

Abstract
It is widely recognized that Th2 cytokines derived from T cells play a major role in the development of allergic lung inflammation that causes most asthma. Beta-agonists are important rescue and maintenance therapies for asthma, yet our understanding of beta-agonist effects on T cell biology is surprisingly poor. Recent studies using both cell culture and more integrative models are beginning to reveal beta-agonist regulation of T cell signaling and function that may be important in the pathogenesis and treatment of asthma and possibly other inflammatory diseases. Here we provide a comprehensive review of the literature concerning beta-agonist effects on T cells, and discuss the relevance of emerging paradigms of beta-adrenergic receptor signaling to T cell function.
Keywords: beta-agonist, T cell, Asthma, Inflammation, Interferon, Interleukin, Airway Hyperresponsiveness, NF-kappaB, JAK, Beta-2-Adrenergic Receptor, Th1, Th2, receptor, AKAP, MAPK, NF-AT, ZAP-70, CD3, Bronchoalveolar Lavage, Prostaglandin, Review
2. INTRODUCTION
T cells are a major focus of asthma research. It is widely recognized that Th2 cytokines derived from T cells are important if not required pathogenic agents of atopic asthma. Given the importance of beta-agonists in asthma therapy, one might suspect that the topic of beta-agonist-mediated regulation of T cell function in asthma would be exhaustively studied and well understood, but such is not the case. Most of the frustration in addressing this topic stems from the complex nature of those systems in which T cells are studied. Here we review the literature concerning beta-agonist effects on T cells that occur in vivo and in culture, and discuss the relevance of emerging paradigms of beta-adrenergic receptor signaling to T cell function.
3. AT THE CELLULAR/MOLECULAR LEVEL: T CELL SIGNALING
3.1. Antigen-dependent signaling
Many important T cell functions, such as proliferation, survival, and cytokine production, are regulated by signaling via the T cell receptor (TCR)/CD3 complex, which is activated naturally by antigenic peptides presented by major histocompatibility complexes (MHCs). Experimentally, agonistic antibodies to CD3 (and usually also the co-stimulatory molecule CD28) or mitogens that agglutinate the TCR/CD3 complex, such as phytohemagglutinen-L (PHA) are frequently used to simulate antigenic stimulation. Such non-specific stimulations are often required in the human system to control for the diverse cognate antigenic repertoire of the T cell populations among individuals.
Provided below is a brief summary of the salient features of antigen-dependent TCR signaling (for a more detailed description, refer to (1–3), and references therein). The T cell receptor is actually a complex comprised of two TCR chains (TCRalpha and TCRbeta, or TCRgamma and TCRdelta), which recognize antigenic peptides presented by MHC molecules, and the CD3 subunits (gamma, delta, epsilon, eta/zeta), which are required to transduce the signals to the cytoplasm when the TCR engages its cognate antigenic peptide. Co-receptor molecules (CD4 or CD8, depending on the T cell subset) and co-stimulatory molecules (e.g., CD28) also may be present in the complex during or after initial engagement of the TCR with peptide/MHC. Proximal TCR signaling (Figure 1) involves the TCR “recognizing” its cognate peptide antigen presented by MHC molecules. When the TCR binds its cognate antigenic peptide, this “recognition” is sensed by CD3 complex molecules, leading to recruitment and auto-activation of the Src family members Lck and Fyn. These two proteins activate CD3zeta/eta subunits, which in turn recruit zeta-chain-associated protein kinase 70 (ZAP-70) via their immunoreceptor tyrosine-based activation motifs, allowing Lck to phosphorylate and activate ZAP-70.
Figure 1.
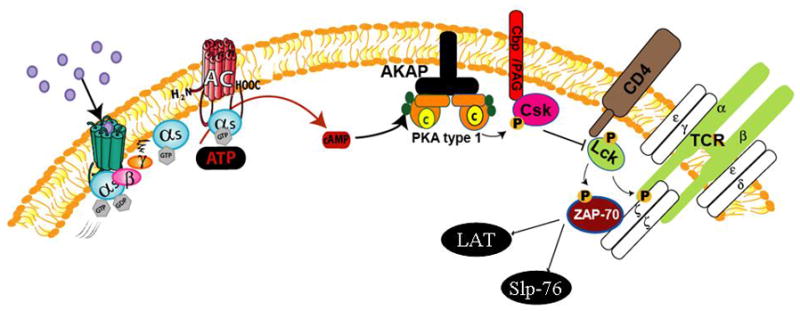
Proximal TCR signaling and regulation by Gs-coupled receptors and PKA. Engagement of TCR with cognate peptide antigen presented by MHC molecules promotes the membrane recruitment and activation of the Src kinases Lck and Fyn (Fyn not shown), phosphorylation of immunoreceptor tyrosine-based activation motifs within the CD3 complex, and ultimate recruitment and activation of ZAP-70. Active ZAP-70 then leads to activation of multiple downstream signaling cascades via activation of LAT and SLP-76 (as depicted in Figure 2). Agonist-activated Gs-coupled receptors such as the Beta2AR promote adenylyl cyclase activity resulting in hydrolysis of ATP into cAMP. cAMP binds the regulatory subunits of the cAMP-dependent protein kinase (PKA, shown docking on an AKAP) which induce release and activation of PKA catalytic subunits. The Src kinase Csk phosphorylates Lck and inhibits its activity. Csk resides in lipids rafts in association with Cbp/PAG in resting T cells, but is transiently displaced to the cytosol upon T cell activation. PKA phosphorylates Csk on Ser 364 to increase Csk activity, which results in Lck inhibition, and inhibition of TCR zeta–chain phosphorylation, and proximal TCR signaling.
