-PP1 (16.6 ng/g) 15 min prior the recall tests exhibited normal retrieval in comparison to that of wild-type mice (n = 11) in one-month contextual fear conditioning (C) and cued fear conditioning tests (D). All values are mean ± SEM.
To determine whether the elevated CaMKII activity affects memory acquisition and consolidation in the transgenic mice, we trained the mice using fear conditioning paradigm and maintained them on regular water between −2 and 30 days (see time-bar above Figure 3C). This protocol allowed both learning and consolidation processes in the transgenic mice to occur in the presence of CaMKII-F89G overexpression. However, 15 min prior to the recall tests on day 30, both groups of mice received i.p. injections of NM-PP1 so that CaMKII-F89G in the transgenic mice was suppressed at the time of memory recall. We observed that these transgenic mice performed normally in one-month long-term contextual recall (Figure 3C) and cued fear memory recall (Figure 3D), suggesting that the continuous presence of αCaMKII-F89G activity during learning and the following post-learning weeks does not alter functions of the memory systems. Furthermore, this evidence strongly indicates that the performance deficits observed in Figures 3A and 3B were likely due to the detrimental effects of αCaMKII-F89G overexpression on the retrieval of old fear memories.
To determine whether the performance deficits in the transgenic mice were, indeed, a result of retrievals deficit from the CaMKII-F89G overexpression during recall, we carried out the elevated CaMKII during recall paradigm and conducted those experiments using a third group of wild-type littermates and transgenic mice. Our previous experiments have shown that long-term inhibition of CaMKII-F89G activity in vivo can be readily achieved via noninvasive chronic oral intake (5 μM of NM-PP1 in drinking water) (Wang et al., 2003). Moreover, withdrawal of NM-PP1 after a one-month oral treatment could quickly restore the αCaMKII-F89G activity in the transgenic brains. In addition, chronic treatment of NM-PP1 on wild-type mice had no observable side effects (Wang et al., 2003).
Therefore, we pretreated both transgenic and their littermate control mice with NM-PP1 (5 μM in the drinking water) 2 days prior to fear conditioning training and continued the drug treatment until posttraining day 28 so that memory acquisition and consolidation occurred with normal amounts of CaMKII in the brain of transgenic mice. Again, the transgenic mice with the overexpressed CaMKII-F89G activity exhibited normal freezing behavior as assessed from the amount of immediate freezing (data not shown). NM-PP1 withdrawal began on posttraining day 28 such that αCaMKII-F89G would be fully active at the time for retrieving one-month fear memories. Similar to the retrieval deficits observed in short-term memory tests, we again found that transgenic mice exhibited significantly less freezing time than wild-type mice in both contextual recall (Figure 4A, histogram on the left) and cued fear recall (Figure 4A, histogram on the right).
Figure 4. Transient Presence of CaMKII-F89G Selectively during the Retention Stage Impaired Retrievals of One-Month Long-Term Fear Memories.
(A) Excessive αCaMKII activity during the recall stage disrupts the retrieval of one-month long-term contextual and cued fear memories. NM-PP1 was withdrawn 2 days prior retrieval. Significant deficits in either contextual freezing or cued freezing were observed in Tg mice (n = 12) compared with wild-type (n = 12) (p < 0.05).
(B) Suppression of αCaMKII-F89G during the recall stage rescued retrieval deficits. The same NM-PP1 treatment was delivered to wild-type and Tg groups as in (A) except that NM-PP1 (16.6 ng/g) was administered 15 min (via i.p.) prior to the retrieval tests. No significant deficits in either contextual freezing or cued freezing in the retrieval test were observed in Tg mice (n = 9) compared with wild-type (n = 8) (p > 0.05). All values are mean ± SEM; *p < 0.05.
Furthermore, we used another group of mice and conducted the similar protocol as in Figure 4A but with the exception that we injected NM-PP1 15 min prior to the one-month retention tests. This injection protocol would allow us to examine whether the recall impairment seen in Figure 4A was due to the overexpressed enzymatic activity of CaMKII or the mere presence of transgenic enzyme complex. Interestingly, we found that the transgenic mice exhibited normal recall of both one-month contextual fear memory (Figure 4B, histogram on the left) and one-month cued fear memory (Figure 4B, histogram on the right).
Thus, the above results collectively suggest that memory retrievals (regardless of newly formed memories or old memories) were specifically and reversibly disrupted by transient expression of αCaMKII-F89G enzymatic activity.
Recall-Induced Erasure of the Memory Is Specific to the One Being Actively Retrieved
The transient CaMKII-F89G overexpression-mediated retrieval deficits can reflect either the rapid erasure/degradation of the memories being retrieved or the difficulty in having access to otherwise intact memories. To distinguish those two scenarios, we used the same one-month fear memory training paradigm but designed the “first recall with CaMKII-F89 expression and second recall with CaMKII-F89G suppressed” protocol. The rationale for such experiments is that, if the retrieval deficits during the first recall were due to the difficulty in gaining access to the memory as a result of excessive CaMKII activity, one would expect that, upon the inhibition of αCaMKII-F89G, mice should be able to retrieve memory normally at the second recall. On the other hand, if the retrieval deficits during the first recall were caused by memory erasure, the mice should still not be able to recall even after αCaMKII-F89G has been inhibited at the time of second recall.
Thus, we treated another group of transgenic and control mice and measured their performance at the end of one-month retention. Consistent with our previous measurements, we found that the transgenic mice failed to retrieve the one-month contextual fear memory (Figure 5A, histogram on the left), as well as the one-month cued fear memories (Figure 5A, histogram on the right). After finishing the first recall tests, the mice were returned to home cages (for 1 hr). At 15 min before the second recall, those mice received NM-PP1 injection, and those mice were then subjected to the second recall tests. Interestingly, even with the inhibition of the CaMKII-F89G activity, the transgenic mice still exhibited profound retrieval deficits in the second retention tests (Figure 5B). Thus, this experiment suggests that retrieval deficits in the transgenic mice were not simply a memory accessibility problem or the impaired motor expression with elevated CaMKII activity, but likely resulted from the recall-induced rapid erasure of those fear memory traces being retrieved.
Figure 5. The Retrieval-Induced Memory Erasure in the Brains of the Transgenic Mice.