From ZAP-70, multiple downstream effector signaling pathways are activated, including p42/p44 mitogen-activated protein kinase (MAPK), p38 MAPK, c-Jun N-terminal kinase (JNK), phosphoinositide 3-kinase (PI3K), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kappaB), and Ca2+/nuclear factor for activated T-cells (NF-AT) pathways, as illustrated in Figure 2. ZAP-70 activates LAT (Linker for activation of T cells), which is responsible for activating the Grb2/SOS complex and phospholipase C (PLC) –gamma. The first complex leads to activation of Ras and the downstream p42/p44 MAPK pathway, as well as connecting to the PI3K pathway. PLC-gamma releases diacylglyceride (DAG) and inositol-triphosphate (IP3) from phosphoinositol-diphosphate. DAG activates protein kinase C (PKC) theta, which transduces activating signals to the NF-kappaB and MAPK pathways. IP3 release leads to elevation of cytoplasmic Ca2+ levels. Ca2+-bound calmodulin stimulates calcineurin’s phosphatase activity, which activates the transcription factor NF-AT via dephosphorylation of its regulatory domain. ZAP-70 also activates SH2 domain containing leukocyte protein of 76kDa (SLP-76). SLP-76 mediates activation of Vav, which via Rac1, leads to activation of the p38 MAPK and JNK pathways. SLP-76 also connects to the actin reorganization machinery via Vav/Nck for Cdc42/Wiskott- Aldrich syndrome protein-mediated actin reorganization and TCR clustering. SLP-76 and Fyn stimulate degranulation promoting adaptor protein (ADAP) to recruit VASP, which directs actin-dependent clustering of integrins.
Figure 2.
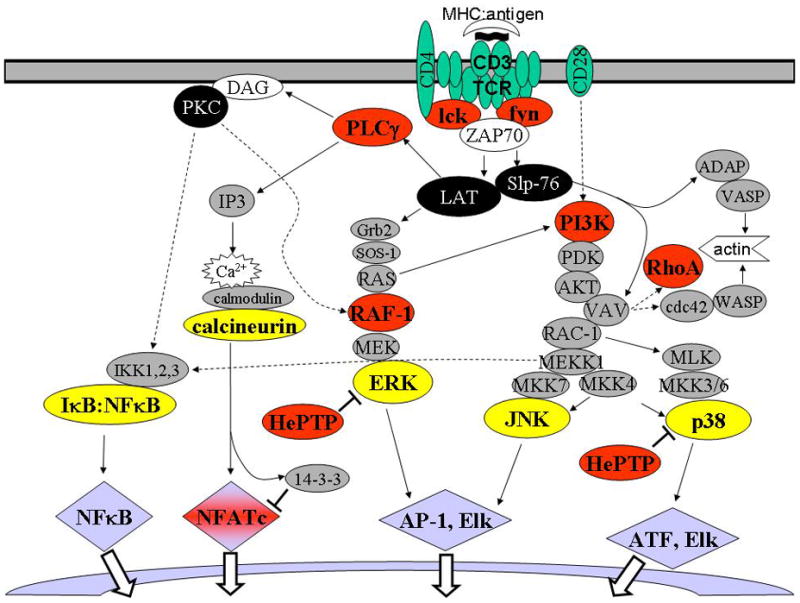
Downstream T-cell signaling events and impact of PKA. Major downstream signaling cascades resulting from MHC:cognate peptide stimulation of TCR:CD3 complex are depicted. Critical signaling molecules transmitting upstream signals from ZAP-70 to multiple major effector signaling pathways (e.g., MAPK, NF-kappaB, Ca2+/NF-AT pathways) are represented by black shading. Intermediate signaling moieties and adaptors are shaded gray. Terminal effector molecules (or complexes), which are responsible for the activation/nuclear translocation of transcription factors, are shaded yellow. Select transcription factors activated as a result of TCR/CD3-mediated stimulation are shaded blue. Inhibition events are indicated by T. Signaling molecules reported to be inhibited by catalytically active PKA are shaded red. This schematic is intended to present major signaling events in antigen-activated T cells, and may not include all signaling molecules, adaptors, and transcription factors involved in this complex system.
The co-receptors CD4 and CD8 enhance activation of downstream signaling pathways by recruiting and activating Lck to enhance ZAP-70 activation. Co-stimulatory molecules, such as CD28 and inducible costimulatory protein (ICOS), mainly contribute by strongly activating the PI3K pathway, dependent upon TCR/CD3- and co-receptor- activated Lck. Overall, the downstream effector signaling from the TCR/CD3 complex leads to activation and nuclear translocation of numerous transcription factors (including NF-κB, NF-AT, AP1 (fos/jun), Atf-1, Elk-1, 3′-5′-cyclic adenosine monophosphate (cAMP) response element binding, and c-Myc, among others), and reorganization of the actin network.
3.2. Antigen-independent signaling
Conversely, “antigen-independent” signaling, mediated by agents such as IL-2 and IL-15, may also regulate T cell functions under various conditions. Interleukin (IL) -2 or IL-15 preferentially induces proliferation of IL-13+ T cells, relative to IL-13 non-producers, in peripheral blood lymphocytes. The major source of IL-2 is antigen-stimulated T cells. Sources of IL-15 are monocytes, macrophages, and dendritic cells, which produce IL-15 upon activation by a variety of stimuli including pathogens (e.g., virus, bacteria) and tissue damage (e.g., products of necrotic cells, endothelins released by endothelia or epithelia, platelet-derived factors). A comprehensive review of antigen-independent signaling, and associated functional consequences, is provided in Loza et al. (4).
Antigen-independent signaling bypasses the TCR complex to transduce mitogenic and pro-survival signaling. For example, IL-2-mediated signaling feeds into the canonical TCR signaling pathways at the levels of Grb2 and PI3K. IL-2Rbeta chain activates Janus kinase (JAK) -3, which leads to activation of Grb2 via Shc. As discussed above for TCR/CD3-mediated signaling, Grb2/Son of sevenless protein activates Ras and the downstream p42/p44 MAPK pathway. JAK1 is also activated by IL-2Rbeta and transduces signaling to the PI3K/Akt pathway, contributing to IL-2-dependent increases in cell survival.