Retrieval-induced degradation of the stored long-term fear memory in transgenic (Tg) mice revealed by first recall (with CaMKII-F89G on) and second recall (with CaMKII-F89G off) protocol. At 1 month after training, both groups of mice were subjected to the first recall tests without any NM-PP1 treatment. Tg mice showed significant deficits in both contextual and cued freezing responses (n = 9) compared with wild-type (n = 10) (p < 0.01) (A). At 1 hour later, the second recall tests were conducted (B). At 15 min prior to the second recall tests, NM-PP1 was injected via i.p. As shown here, these Tg mice (even with the normal level of αCaMKII activity) at this stage still showed significant deficits in both contextual and cued freezing responses (n = 9) compared with wild-type (n = 10) (p < 0.01). All values are mean ± SEM; **p < 0.01. This indicates that the expression of CaMKII at the first recall have erased those fear memories. (C–D) A 2 week interval between the first and second recall tests was used for further assessment of memory erasure. The Tg mice exhibited profound deficits in fear memory recall in both the first and second recall tests (n = 12) compared with their controls (n = 12). All values are mean ± SEM; **p < 0.01, *p < 0.05.
To examine whether the observed memory deficits are resistant from spontaneous recovery (Miller and Springer, 1974; Lattal and Abel, 2004), we used another group of mice and repeated the above experimental protocol but with a longer interval (2 week interval) between the first recall tests and second recall tests. Once again, we found that there were significant differences in the recall performance between the transgenic and controls during the first recall tests (Figure 5C). Most importantly, under the condition of transgenic CaMKII activity being suppressed at the time of the second recall, the transgenic mice still exhibited significant impairment during the second retention tests in comparison to those of their littermates in both contextual and cued retention tests, even after the interval increased to 2 weeks of duration (Figure 5D). This revealed that there was no spontaneous memory recovery from the memory degradation occurring at the first recall.
To further determine whether such memory erasure is specific to the memory being retrieved or reflects the global degradation of all stored memories, we designed additional “sequential memory recall” experiments. The idea is to subject the mice to the active retrieval of one type of fear memories, for example, contextual memory, in the presence of CaMKII-F89G overexpression. From our previous experiments, we expect that this would initiate the erasure of contextual memory. After the contextual recall, the mice will be returned to the home cages and receive NM-PP1 injection so that CaMKII-F89G activity in Tg mice would be suppressed by the time the cued recall test is carried out 15 min later. If the contextual memory erasure reflects the global degradation of many memories, one should expect that cued recall will also be impaired even with CaMKII-F89G being inhibited. However, if the deficit in recalling contextual memory represents a highly specific process that only occurs to the contextual memory, the cued fear memory should remain intact and can be readily retrieved.
Thus, we trained yet another group of transgenic and control mice with the paired contextual and cued conditioning tests. Those mice were first subjected to the one-month contextual memory recall tests in the presence of CaMKII-F89G overexpression. Once again, the transgenic mice showed retrieval deficits in retrieving contextual fear memory in comparison to that of wild-type littermates (Figure 6A, histogram on the left). Then, after the completion of contextual recall tests, the mice were placed back in the home cages and injected with NM-PP1. At 15 min later, the mice underwent the cued memory recall tests. We found that these transgenic mice were able to retrieve cued fear memory normally (Figure 6A, histogram on the right), suggesting that the cued memory remained intact and was not degraded during contextual recall.
Figure 6. Recall-Induced Erasure Was Specific to the Memory Being Actively Retrieved during the CaMKII-F89G Expression.

(A) One month after training, control mice and Tg mice (with elevated CaMKII-F89G during recall) were subjected to the first recall test (contextual recall). Significant deficits in recall contextual memory were observed in Tg mice (n = 25) compared with control mice (n = 19) (p < 0.01). We then split the mice into two groups for measuring their second recall performance (cued recall) using either a 15 min interval or a 2 week interval. As shown, the Tg mice retrieved cued memory normally with 15 min interval (NM-PP1 was injected 15 min prior to the second cued recall test). The number of mice used in the 15 min interval test: Tg, n = 9; WT, n = 8. The number of mice used in the 2 week interval test (see Figure 6C): Tg, n = 16; WT, n = 11.
(B) The identical treatment was introduced to another group of Tg (n = 23) and control mice (n = 18) except that the cued fear memory was first recalled. We then split these mice into two groups for measuring their second recall performance (contextual recall) using either 15 min interval or 2 week interval (see Figure 6D). The Tg mice showed significant deficit in the first cued recall tests compared to the WT mice (P < 0.01). However, the Tg mice can still recall contextual memory normally (with 15 min interval). The number of mice used in the 15 min interval test is: Tg, n = 8; WT, n = 8. The number of mice used in the 2 week interval batch is: Tg, n = 15; n = 10 (see Figure 6D).
(C) Using a 2 week interval between the first (contextual) recall and second (cued) recall, we found that cued memory can still be retrieved normally in the Tg mice.
(D) Under a 2 week interval between the first recall (cued) and second recall (contextual) condition, there is no statistically significant difference between the Tg mice and control group. All values are mean ± SEM. **p < 0.01.
To further examine the relationship between the selective memory erasures and recall orders, we trained one more group of transgenic and control mice and tested them in reverse order. At the end of one-month retention, those mice were first subjected to the one-month cued fear memory recall tests in the presence of CaMKII-F89G overexpression. As expected, the transgenic mice showed retrieval deficits in cued fear memory in comparison to that of the wild-type littermates (Figure 6B, histogram on the left). Then, after the completion of cued recall tests, mice were brought back in the home cages and injected with NM-PP1. At 15 min later, the mice were brought back to the initial fear conditioning chamber and underwent the contextual memory recall tests. Our measurements showed that these transgenic mice retrieved contextual fear memory normally (Figure 6B, histogram on the right). Thus, CaMKII overexpression-mediated memory erasure appears to be highly specific to the memory that is being actively retrieved. In addition, we also tested the effects of longer intervals between the first recall and second recalls by using a 2 week interval. Again, transgenic mice exhibited indistinguishable performances compared with their wild-type controls in the second recall tests (Figures 6C for second cued recall and 6D for the second contextual recall). This suggests that memory erasure is selective to the retrieved memories and is also independent of test time intervals.