3.3. Beta-agonist effects on T cell signaling
The effects of beta-agonist (and other Gs-coupled receptor agonists such as prostaglandin E2 (PGE2)) on both TCR and antigen-independent signaling are presumed to be mediated via the cAMP-dependent protein kinase (PKA)(5;6). Previous studies have asserted PKA-dependent effects in T cells by either assessing agonist-stimulated PKA activity in an in vitro assay or demonstrating actions of pharmacologic PKA inhibitors and activators (7–14). Gs-coupled G protein-coupled receptors (GPCRs) such as the beta-2-adrenergic receptor (Beta2AR) and the EP2 receptor activate adenylyl cyclase, which hydrolyzes adenosine-5′-triphosphate (ATP) into cAMP, and cAMP in turn activates PKA. cAMP-independent and cAMP-dependent/PKA-independent actions of the Beta2AR have also been identified in some cells types, but not in T cells (15). In fact, the Rap1 guanine exchange factor Exchange Protein directly Activated by Cyclic AMP (EPAC), which can be activated by cAMP in both a PKA –dependent and –independent manner, does not appear to be expressed in T cells (unpublished observations and (16)).
Junctures at which Beta2AR-mediated PKA activation can regulate T cell signaling are depicted in both Figures 1 and 2. Regulation of Csk appears to the principal means by which PKA regulates proximal TCR signaling. Csk is a Src family kinase that phosphorylates Lck at Tyr-505 to inhibit Lck activity, TCR zeta–chain phosphorylation, and TCR-mediated signaling. PKA type 1 phosphorylates Csk at Ser-364 and increases its activity 2–4 fold (17). This phosphorylation/activation is facilitated by the formation of a complex of PKA type 1, Csk, sodium-hydrogen exchanger regulatory factor-1, and Csk-binding protein/phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG). The A Kinase Anchoring Protein (AKAP) Ezrin (18) serves as a scaffold to coordinate the assembly of this complex, thus enabling efficient targeting of Csk to its substrate Lck.
Numerous downstream intermediates are also regulated by PKA. PKA phosphorylation of Ser-43, Ser-233, and Ser-259 on Raf-1 disrupts Ras-Raf-l association to inhibit downstream activation of p42/p44 (19). Conversely, Ser-23 phosphorylation of the hematopoietic protein-tyrosine phosphatase (HePTP) causes release of HePTP from p42/p44 and p38 to allow their activation by MAPK/ERK kinases (20;21). cAMP elevation has also been shown to inhibit IL-2-stimulated activation of PI3K and downstream, mammalian target of rapamycin-dependent phosphorylation of p70 s6 kinase in T cells (22).
PKA phosphorylates and inhibits PLC-gamma-1 and -2, causing a reduction of intracellular Ca2+ mobilization and phosphotidylinositol hydrolysis (23;24), and thus possibly serving as a mechanism for inhibiting (Ca2+/calcineurin-mediated) nuclear translocation of the transcription factor NF-AT initiated by TCR signaling. However, a more direct effect of PKA on NF-AT occurs via direct NF-AT phosphorylation. Whereas desphosphorlyation of NF-AT by calcineurin promotes dissociation of NF-AT from 14-3-3 protein and enables nuclear translocation, PKA phosphorylation of NF-AT promotes 14-3-3 binding and retention of NF-AT in the cytosol (25). This competitive regulation of NF-AT by calcineurin and PKA is believed to be important in the regulation of IL-2 gene expression (25–27).
PKA has been shown to regulate the activity of NF-kappaB via numerous mechanisms, many of which appear cell type specific. Numerous members of the NF-kappaB family can be phosphorylated by PKA, resulting in various effects: altered ability to retain heterodimer formation, promotion of nuclear transclocation, DNA binding, and transcriptional activity. Zhong et al. (28) reported PKA phosphorylation of the p65 subunit promotes p65 interaction with the coactivator CBP/p300 to increase p65 transcriptional activity in Jurkat cells. Conversely, Neumann et al. (29) reported that PKA inhibits nuclear translocation of p65 in Jurkat cells by impairing degradation of inhibitory factor kappa B. Takahashi et al. reported PKA activation inhibits transcriptional activity of NF-kappaB in Jurkat cells via an effect involving the p65 C-terminal transactivation domain. These disparate results obtained in the same cell line are possibly explained by differences in experimental design, in particular the use of different reporter constructs as indicators of NF-kappaB activity. Of note, the majority of studies to date have demonstrated inhibitory effects of PKA on NF-kappaB-dependent gene expression.
In summary, activation of the TCR/CD3 complex promotes the stimulation of several major downstream effector signaling pathways. These pathways culminate in the activation and translocation of numerous transcription factors and activation of translation and cell cycle machinery. PKA has the capacity to block TCR/CD3-mediated signaling both at the most proximal (Lck/Fyn block by PKA-dependent localization of Csk) and at various downstream (including Raf-Ras interaction for the p42/p44 pathway, phosphorylation of NF-AT, and inactivation of PLC-gamma) points. Conversely, some degree of PKA activity may be necessary for the full potential of TCR/CD3-stimulated signaling (PKA-dependent release of inhibitory HePTP from p42/p44 and p38 MAPKs).
4. ASTHMA, INFLAMMATION, AND T CELLS
Human asthma is typically associated with airway inflammation as evidenced by the large numbers of inflammatory cells, and their mediators, present in bronchoalveolar lavage (BAL) fluid (BALF) recovered from asthmatics. Although numerous inflammatory cell types probably contribute to the development of inflammation in asthma, most mechanistic studies of allergic lung inflammation have ascribed critical roles to eosinophils, mast cells, and T cells (reviewed in (30;31), Figure 3). T cells participate in allergic lung inflammation by driving allergen sensitization and B cell production of allergen-specific IgE. This critical step in allergic inflammation is mediated by production of type 2 (or Th2-like) cytokines, particularly IL-4 and IL-13, by Th2 cells. Allergen-activated Th2 cells provide “help” to allergen-reactive B cells to promote their survival, expansion, and, in conjunction with IL-4 and IL-13, immunoglobulin class-switching to IgE. After establishment of allergen sensitization, T cells further contribute to allergic inflammation by local production of type 2 cytokines. IL-4 and particularly IL-13 produced in airways lead to growth of airway epithelia and their transformation into mucus-producing goblet cells. IL-5 produced by Th2 cells is an important factor for eosinophil release from bone marrow and for their survival in the periphery.
Figure 3.
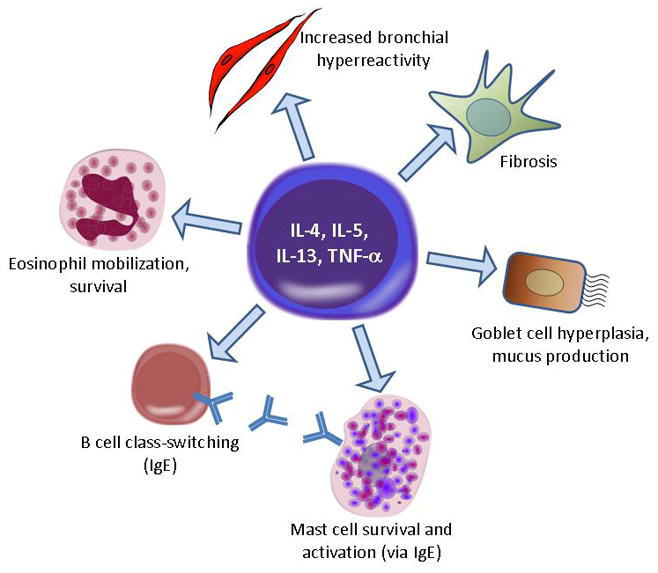
Impact of Th2-derived cytokines on airway inflammation and remodeling.
IL-13, in addition to IL-4, has been shown to be a critical modulator of allergic inflammation. In murine models (reviewed by (32)), introduction of elevated levels of IL-13 in the airways via pulmonary delivery and transgenic expression has been shown to be sufficient to induce both airway inflammation and increased airway resistance and hyperresponsiveness (AHR). In contrast, targeted expression of IL-4 in the airways by transgenic expression leads to increased airway inflammation but not significant AHR. Ablation of the genes encoding IL-13, IL-4Rα, or IL13Ralpha1 (but not the decoy/antagonistic IL-13Ralpha2) or blocking IL-13 by soluble IL-13Ralpha2-Fc largely inhibits the development of allergic inflammation, fibrosis, and AHR induced by allergen. Allergen-induced allergic inflammation and AHR are only weakly blocked, and fibrosis is unaffected, in IL4 knockout mice. Deletion of both IL4 and IL13 genes results in a complete block in allergen-induced inflammation, fibrosis, and AHR. These results underscore the importance of IL-13 in the fibrosis and AHR components, in addition to its overlapping role with IL-4 in inflammation, in murine models of allergic inflammation/asthma.
In humans, IL-13 and IL-4 are elevated in BALF obtained from asthmatics, and segmental antigen challenge of airways of human asthmatics causes large increases in Th2 (IL-4, IL-5, and IL-13) as well as the Th1 cytokine interferon (IFN) -gamma. Overall, the cellular and cytokine profile observed in airways of human asthmatics and in animal models of allergic airway inflammation defines an exaggerated Th2 cytokine response, and has contributed to the “Th2:Th1 hypothesis” in asthma (33). The extent to which Th1 cytokine levels in the asthmatic airway are altered is subject to debate, as is the role of Th1 cytokines in airway inflammation and asthma (34). Although the exact dynamic between Th2 and Th1 T cells in asthma is not clear, it is likely that both subtypes are important in regulating the nature of airway inflammation in asthma.
Tregs, a T cell subset critical for maintaining peripheral tolerance to self-antigen, are reported to be found in reduced numbers in airways of asthmatics, although their numbers in peripheral blood are normal. However, the role of Tregs in the development of allergy and local allergic inflammation is still a matter of ongoing debate (35).
5. EFFECTS OF BETA-AGONISTS ON T CELLS
5.1. Effects of beta-agonists in in vivo models of allergic lung inflammation
It is well accepted that inhaled beta-agonists are effective in reversing or preventing bronchoconstriction via their direct actions on airway smooth muscle (36). Whether effects on other cell types contribute to their therapeutic benefit is subject to debate. Although in vitro studies have identified various inflammatory cell functions inhibited by beta-agonists, results from in vivo studies are mixed and suggest little if any effect of inhaled beta-agonist on lung inflammation associated with asthma. In fact, several studies have indicated that beta-agonist therapy may actually worsen inflammation (37–41).
Because inflammation is a multi-cellular, multi-event, time-dependent process whose presentation is highly variable and is effected by numerous stimuli, integrative approaches attempting to understand T cell function and its regulation must be interpreted with extreme caution. Changes in airway T cell cytokine levels associated with beta-agonist therapy can reflect either direct or indirect effects of beta-agonist on T cells. In addition, analyses of direct effects of beta-agonist on T cells in models of allergic inflammation may be confounded by the capacity of inflammation to desensitize the Beta2AR on T cells, which has been suggested by both in vivo (42–44) and in vitro (45) studies.
A limited number of in vivo studies exist examining the effects of beta-agonists on various indices of inflammation including outcomes suggestive of T cell function. Several of these studies have provided comparisons among racemic and enantiomer formulations of albuterol. Using a guinea pig model, Westerhof et al. (46) noted inhalation of RS-, R-, or S- albuterol had no effect on inflammatory cell infiltration (including lymphocytes) induced by allergen challenge. In a murine model of ovalbumin (OVA) -induced allergic inflammation (47), both R- and S- albuterol delivered by a miniosmotic pump reduced airway eosinophil infiltration, yet neither affected the OVA-induced increase in BAL levels of IL-5 or IL-13. In contrast, using a similar infusion protocol with C57BL/6 mice Ferrada et al. (48) found no effects of S- albuterol on various indices of inflammation. R- and RS-, but not S- albuterol inhibited OVA-induced increases in BAL eosinophils, and the compounds similarly affected serum IgE levels. Collectively, these in vivo studies suggest modest if any effects of beta-agonists on airway inflammation. Studies examining enantiomers are consistent with most of the literature on beta-agonist enantiomer pharmacology, i.e., R- enantiomers are the active component of racemic mixtures and S- enantiomers simply have a null effect.