Memory Erasure Is a Rapid Process with Temporal Dependence
To further examine how rapidly the αCaMKII-F89G overexpression-mediated erasure/degradation of the memories occurs, we trained another set of transgenic and littermate control mice using one-day fear conditioning tests and conducted more detailed analysis by using multiple time points measurement over the course of memory recall. Our analysis clearly shows the temporal dependence of memory erasure (Figures 7A and 7B). That is, the freezing performance of transgenic mice was relatively high early in the recall test (initially 0.5 or 1 min) and then followed by a gradual decay. This is the case for both contextual fear memory (Figure 7A) and cued fear memory (Figure 7B). Generally speaking, the performance peaked in about 0.5–1 min and then decayed very rapidly over the next 2–3 min. This temporal dependence of memory erasure suggests that the action of memory recall under the elevated αCaM-KII-F89G expression triggered a very rapid biochemical and physiological process that results in erasure/degradation of the memories being retrieved.
Figure 7. Time Dependency of Memory Degradation during One-Day Contextual Recall and Cued Recall.

Tg = 12. WT = 12. The amount of freezing over each 30 s was pooled into one time point. The total measurement time was 3 min.
(A and B) For contextual recall at 0.5 min (p = 0.396); at 1 min (p = 0.039); at 1.5 min (p = 0.037); at 2.0 min (p = 0.0067); at 2.5 min (p = 0.0029); and at 3 min (p = 0.039) (A).
For cued recall at 0.5 min (p = 0.728); at 1 min (p = 0.008); at 1.5 min (p = 0.03); at 2.0 min (p = 0.0026); at 2.5 min (p = 0.004); and at 3 min (p = 0.025) (B). Degradation/erasure of the first training day memory evidenced from lack of savings on the second training trials. The first recall took place without NM-PP1 injection. The second and third trainings and recalls occurred under NM-PP1 treatment so that Tg-CaMKII-F89G was suppressed. Freezing at each test reflects the cumulative memory formation from the previous training day(s).
(C and D) Cumulative formation of contextual fear memories from three consecutive days of trainings. For the contextual memory recall, in the ANOVOA by effect of group: first test day F(1,22) = 5.01, p = 0.035; second day F(1,22) = 5.76, p = 0.025, < 0.05. There is also a significant genetic groups × day interaction: F(5,66) = 4.27, p = 0.002, < 0.01. There is no significant difference between the amount of freezing in the day one test of wild-type mice and day two test of transgenic mice, indicating no savings from the first conditioning memory in Tg mice.
(E and F) Cumulative formation of cued fear memories from three consecutive trainings. For the cued freezing, in the ANOVOA by effect of group: first test day F(1,22) = 5.67, p = 0.026; second day F(1,22) = 4.51, p = 0.045, < 0.05. There is also a significant groups × day interaction: F(5,66) = 6.50, p = 0.0006. However, there is no significant difference between the amount of cued freezing in day one test of wild-type mice and day two test of transgenic mice, suggesting erasure of one-day memory in Tg mice.
All values are mean ± SEM.
To further confirm whether memory erasure/degradation has indeed occurred, we performed the Memory Savings experiments that are often used for determining whether suppressed memory performance was due to lack of learning/storage or merely suppression of behavioral expression (Cahill et al., 1999; Fanselow and LeDoux, 1999; Goosens and Maren, 2003). Savings were measured by the rate of reacquisition in Tg mice after the purposed memory erasure occurred during the day 1 recall tests. Our data show that the transgenic mice had their memory erased or degraded since there is no savings from the first training (day 1), as revealed by the lagged memory performance in comparison to the wild-type mice upon additional trainings (second training) in both contextual (Figures 7C and 7D) and cued tests (Figures 7E and 7F). Therefore, these experiments further demonstrate that erasure has indeed occurred during the first recall.
Transgenic Mice Have Normal Ability to Express Freezing and Other Motor Behaviors
Finally, we conducted an additional set of behavioral measurements to measure the basic motor behavior and the freezing ability in the transgenic mice. We first measured locomotor activity in the open-field test and found that the transgenic mice with elevated CaMKII activity showed a normal amount of movement (Figure 8A). In addition, these transgenic mice also exhibited indistinguishable performances on the rotarod test (Figure 8B). Moreover, we further assessed their freezing responses by unexpectedly exposing them to cat odor (Dielenberg et al., 1999). Both transgenic and wild-type control mice exhibited heightened freezing upon exposure to cat odor, and there was no difference in their freezing responses between these two genotypes (Figure 8C). Therefore, we conclude that the elevated CaMKII activity in the transgenic mice did not affect their freezing ability and locomotor activity.
Figure 8. Tg Mice Are Normal in Open-Field Rotarod Tests and Have Normal Freezing Ability.
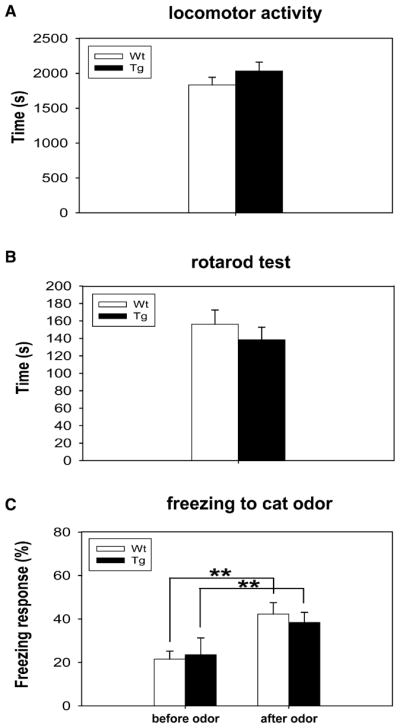
(A) No significant difference was found between the control and transgenic mice groups in the open-field test (WT, n = 18; Tg, n = 16). The data is expressed as mean ± SEM.
(B) Normal motor coordination, as measured by the rotarod test, was also observed in both Tg and control mice.
(C) Both transgenic and wild-type mice exhibited comparable freezing responses to cat odor. Before cat odor stimulus, two groups showed no significant difference in the basic level of freezing response (left column: WT, n = 9, 21.56% ± 3.64%; Tg, n = 9, 23.50% ± 5.29%, p = 0.77). When exposed to predatory odor, both wild-type and transgenic mice showed a greatly increased amount of freezing response (right column: WT, n = 9, 42.23% ± 7.78%; Tg, n = 9, 38.40% ± 4.63%, p = 0.68). However, no significant difference was observed between them. The value is expressed as mean ± SEM. Student’s t tests were used for statistical analysis.
DISCUSSION
Memory retrieval, a rapid reconstructive process involving a recapitulation of the learned information (Sara, 2000; Wagner et al., 2005), has not been explored with the same intensity as has memory acquisition and consolidation or reconsolidation. However, pioneering studies have shown that memory retrieval can be enhanced by the activation of β-adrenergic receptors (Sara and Devauges, 1988; Devauges and Sara, 1991). It has also been shown that learning and memory can be genetically enhanced (Tang et al., 1999; Tang et al., 2001; White and Youngentob, 2004). Thus, it would be important to explore the means by which memories (e.g., traumatic war memories and unwanted fear memories) might be inducibly and selectively erased.
In the current study, we have chosen three different behavioral paradigms, namely novel object recognition memory, contextual fear memory, and cued fear memory, for the following reasons. First, the behavioral expression of novel object recognition memory (via active exploration) is quite opposite to the behavioral expression of fear memories (via freezing); therefore, the observed retrieval deficits were unlikely due to the deficits in motor performance of memory tasks. Second, all of these behavioral tests involve a single training session, and acquisitions of those memories are known to be dependent on NMDA receptor activation (Brown and Aggleton, 2001; Falls et al., 1992; Maren et al., 1996; Rampon et al., 2000). Third, the novel object recognition memory tends to only last for several days (Tang et al., 1999), whereas contextual and cued fear memories in mice can last for at least 10 months (Cui et al., 2004). This information provides an opportunity to compare these memory processes that are known to have distinct temporal durations. Finally, novel object recognition memory is known to engage in different neuronal circuits compared to the fear memories, whereas the contextual fear memory and cued fear memory share many of the fundamental circuits (Davis et al., 1987), thereby allowing us to measure and directly compare whether the retrieval of those short-term and long-term memories shares any commonalities.
Our behavioral analyses illustrate that rapid memory retrieval can be unexpectedly manipulated by altering CaMKII activity using our chemical genetic method. Our previous biochemical studies show that NM-PP1 is highly specific to the genetically sensitized CaMKII-F89G and was completely ineffective against wild-type kinases, including αCaMKII (IC50 > 800 μM), CaMKIV (IC50 > 800 μM), PKC (IC50 > 800 μM), and CDK5 (IC50 > 300 μM) (Wang et al., 2003; Cho et al., 2007). Thus, this chemical genetic method has integrated the molecular and regional specificity of genetics with the high temporal resolution of chemical inhibition and is highly suitable for the analysis of memory retrieval.
The high-resolution, temporally controlled expression of αCaMKII-F89G activity during recall impairs the retrievals of both short-term and long-term memories, suggesting a common molecular process despite obvious differences in their synaptic and neuronal status (Gerard, 1949; Squire, 1987). Moreover, the retrieval deficits have been observed in several different memory tests, such as the novel object memory and fear memory tests, which are also likely to use somewhat different neural circuits to store and retrieve memory (Brown and Aggleton, 2001; Rudy et al., 2005). This further indicates a shared molecular process that is sensitive to the disruption by excessive CaMKII activity. More importantly, despite overlapping circuits involved in encoding contextual and cued fear memories, the selective erasure to contextual, but not cued, memory (or vice versa) strongly suggests that the erasure occurs at a highly specific set of synapses and circuits that define the specific features for contextual or cued memories.
The observed retrieval deficits are unlikely attributed to the potential locomotor-related performance problem caused by elevated CaMKII activity for the following reasons. First, the transgenic mice performed normally in the open-field measurement and rotarod tests. Second, the retrieval deficits in the transgenic mice have been assessed by two completely different behavioral measurements (e.g., novel object recognition memory, which involves the measurement of the preference in exploring the new or old objects, versus fear conditioning memories, which are measured by the amount of freezing time during recall). Third, in the case of fear conditioning, the freezing behaviors in the transgenic mice seem to be the same as that of the wild-type controls since the amount of immediate freezing is indistinguishable between the transgenic and control mice. Fourth, as shown in the cat odor exposure experiments, the transgenic mice indeed exhibit the same amount of fear freezing in comparison to that of their wild-type littermates. Finally, as shown in Figure 5, Tg mice still had exhibited performance deficits in the second recall tests even when the transgenic CaMKII level was suppressed by NM-PP1 injection. This again suggests that retrieval impairment is not due to the motor/performance problems caused by CaMKII overexpression. Therefore, the above evidence collectively supports the interpretation that the presence of transgenic CaMKII-F89G activity is likely caused by the recall-induced erasure/degradation of the memory traces that are being retrieved.
In the literature, some memory deficits (e.g., induced by protein synthesis inhibitors) prove to be only temporary, and spontaneous recovery from initial memory deficits has been reported over the time course of several days or week(s) (Miller and Springer, 1974; Lattal and Abel, 2004). Interestingly, the memory degradation described here seems to be long lasting since the poor performances were still observed even when the interval between the first and second recall tests were increased to the 2 week duration (Figures 5C and 5D).
In another recent report, Shema et al. described a very interesting phenomenon in which posttraining injection of an inhibitor of the protein kinase M zeta into the insular cortex caused retrieval deficits of one-month conditioned taste aversion memory (Shema et al., 2007). In that study, the authors show that application of the inhibitor at various time points (e.g., 3 days, 7 days, or 25 days after learning) is all capable of causing performance deficits in one-month retention tests (Shema et al., 2007). In our case, memory retrieval deficit only occurs if transgenic αCaM-KII-F89G is expressed at the time of recall. On the other hand, the transient presence of transgenic αCaMKII-F89G enzyme at second, third, or fourth posttraining weeks had no effect on the recall of one-month contextual and cued fear memories as long as retrieval took place within the suppression of transgenic αCaMKII-F89G activity (Wang et al., 2003). Furthermore, the inhibitor of protein kinase M zeta seems to need at least 2 hr to exert its effects on the stored memory, whereas the memory erasure described here is an extremely rapid process (within 1–3 min) at the time of memory recall. Finally, unlike the general effects of the protein kinase M zeta inhibitor on the multiple memories stored in the same region (two conditioned tests were used), the effects we observed here are highly restricted to the one that is undergoing active retrieval while leaving the unretrieved memory intact. Therefore, transgenic αCaMKII-F89G overexpression-mediated memory erasure takes place only when that memory is undergoing active recall.