5.2. EFFECTS OF BETA-AGONISTS ON T CELLS USING CELL CULTURE MODELS
Unlike integrative models, studies using T cell cultures offer the opportunity to assess direct effects of beta-agonists on T cells, with the caveat that indirect effects on T cell subtypes are possible in populations of mixed cells (e.g., Peripheral Blood Mononuclear Cells (PBMC)). Beta-agonist activation of the Beta2AR has been reported to block TCR-mediated signaling to inhibit both cytokine production (IFN-gamma and IL-2) and proliferation of T cells (49;50). Although there appears to be a consensus regarding the ability of beta-agonists to directly regulate Th1 T cells, disparate results exist with respect to Th2 T cells. Two studies have reported beta-agonist-mediated inhibition of IL-4 and IL-13 production in freshly isolated human T cells (51;52). However, several studies examining various primary and clonal populations and polyclonal lines of T cells in murine and human systems have reported that beta-agonists do not directly affect type 2 T cells (53). Recently, to clarify this issue we analyzed beta-agonist-mediated signaling events at the single cell level (6). In both CD56+ (Th1) and cells expressing Chemoattractant-receptor homologous molecule expressed on Th2 cells (CRTH2+) (Th2) in freshly isolated peripheral blood lymphocytes, and in a near homogenous population of Th2 cells (CD2−/lo cultures (54)) beta-agonist induced the phosphorylation of the intracellular PKA substrate vasodilator-stimulated phosphoprotein (VASP). Moreover, in CRTH2+ cells beta-agonist inhibited the induction of CD25, and this effect was reversed by inclusion of a Beta2AR-selective antagonist. Thus, both human Th1 and Th2 T cells appear to express functional Beta2ARs.
Regarding effects of beta-agonist enantiomers, Ferrada et al. (48) recently reported that after 12 h stimulation of the T cell line EL-4 or murine splenocytes with Con A and phorbol 12-myristate 13-acetate (PMA), treatment with R- but not S-albuterol reduced levels of induced IL-2 and IL-13 mRNA, and effects of RS- albuterol were similar to those of R- albuterol. In parallel experiments in EL-4 cells, R- and RS- but not S- albuterol inhibited the induction of Con A + PMA- stimulated NF-κB reporter activity.
Borish and colleagues have examined both albuterol (55) and formoterol (56) enantiomers in antigen-specific T cell lines generated by culture of PBMC in tetanus and IL-2. R- albuterol and RR formoterol inhibited whereas the S-/SS-enantiomers had no effect on PMA-induced proliferation. Inhibitory effects on cytokine production were also observed for R- albuterol and RR- formoterol. In these studies, however, racemic mixtures were less effective in inhibiting proliferation and cytokine production than were R-/RR- enantiomers.
6. WHAT’S NEXT: FUTURES STUDIES EXAMINING BETA-AGONIST EFFECTS ON T CELLS
Although recent studies have been helpful, numerous questions remain regarding the effects of beta-agonists on T cell signaling and function in inflammatory lung disease. Important topics include:
6.1. The fundamental question of the relevance of beta-agonist regulation of T cells
Although in vitro studies suggest that beta-agonist can stimulate the Beta2AR on T cells to modulate T cell proliferation, survival, and cytokine production, the extent to which this occurs in vivo and whether it is of any physiological consequence is essentially unknown. Circulating catecholamines, especially when levels are altered such as with stress or heart failure, could conceivably affect T cell numbers and function and impact the pathogenesis of various inflammatory diseases. However, it is difficult to speculate how findings from in vitro studies relate to the effects of inhaled beta-agonist on infiltrating (and activated) T cells in the asthmatic lung. Complicating the issue is the need to address these questions in the human lung, in which experimental strategies for assessing T cell function are limited, and the mechanisms of allergic inflammation and T cell function are in many ways dissimilar to those in the mouse.
6.2. Agonism properties of various beta-agonists
The majority of in vitro studies have noted a profound difference between beta-agonists and PGE2 (both Gs-coupled receptor agonists) with respect to their ability to regulate T cell proliferation, survival, and cytokine production. We determined that in human type 2 T cells, beta-agonists are weak activators of PKA, whereas PGE2 is relatively efficacious (6;57;58). This disparity is associated with differences in the inhibitory effect on IL-2 and IL-13 production, p38, p42/p44, and NF-kappaB activity, and cell proliferation, and in the ability of beta-agonists to promote increased survival of IL-2-stimulated type 2 T cells (6;57). These results suggest agonism is an important characteristic of beta-agonists, and that an increased ability to generate cAMP and stimulate PKA may enable beta-agonists to function more like PGE2 and promote a greater anti-inflammatory effect. Such an increased ability may occur by relieving mechanisms of Beta2AR desensitization or by developing new beta-agonists with increased intrinsic activity or capacity for allosteric modulation (36;59). Of note is a recent study by Oostendorp et al. (60) in which differences in Beta2AR desensitization among cells expressing different ADRB2 gene haplotypes were associated differences in beta-agonist inhibition of lymphocyte IFN-gamma and IL-5 production.
Alternatively, beta-agonists capable of qualitatively distinct signaling (functional selectivity or biased agonism (59)) properties with superior anti-inflammatory features may be developed (or identified). Such development will require a more expansive screening strategy for pipeline Beta2AR agonists that considers numerous, diverse outcomes of Beta2AR signaling in T cells.
It is also important to note the possibility that Beta2AR agonism per se in T cells promotes inflammation. A recent study by Bond and colleagues (61) demonstrated that either ADBR2 gene knockout, or treatment of mice with inverse agonists of the Beta2AR, attenuated both airway inflammation and AHR promoted by sensitization and challenge with antigen. Although the cells that participate in this effect of Beta2AR antagonism remain to be identified, T cells are an obvious candidate.