What might be the cellular and physiological explanations that can account for the observed recall-induced retrieval deficits? It is interesting to note that the transgenic overexpression of αCaMKKII-F89G can alter the bidirectional synaptic plasticity response curve (Wang et al., 2003). This is consistent with the role of the CaMKII in regulating LTP (Malinow et al., 1989; Silva et al., 1992; Barria et al., 1997; Malenka and Nicoll, 1999; Mayford et al., 1995). Our previous measurement of synaptic physiology has revealed that, in addition to the larger LTP in response to 10 or 100 Hz stimulation, the transgenic hippocampus can produce a profound shift of LTD peak from 1 to 3 Hz. This shift in LTD peak response can be readily reversed by the addition of 5 μM NM-PP1 in the recording solution (Wang et al., 2003).
It is tempting to speculate that the relation between the rapid induction of LTD response by 3 Hz frequency may play a role in memory erasure observed in the transgenic mice. The 3 Hz frequency stimulation usually does not produce changes in synaptic efficacy in the wild-type brain (Bear and Malenka, 1994; Korz and Frey, 2005). Many in vivo studies report that neurons often fire in the range of 3–12 Hz during recognition of spatial and nonspatial cues (O’Keefe and Dostrovsky, 1971; Deadwyler et al., 1996; Wood et al., 1999; Lin et al., 2005; Lin et al., 2006; Quirk et al., 1992; Sharp, 1999). Therefore, it would be interesting to entertain the idea that, in the presence of the elevated CaMKII activity, 3 Hz-like firing patterns embedded in the retrieval neural process might have induced abnormal LTD-like responses in those memory circuits of the transgenic brains, leading to aberrant and selective degradation of the recalled memory traces. While this explanation is attractive, we currently do not know whether such altered LTD responses have also occurred in other brain regions.
It might be also possible that CaMKII overexpression may alter other biochemical pathways implicated in memory retrieval (Devauges and Sara, 1991; Murchison et al., 2004; Chen et al., 2005; Moulds and Bryant, 2005) or synaptic protein degradation (Lee et al., 2008). For example, it might be worthwhile to examine whether CaMKII overexpression during recall period triggers some kind of rapid synaptic protein degradation via abnormally activating a polyubiquitination pathway (Lee et al., 2008).
It is also prudent to consider the theoretical possibility that increasing αCaMKII activity at the time of encoding may produce some sort of neurotoxicity to the cells involved in forming the memory trace. Our data seem to argue against this scenario: in both one-hour and one-month memory tests (see Figures 1D–1F, 3C, and 3D), the constitutive presence of transgenic αCaMKII-F89G at the time of learning and postlearning periods did not impair memory performance in the transgenic mice.
One future effort might be to define further the critical subregions in the forebrain where those memories might be stored and retrieved (Cui et al., 2004; Wagner et al., 2005; Frankland and Bontempi, 2005; Rudy et al., 2005). For example, it would be most ideal if our manipulation could be further restricted to a set of specific subsites, say, the lateral amygdala versus central amygdala, the anterior cingular cortex versus perirhinal cortex, etc. However, it is worthy to note that memory may require multiple regions to participate various aspects of memory components (Ribeiro et al., 2004; Euston et al., 2007; Tsien, 2007), thereby requiring simultaneous manipulation of the transgenic activity in those subregions.
Given the fact that so many war veterans coming back from war zones often suffer from reoccurring traumatic memory replays, our report of selective erasure of fear memories in an inducible and rapid way suggests the existence of molecular paradigm(s) under which some traumatic memories can be erased or degraded while preserving other memories in the brain. However, we do not think that our chemical genetic approach in its current form can be applied directly as a clinical strategy since it would require the development of some sorts of αCaMKII-specific activators. On the other hand, it might be useful to further identify the downstream drugable targets through which overexpressed αCaMKII produces such an effect. In conclusion, we have shown that transient elevation of transgenic αCaMKII activity at the time of memory recall can cause rapid erasure of memory being retrieved. Our experiments have illustrated a molecular genetic paradigm through which a selected set of memories, such as new and old fear memories, can be rapidly and specifically erased in a controlled and inducible manner while leaving other memories intact in the brain.
EXPERIMENTAL PROCEDURES
Production and Basic Characterizations of Transgenic Mice
The transgenic mice were produced and maintained on the BCF1 hybrid background (Wang et al., 2003). Southern blot was used for determining the copy number of the transgene, which is estimated to be approximately five copies in the transgenic mice. Routine genotyping of the CaMKII F89G transgene was conducted by PCR. Tail DNAs were amplified 35 cycles (94°C, 50 s; 55°C, 45 s; and 72°C, 50 s) on a thermal cycler using primers specific to the SV40 polyA (5′-GCTAGAGGATCTTTGTGAAGGAAC-3′ and 5′-GGAAAGTCC TTGGGGTCTTCTACCT-3′, resulting in a single 319 base pair band). The biochemical assays of CaMKII activity and NM-PP1 pharmacokinetics were conducted as previously described (Wang et al., 2003). NM-PP1 was dissolved in DMSO (molecular biology grade) first and then diluted in water (1:1000 dilution for drinking) or sterile PBS (for IP injection), as noted by Brian Jones’ message. Dilution was done with vigorous mixing in order to prevent NM-PP1 precipitation.
Behavioral Measurements
Mice were maintained in a temperature-controlled environment on a 12 hr light/dark cycle. Adult CaMKII F89G transgenic and wild-type littermate mice (2- to 4-months old) were used throughout all behavioral tests. For NM-PP1 treatment, the dose of 16.57 ng per gram of body weight per mouse was used via i.p. injection (5 μM). NM-PP1 was dissolved in DMSO with final concentration of 0.02% of DMSO in the solution. All of the control animals (wild-type littermates) received the same drug treatment as the transgenic animals during the behavioral experiments.
Novel Object Recognition Task
The experimental protocol was the same as described previously (Rampon et al., 2000; Tang et al., 1999). Briefly, mice were individually habituated to an open-field box (20 × 20 × 10 high inches) for 3 days. During the training sessions, two novel objects were placed in the open field, and the animal was allowed to explore for 15 min. The time spent exploring each object was recorded. During the one-hour recall tests, the animal was placed back into the same box, in which one of the familiar objects during training was replaced by a novel object and allowed to explore freely for 15 min. A preference index, a ratio of the time spent exploring any one of the two objects (training session) or the novel one (retention session) over the total time spent exploring both objects, was used to measure recognition memory. Data were calculated as mean ± SEM. Student’s t test was used to determine genotype effects on the behavioral responses.
Fear Conditioning Task
Protocols were the same as those described previously (Rampon et al., 2000; Tang et al., 1999). A fear conditioning shock chamber (10 × 10 × 15 high inches) and TruScan multiparameter activity monitors (Coulbourn Instrument) were used. The mice (2- to 4-months old) were handled for 3 days and then habituated to the training chamber for 5 min 1 day before the training began. The conditioned stimulus (CS) used was an 85 dB sound at 2800 Hz, and the unconditioned stimulus (US) was one time-continuous scrambled foot shock at 0.75 mA for 2 s. After the CS/US paring, the animal was allowed to stay in the chamber for another 30 s for the measurement of immediate freezing. Freezing was judged as a complete immobility of the body except for respiratory movements. The animals were then returned to their home cages for either 1 hr or 1 month as indicated in the experiments. During the recall test, each mouse was placed back into the same chamber, and the freezing responses were recorded and scored every 5 s for 5 min (contextual freezing). Subsequently, the mice were put into a novel chamber and monitored for 2 min (pretone freezing) and then subjected to 3 min of CS tone exposure, during which their freezing responses were measured using a time-sample method every 5 s. Data were calculated as mean ± SEM. Student’s t test was used to determine genotype effects on the behavioral responses.
Open-Field and Rotarod Tests
The protocols were the same as described (Cui et al., 2005). For the measurement of the open-field activity, mice were placed in an open field, made of a 14 × 14 inches black box. The box was marked by a 2 × 2 inches small square grid (7 squares by 7 squares, with 49 squares in total). The open-field activity of animals was measured by the number of crosses that the mice have passed within the 3 min period. For the measurement of rotarod test, the mice were placed to an accelerating rotating wood rod. The rod is 12 inches long and 1 inch in diameter. The initial rotation speed was set at 4 rpm and then steadily accelerated to 40 rpm. The performance was measured by the amount of time (in seconds) that mice managed to remain on the rotating rod. Student’s t test was used to determine the significance of those behavioral measurements.
Freezing Responses to Cat Odor Exposure
This experiment was performed according to the method described previously (Dielenberg et al., 1999). A chamber of perspex (36 cm × 21cm × 30 cm) was used, and cat collars (with or without cat order) were pasted on each wall (15 cm above the basic floor of box) to prevent mice from touching the collars. Those cat collars without cat odor were new and not worn by a cat previously. Those cat collars with cat odor had been worn by domestic cats for 1 month before the experiment. The collar was cut into several equivalent pieces, which were stored in sealed plastic bag at 4°C.
The experiment consisted of two phases: habituation and test phase. During habituation phase, mice were individually adapted to the Exposure Chamber for 3 days. Every day, the mouse was placed into the center of the chamber with the unused cat collars (without cat odor stimulus) for 10 min. On the third day of habituation, the basic level of freezing response was recorded in the absence of predatory odor (before cat odor exposure). During the test phase, each mouse was put into the chamber containing the used cat collars for 10 min. The amount of freezing responses was recorded as their responses during cat odor exposure. Student’s t tests were used for statistical analysis.
Supplementary Material
Acknowledgments
We thank Monica Meyer for editing our manuscript. We are indebted to Dr. Kevan Shokat of UCSF for providing the NM-PP1 inhibitor. H.W. was supported by an NSRA fellowship, and X.C. and B.M. were supported in part by 973 program (#2003CB716605 and #2003CB716602) and NSF and National Natural Science Fund of China (#30670682) and funds from SSTC (06DZ19003). This research was supported by funds from NIMH and NIA (to J.Z.T.).
Footnotes
The Supplemental Data include one figure and can be found with this article online at http://www.neuron.org/supplemental/S0896-6273(08)00768-X.