6.3. Compartmentalization of Beta2AR signaling in T cells
Numerous studies have demonstrated that both T cell receptor and cAMP-mediated signaling in T cells is highly compartmentalized (reviewed in (62). Proximal TCR signaling occurs within lipid rafts. cAMP-dependent signaling in T cell also initiates in lipids rafts and is further compartmentalized by AKAPs. In numerous elegant studies, Tasken and colleagues have demonstrated that both AKAPs and lipid rafts in T cells help to coordinate, dependent on the extracellular stimulus, the formation of specific signaling complexes into discrete microdomains for the purpose of localizing signaling events to either propagate signals or regulate feedback processes (15;17;18;63–67). Although expansion on these studies is beyond the scope of this review, AKAPs and compartmentalized signaling represent another layer of regulation of Gs-coupled receptors that might contribute to differential effects among Gs-coupled receptor ligands on T cells.
Clarification of the questions proposed above will be important to our understanding of T cell biology and have obvious therapeutic implications as well. A critical issue to current asthma therapy involves the limitations, and possible deleterious effects, of inhaled beta-agonists. It has been speculated that a failure of beta-agonists to treat inflammation underlies the adverse clinical outcomes associated with chronic beta-agonist therapy. Should this be the case, the possibility exists that new generations of beta-agonists (possessing distinct signaling capabilities) could regulate T cell or other inflammatory cell functions to promote positive therapeutic benefits. Alternatively, understanding the limitations or negative consequences of beta-agonists with respect to inflammation/inflammatory cells will assist in identifying necessary adjunct therapies.
Acknowledgments
Supported by HL58506 (R.B.P.)
Abbreviations
- ADAP
degranulation promoting adaptor protein
- AHR
airway hyperresponsiveness
- AKAP
A kinase anchoring protein
- ATP
adenosine-5′-triphosphate
- Beta2AR
beta-2-adrenergic receptor
- BAL
bronchoalveolar lavage
- BALF
bronchoalveolar lavage fluid
- cAMP
3′-5′-cyclic adenosine monophosphate
- CRTH2
Chemoattractant-receptor homologous molecule expressed on Th2 cells
- DAG
diacylglyceride
- EPAC
exchange protein directly activated by cyclic AMP
- GPCR
G protein-coupled receptor
- HePTP
hematopoietic protein-tyrosine phosphatase
- IFN
interferon
- IL
interleukin
- IP3
inositol-triphosphate
- JAK
Janus kinase
- JNK
c-Jun N-terminal kinase
- LAT
Linker for activation of T cells
- MAPK
mitogen-activated protein kinase
- MHC
major histocompatibility complex
- PAG
phosphoprotein associated with glycosphingolipid-enriched microdomains
- PBMC
peripheral blood mononuclear cells
- PHA
phytohemagglutinen-L
- PGE2
prostaglandin E2
- PI3K
phosphoinositide 3-kinase
- PKA
cAMP-dependent protein kinase
- PKC
protein kinase C
- PLC
phospholipase C
- PMA
phorbol 12-myristate 13-acetate
- NF-AT
nuclear factor for activated T-cells
- NF-kappaB
nuclear factor kappa-light-chain-enhancer of activated B cells
- OVA
ovalbumin
- SLP-76
SH2 domain containing leukocyte protein of 76kDa
- TCR
T cell receptor
- VASP
vasodilator-stimulated phosphoprotein
- ZAP-70
zeta-chain-associated protein kinase 70
References
- 1.Griffiths EK, Penninger JM. Communication between the TCR and integrins: role of the molecular adapter ADAP/Fyb/Slap. Curr Opin Immunol. 2002;14:317–322. doi: 10.1016/s0952-7915(02)00334-5. [DOI] [PubMed] [Google Scholar]
- 2.Nel AE. T-cell activation through the antigen receptor. Part 1: signaling components, signaling pathways, and signal integration at the T-cell antigen receptor synapse. J Allergy Clin Immunol. 2002;109:758–770. doi: 10.1067/mai.2002.124259. [DOI] [PubMed] [Google Scholar]
- 3.Nel AE, Slaughter N. T-cell activation through the antigen receptor. Part 2: role of signaling cascades in T-cell differentiation, anergy, immune senescence, and development of immunotherapy. J Allergy Clin Immunol. 2002;109:901–915. doi: 10.1067/mai.2002.124965. [DOI] [PubMed] [Google Scholar]
- 4.Loza MJ, Peters SP, Penn RB. Atopy, asthma, and experimental approaches based on the linear model of T cell maturation. Clin Exp Allergy. 2005;35:8–17. doi: 10.1111/j.1365-2222.2004.02148.x. [DOI] [PubMed] [Google Scholar]
- 5.Borger P, Postma DS, Vellenga E, Kauffman HF. Regulation of asthma-related T-cell cytokines by the cyclic AMP-dependent signalling pathway. Clin Exp Allergy. 2000;30:920–926. doi: 10.1046/j.1365-2222.2000.00794.x. [DOI] [PubMed] [Google Scholar]
- 6.Loza MJ, Foster S, Peters SP, Penn RB. Beta-agonists modulate T cell functions via direct actions on type 1 and type 2 cells. Blood. 2006;107:2052–2060. doi: 10.1182/blood-2005-08-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aandahl EM, Moretto WJ, Haslett PA, Vang T, Bryn T, Tasken K, Nixon DF. Inhibition of antigen-specific T cell proliferation and cytokine production by protein kinase A type I. J Immunol. 2002;169:802–808. doi: 10.4049/jimmunol.169.2.802. [DOI] [PubMed] [Google Scholar]
- 8.Bartik MM, Bauman GP, Brooks WH, Roszman TL. Costimulatory signals modulate the antiproliferative effects of agents that elevate cAMP in T cells. Cell Immunol. 1994;158:116–130. doi: 10.1006/cimm.1994.1261. [DOI] [PubMed] [Google Scholar]