References
- Alberini CM. Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trends Neurosci. 2005;28:51–56. doi: 10.1016/j.tins.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Biedenkapp JC, Rudy JW. Context memories and reactivation: constraints on the reconsolidation hypothesis. Behav Neurosci. 2004;118:956–964. doi: 10.1037/0735-7044.118.5.956. [DOI] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray N, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Morgan DO, Shokat KM. A chemical switch for inhibitor-sensitive alleles of any kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Brown MW, Aggleton JP. Recognition memory: what are the roles of the perirhinal cortex and hippocampus? Nat Rev Neurosci. 2001;2:51–61. doi: 10.1038/35049064. [DOI] [PubMed] [Google Scholar]
- Cahill L, Weinbergerm NM, Roosendaal B, McGaugh JL. Is the amygdale a locus of “conditioned fear”? Some questions and caveats. Neuron. 1999;23:227–228. doi: 10.1016/s0896-6273(00)80774-6. [DOI] [PubMed] [Google Scholar]
- Cao X, Cui Z, Feng R, Tang YP, Qin Z, Mei B, Tsien JZ. Maintenance of superior learning and memory function in NR2B transgenic mice during aging. Eur J Neurosci. 2007;25:1815–1822. doi: 10.1111/j.1460-9568.2007.05431.x. [DOI] [PubMed] [Google Scholar]
- Chan CS, Weeber EJ, Kurup S, Sweatt JD, Davis RL. Integrin requirement for hippocampal synaptic plasticity and spatial memory. J Neurosci. 2003;18:7107–7116. doi: 10.1523/JNEUROSCI.23-18-07107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Cho MH, Cao X, Wang D, Tsien JZ. Dentate gyrus-specific manipulation of beta-Ca2+/calmodulin-dependent kinase II disrupts memory consolidation. Proc Natl Acad Sci USA. 2007;104:16317–16322. doi: 10.1073/pnas.0703344104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RE, Zola SM, Squire LR. Impaired recognition memory in rats after damage to the hippocampus. J Neurosci. 2000;20:8853–8860. doi: 10.1523/JNEUROSCI.20-23-08853.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Wang H, Tan Y, Zaia KA, Zhang S, Tsien JZ. Inducible and reversible NR1 knockout reveals crucial role of the NMDA receptor in preserving remote memories in the brain. Neuron. 2004;41:781–793. doi: 10.1016/s0896-6273(04)00072-8. [DOI] [PubMed] [Google Scholar]
- Cui Z, Lindl KA, Mei B, Zhang S, Tsien JZ. Requirement of NMDA receptor reactivation for consolidation and storage of nondeclarative taste memory revealed by inducible NR1 knockout. Eur J Neurosci. 2005;22:755–763. doi: 10.1111/j.1460-9568.2005.04257.x. [DOI] [PubMed] [Google Scholar]
- Davis M, Hitchcock J, Rosen JB. Anxiety and amygdala: pharmacological and anatomical analysis of the fear-potentiated startle paradigm. In: Bower GH, editor. The Psychology of Learning and Memory. New York: Academic; 1987. [Google Scholar]
- Davis M, Walker DL, Myers KM. Role of the amygdala in fear extinction measured with potentiated startle. Ann NY Acad Sci. 2003;985:218–232. doi: 10.1111/j.1749-6632.2003.tb07084.x. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, Bunn T, Hampson RE. Hippocampal ensemble activity during spatial delayed-nonmatch-to-sample performance in rats. J Neurosci. 1996;16:354–372. doi: 10.1523/JNEUROSCI.16-01-00354.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devauges V, Sara SJ. Memory retrieval enhancement by locus coeruleus stimulation: evidence for mediation by beta-receptors. Behav Brain Res. 1991;43:93–97. doi: 10.1016/s0166-4328(05)80056-7. [DOI] [PubMed] [Google Scholar]
- Dielenberg RA, Arnold JC, McGregor IS. Low-dose midazolam attenuates predatory odor avoidance in rats. Pharmacol Biochem Behav. 1999;62:197–201. doi: 10.1016/s0091-3057(98)00064-1. [DOI] [PubMed] [Google Scholar]
- Elgersma Y, Fedorov NB, Ikonen S, Choi ES, Elgersma M, Carvalho OM, Giese KP, Silva AJ. Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron. 2002;36:493–505. doi: 10.1016/s0896-6273(02)01007-3. [DOI] [PubMed] [Google Scholar]
- Euston DR, Tatsuno M, McNaughton BL. Fast-forward playback of recent memory sequences in prefrontal cortex during sleep. Science. 2007;318:1147–1150. doi: 10.1126/science.1148979. [DOI] [PubMed] [Google Scholar]
- Falls WA, Miserendino MJD, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, LeDoux JE. Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdale. Neuron. 1999;23:229–232. doi: 10.1016/s0896-6273(00)80775-8. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- Gerard RW. Physiology and psychiatry. Am J Psychiatry. 1949;105:161–173. doi: 10.1176/ajp.106.3.161. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Maren S. Pretraining NMDA receptor blockade in the basolateral complex, but not the central nucleus, of the amygdala prevents savings of conditional fear. Behav Neurosci. 2003;117:738–750. doi: 10.1037/0735-7044.117.4.738. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annu Rev Biochem. 1992;61:559–601. doi: 10.1146/annurev.bi.61.070192.003015. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Eichenbaum H. Hippocampal mechanisms for the context-dependent retrieval of episodes. Neural Netw. 2005;18:1172–1190. doi: 10.1016/j.neunet.2005.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Hunter T. 1001 protein kinases redux–towards 2000. Semin Cell Biol. 1994;5:367–376. doi: 10.1006/scel.1994.1044. [DOI] [PubMed] [Google Scholar]
- Kensinger EA, Schacter DL. Retrieving accurate and distorted memories: neuroimaging evidence for effects of emotion. Neuroimage. 2005;27:167–177. doi: 10.1016/j.neuroimage.2005.03.038. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Jung MW. Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev. 2006;30:188–202. doi: 10.1016/j.neubiorev.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korz V, Frey JU. Bidirectional modulation of hippocampal long-term potentiation under stress and no-stress conditions in basolateral amygdala-lesioned and intact rats. J Neurosci. 2005;25:7393–7400. doi: 10.1523/JNEUROSCI.0910-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Abel T. Behavioral impairments caused by injections of the protein synthesis inhibitor anisomycin after contextual retrieval reverse with time. Proc Natl Acad Sci USA. 2004;101:4667–4672. doi: 10.1073/pnas.0306546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Lee SH, Kim H, Kaang BK. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Lin L, Osan R, Shoham S, Jin W, Zuo W, Tsien JZ. Identification of network-level coding units for real-time representation of episodic experiences in the hippocampus. Proc Natl Acad Sci USA. 2005;102:6125–6130. doi: 10.1073/pnas.0408233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Osan R, Tsien JZ. Organizing principles of real-time memory encoding: Neural clique assemblies and universal neural codes. Trends Neurosci. 2006;29:48–56. doi: 10.1016/j.tins.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Liu Y, Bishop A, Witucki L, Kraybill B, Shimizu E, Tsien J, Ubersax J, Blethrow J, Morgan DO, Shokat KM. Structural basis for selective inhibition of Src family kinases by PP1. Chem Biol. 1999;6:671–678. doi: 10.1016/s1074-5521(99)80118-5. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation–a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Stote DL, Fanselow MS. N-methyl-D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O’Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- Miller RR, Springer AD. Implications of recovery from experimental amnesia. Psychol Rev. 1974;81:470–473. doi: 10.1037/h0036951. [DOI] [PubMed] [Google Scholar]