- 9.Bauman GP, Bartik MM, Brooks WH, Roszman TL. Induction of cAMP-dependent protein kinase (PKA) activity in T cells after stimulation of the prostaglandin E2 or the beta-adrenergic receptors: relationship between PKA activity and inhibition of anti-CD3 monoclonal antibody-induced T cell proliferation. Cell Immunol. 1994;158:182–194. doi: 10.1006/cimm.1994.1266. [DOI] [PubMed] [Google Scholar]
- 10.Benbernou N, Esnault S, Shin HC, Fekkar H, Guenounou M. Differential regulation of IFN-gamma, IL-10 and inducible nitric oxide synthase in human T cells by cyclic AMP-dependent signal transduction pathway. Immunology. 1997;91:361–368. doi: 10.1046/j.1365-2567.1997.00260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganapathy V, Gurlo T, Jarstadmarken HO, von Grafenstein H. Regulation of TCR-induced IFN-gamma release from islet-reactive non-obese diabetic CD8(+) T cells by prostaglandin E(2) receptor signaling. Int Immunol. 2000;12:851–860. doi: 10.1093/intimm/12.6.851. [DOI] [PubMed] [Google Scholar]
- 12.Laxminarayana D, Berrada A, Kammer GM. Early events of human T lymphocyte activation are associated with type I protein kinase A activity. J Clin Invest. 1993;92:2207–2214. doi: 10.1172/JCI116823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumann M, Grieshammer T, Chuvpilo S, Kneitz B, Lohoff M, Schimpl A, Franza BR, Jr, Serfling E. RelA/p65 is a molecular target for the immunosuppressive action of protein kinase A. EMBO J. 1995;14:1991–2004. doi: 10.1002/j.1460-2075.1995.tb07191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gjertsen BT, Mellgren G, Otten A, Maronde E, Genieser HG, Jastorff B, Vintermyr OK, McKnight GS, Doskeland SO. Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1 beta action. J Biol Chem. 1995;270:20599–20607. doi: 10.1074/jbc.270.35.20599. [DOI] [PubMed] [Google Scholar]
- 15.Abrahamsen H, Baillie G, Ngai J, Vang T, Nika K, Ruppelt A, Mustelin T, Zaccolo M, Houslay M, Tasken K. TCR- and CD28-mediated recruitment of phosphodiesterase 4 to lipid rafts potentiates TCR signaling. J Immunol. 2004;173:4847–4858. doi: 10.4049/jimmunol.173.8.4847. [DOI] [PubMed] [Google Scholar]
- 16.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 17.Vang T, Torgersen KM, Sundvold V, Saxena M, Levy FO, Skalhegg BS, Hansson V, Mustelin T, Tasken K. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J Exp Med. 2001;193:497–507. doi: 10.1084/jem.193.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruppelt A, Mosenden R, Gronholm M, Aandahl EM, Tobin D, Carlson CR, Abrahamsen H, Herberg FW, Carpen O, Tasken K. Inhibition of T cell activation by cyclic adenosine 5′-monophosphate requires lipid raft targeting of protein kinase A type I by the A-kinase anchoring protein ezrin. J Immunol. 2007;179:5159–5168. doi: 10.4049/jimmunol.179.8.5159. [DOI] [PubMed] [Google Scholar]
- 19.Dumaz N, Marais R. Protein kinase A blocks Raf-1 activity by stimulating 14–3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem. 2003;278:29819–29823. doi: 10.1074/jbc.C300182200. [DOI] [PubMed] [Google Scholar]
- 20.Blanco-Aparicio C, Torres J, Pulido R. A novel regulatory mechanism of MAP kinases activation and nuclear translocation mediated by PKA and the PTP-SL tyrosine phosphatase. J Cell Biol. 1999;147:1129–1136. doi: 10.1083/jcb.147.6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saxena M, Williams S, Tasken K, Mustelin T. Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase. Nat Cell Biol. 1999;1:305–311. doi: 10.1038/13024. [DOI] [PubMed] [Google Scholar]
- 22.Monfar M, Lemon KP, Grammer TC, Cheatham L, Chung J, Vlahos CJ, Blenis J. Activation of pp70/85 S6 kinases in interleukin-2-responsive lymphoid cells is mediated by phosphatidylinositol 3-kinase and inhibited by cyclic AMP. Mol Cell Biol. 1995;15:326–337. doi: 10.1128/mcb.15.1.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alava MA, DeBell KE, Conti A, Hoffman T, Bonvini E. Increased intracellular cyclic AMP inhibits inositol phospholipid hydrolysis induced by perturbation of the T cell receptor/CD3 complex but not by G-protein stimulation. Association with protein kinase A-mediated phosphorylation of phospholipase C-gamma 1. Biochem J. 1992;284(Pt 1):189–199. doi: 10.1042/bj2840189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park DJ, Min HK, Rhee SG. Inhibition of CD3-linked phospholipase C by phorbol ester and by cAMP is associated with decreased phosphotyrosine and increased phosphoserine contents of PLC-gamma 1. J Biol Chem. 1992;267:1496–1501. [PubMed] [Google Scholar]
- 25.Chow CW, Davis RJ. Integration of calcium and cyclic AMP signaling pathways by 14-3-3. Mol Cell Biol. 2000;20:702–712. doi: 10.1128/mcb.20.2.702-712.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chow CW, Rincon M, Davis RJ. Requirement for transcription factor NFAT in interleukin-2 expression. Mol Cell Biol. 1999;19:2300–2307. doi: 10.1128/mcb.19.3.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torgersen KM, Vang T, Abrahamsen H, Yaqub S, Tasken K. Molecular mechanisms for protein kinase A-mediated modulation of immune function. Cell Signal. 2002;14:1–9. doi: 10.1016/s0898-6568(01)00214-5. [DOI] [PubMed] [Google Scholar]
- 28.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 29.Neumann M, Grieshammer T, Chuvpilo S, Kneitz B, Lohoff M, Schimpl A, Franza BR, Jr, Serfling E. RelA/p65 is a molecular target for the immunosuppressive action of protein kinase A. Embo J. 1995;14:1991–2004. doi: 10.1002/j.1460-2075.1995.tb07191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finkelman FD, Vercelli D. Advances in asthma, allergy mechanisms, and genetics in 2006. J Allergy Clin Immunol. 2007;120:544–550. doi: 10.1016/j.jaci.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 31.Holgate ST. Pathogenesis of asthma. Clin Exp Allergy. 2008;38:872–897. doi: 10.1111/j.1365-2222.2008.02971.x. [DOI] [PubMed] [Google Scholar]