- Moulds ML, Bryant RA. An investigation of retrieval inhibition in acute stress disorder. J Trauma Stress. 2005;18:233–236. doi: 10.1002/jts.20022. [DOI] [PubMed] [Google Scholar]
- Murchison CF, Zhang XY, Zhang WP, Ouyang M, Lee A, Thomas SA. A distinct role for norepinephrine in memory retrieval. Cell. 2004;117:131–143. doi: 10.1016/s0092-8674(04)00259-4. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE. The labile nature of consolidation theory. Nat Rev Neurosci. 2000;1:216–219. doi: 10.1038/35044580. [DOI] [PubMed] [Google Scholar]
- O’Keefe J, Dostrovsky J. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res. 1971;34:171–175. doi: 10.1016/0006-8993(71)90358-1. [DOI] [PubMed] [Google Scholar]
- Ouyang Y, Kantor D, Harris KM, Schuman EM, Kennedy MB. Visualization of the distribution of autophosphorylated calcium/cal-modulin-dependent protein kinase II after tetanic stimulation in the CA1 area of the hippocampus. J Neurosci. 1997;17:5416–5427. doi: 10.1523/JNEUROSCI.17-14-05416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Muller RU, Kubie JL, Ranck JB., Jr The positional firing properties of medial entorhinal neurons: description and comparison with hippocampal place cells. J Neurosci. 1992;12:1945–1963. doi: 10.1523/JNEUROSCI.12-05-01945.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampon C, Tang YP, Goodhouse J, Shimisu E, Maureen K, Tsien JZ. Enrichment promotes structural changes and rescues nonspatial memory deficits in CA1-NMDAR1-knockout mice. Nat Neurosci. 2000;3:238–244. doi: 10.1038/72945. [DOI] [PubMed] [Google Scholar]
- Ribeiro S, Gervasoni D, Soares ES, Zhou Y, Lin SC, Pantoja J, Lavine M, Nicolelis MA. Long-lasting novelty-induced neuronal reverberation during slow-wave sleep in multiple forebrain areas. PLoS Biol. 2004;2:E24. doi: 10.1371/journal.pbio.0020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy JW, Biedenkapp JC, O’Reilly RC. Prefrontal cortex and the organization of recent and remote memories: an alternative view. Learn Mem. 2005;12:445–446. doi: 10.1101/lm.97905. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Biedenkapp JC, Moineau J, Bolding K. Anisomycin and the reconsolidation hypothesis. Learn Mem. 2006;13:1–3. doi: 10.1101/lm.157806. [DOI] [PubMed] [Google Scholar]
- Sara SJ. Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem. 2000;7:73–84. doi: 10.1101/lm.7.2.73. [DOI] [PubMed] [Google Scholar]
- Sara SJ, Devauges V. Priming stimulation of locus coeruleus facilitates memory retrieval in the rat. Brain Res. 1988;438:299–303. doi: 10.1016/0006-8993(88)91351-0. [DOI] [PubMed] [Google Scholar]
- Sharp PE. Subicular place cells expand or contract their spatial firing pattern to fit the size of the environment in an open field but not in the presence of barriers: comparison with hippocampal place cells. Behav Neurosci. 1999;113:643–662. doi: 10.1037//0735-7044.113.4.643. [DOI] [PubMed] [Google Scholar]
- Shema R, Sacktor TC, Dudai Y. Rapid erasure of long-term memory association in the cortex by an inhibitor of PKM zeta. Science. 2007;317:951–953. doi: 10.1126/science.1144334. [DOI] [PubMed] [Google Scholar]
- Shimizu E, Tang YP, Rampon C, Tsien JZ. NMDA receptor-dependent synaptic reinforcement as a crucial process for memory consolidation. Science. 2000;290:1170–1174. doi: 10.1126/science.290.5494.1170. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Squire LR. Memory and Brain. New York: Oxford University Press; 1987. [Google Scholar]
- Sutherland K, Bryant RA. Self-defining memories in post-traumatic stress disorder. Br J Clin Psychol. 2005;44:591–598. doi: 10.1348/014466505X64081. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Tang YP, Wang H, Feng R, Kyin M, Tsien JZ. Differential effects of enrichment on learning and memory function in NR2B transgenic mice. Neuropharmacology. 2001;41:779–790. doi: 10.1016/s0028-3908(01)00122-8. [DOI] [PubMed] [Google Scholar]
- Torras-Garcia M, Lelong J, Tronel S, Sara SJ. Reconsolidation after remembering an odor-reward association requires NMDA receptors. Learn Mem. 2005;12:18–22. doi: 10.1101/lm.80905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ. The memory code. Sci Am. 2007;297:52–59. doi: 10.1038/scientificamerican0707-52. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta P, Tonegawa S. Essential role for the hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial learning. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Wagner AD, Shannon BJ, Kahn I, Buckner RL. Parietal lobe contributions to episodic memory retrieval. Trends Cogn Sci. 2005;9:445–453. doi: 10.1016/j.tics.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Wang H, Shimizu E, Tang Y, Cho M, Alaimo PJ, Morimoto H, Zhuo M, Shokat KM, Tsien JZ. Conditional protein knockout reveals temporal requirement of CaMKII reactivation for long-term memory formation in the brain. Proc Natl Acad Sci USA. 2003;100:4287–4292. doi: 10.1073/pnas.0636870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TL, Youngentob SL. The effect of NMDA-NR2B receptor subunit overexpression on olfactory memory task performance in the mouse. Brain Res. 2004;1021:1–7. doi: 10.1016/j.brainres.2004.05.114. [DOI] [PubMed] [Google Scholar]
- Wittenberg GM, Tsien JZ. An emerging molecular and cellular framework for memory processing by the hippocampus. Trends Neurosci. 2002;25:501–505. doi: 10.1016/s0166-2236(02)02231-2. [DOI] [PubMed] [Google Scholar]
- Wood ER, Dudchenko PA, Eichenbaum H. The global record of memory in hippocampal neuronal activity. Nature. 1999;397:613–616. doi: 10.1038/17605. [DOI] [PubMed] [Google Scholar]
- Zola-Morgan SM, Squire LR. The primate hippocampal formation: Evidence for a time-limited role in memory storage. Science. 1990;250:288–290. doi: 10.1126/science.2218534. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.