- 32.Kasaian MT, Miller DK. IL-13 as a therapeutic target for respiratory disease. Biochem Pharmacol. 2008;76:147–155. doi: 10.1016/j.bcp.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 33.Colavita AM, Reinach AJ, Peters SP. Contributing factors to the pathobiology of asthma. The Th1/Th2 paradigm. Clin Chest Med. 2000;21:263–277. viii. doi: 10.1016/s0272-5231(05)70265-3. [DOI] [PubMed] [Google Scholar]
- 34.Holtzman MJ, Sampath D, Castro M, Look DC, Jayaraman S. The one-two of T helper cells: does interferon-gamma knock out the Th2 hypothesis for asthma? Am J Respir Cell Mol Biol. 1996;14:316–318. doi: 10.1165/ajrcmb.14.4.8600934. [DOI] [PubMed] [Google Scholar]
- 35.Broide DH. Immunologic and inflammatory mechanisms that drive asthma progression to remodeling. J Allergy Clin Immunol. 2008;121:560–570. doi: 10.1016/j.jaci.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deshpande DA, Penn RB. Targeting G protein-coupled receptor signaling in asthma. Cell Signal. 2006;18:2105–2120. doi: 10.1016/j.cellsig.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 37.Agarwal N, Nir I, Papermaster DS. Loss of diurnal arrestin gene expression in rds mutant mouse retinas. Exp Eye Res. 1994;58:1–8. doi: 10.1006/exer.1994.1189. [DOI] [PubMed] [Google Scholar]
- 38.Sears MR, Taylor DR, PCG, Lake DC, Li Q, Flannery EM, Yates DM, Lucas MK, Herbison GP. Regular inhaled beta-agonist treatment in bronchial asthma. Lancet. 1990;336:1391–1396. doi: 10.1016/0140-6736(90)93098-a. [DOI] [PubMed] [Google Scholar]
- 39.Spitzer WO, Suissa S, Ernst P, Horwitz RI, Habbick B, Cockroft D, Boivin JF, McNutt M, Buist AS, Rebuck AS. The use of β-agonists and the risk of death and near-death from asthma. N Engl J Med. 1992;326:501–506. doi: 10.1056/NEJM199202203260801. [DOI] [PubMed] [Google Scholar]
- 40.Taylor DR, Town GI, Herbison GP, Boothman-Burrell D, Flannery EM, Hancox B, Harre E, Laubscher K, Linscott V, Ramsay CM, Richards G. Asthma control during long-term treatment with regular inhaled salbutamol and salmeterol. Thorax. 1998;53:744–752. doi: 10.1136/thx.53.9.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wooltorton E. Salmeterol (Serevent) asthma trial halted early. Cmaj. 2003;168:738. [PMC free article] [PubMed] [Google Scholar]
- 42.Heijink IH, van den BM, Vellenga E, de Monchy JG, Postma DS, Kauffman HF. Altered beta2-adrenergic regulation of T cell activity after allergen challenge in asthma. Clin Exp Allergy. 2004;34:1356–1363. doi: 10.1111/j.1365-2222.2004.02037.x. [DOI] [PubMed] [Google Scholar]
- 43.Meurs H, Koeter GH, de VK, Kauffman HF. Dynamics of the lymphocyte beta-adrenoceptor system in patients with allergic bronchial asthma. Eur J Respir Dis Suppl. 1984;135:47–61. [PubMed] [Google Scholar]
- 44.Meurs H, Kauffman HF, Timmermans A, de Monchy JG, Koeter GH, de VK. Specific immunological modulation of lymphocyte adenylate cyclase in asthmatic patients after allergenic bronchial provocation. Int Arch Allergy Appl Immunol. 1986;81:224–229. doi: 10.1159/000234138. [DOI] [PubMed] [Google Scholar]
- 45.Heijink IH, Vellenga E, Oostendorp J, de Monchy JG, Postma DS, Kauffman HF. Exposure to TARC alters beta2-adrenergic receptor signaling in human peripheral blood T lymphocytes. Am J Physiol Lung Cell Mol Physiol. 2005;289:L53–L59. doi: 10.1152/ajplung.00357.2004. [DOI] [PubMed] [Google Scholar]
- 46.Westerhof F, Timens W, van Oosten A, Zuidhof AB, Nauta N, Schuiling M, Vos JT, Zaagsma J, Meurs H, Coers W. Inflammatory cell distribution in guinea pig airways and its relationship to airway reactivity. Mediators Inflamm. 2001;10:143–154. doi: 10.1080/09629350124877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sartori C, Fang X, McGraw DW, Koch P, Snider ME, Folkesson HG, Matthay MA. Selected contribution: long-term effects of beta(2)-adrenergic receptor stimulation on alveolar fluid clearance in mice. J Appl Physiol. 2002;93:1875–1880. doi: 10.1152/japplphysiol.00275.2002. [DOI] [PubMed] [Google Scholar]
- 48.Ferrada MA, Gordon EL, Jen KY, He HZ, Lu X, Barone LM, Amirifeli S, Perkins DL, Finn PW. (R)-albuterol decreases immune responses: role of activated T cells. Respir Res. 2008;9:3. doi: 10.1186/1465-9921-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borger P, Hoekstra Y, Esselink MT, Postma DS, Zaagsma J, Vellenga E, Kauffman HF. Beta-adrenoceptor-mediated inhibition of IFN-gamma, IL-3, and GM-CSF mRNA accumulation in activated human T lymphocytes is solely mediated by the beta2-adrenoceptor subtype. Am J Respir Cell Mol Biol. 1998;19:400–407. doi: 10.1165/ajrcmb.19.3.2765. [DOI] [PubMed] [Google Scholar]
- 50.Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol. 1997;158:4200–4210. [PubMed] [Google Scholar]
- 51.Goleva E, Dunlap A, Leung DY. Differential control of TH1 versus TH2 cell responses by the combination of low-dose steroids with beta2-adrenergic agonists. J Allergy Clin Immunol. 2004;114:183–191. doi: 10.1016/j.jaci.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 52.Heijink IH, Vellenga E, Borger P, Postma DS, Monchy JG, Kauffman HF. Polarized Th1 and Th2 cells are less responsive to negative feedback by receptors coupled to the AC/cAMP system compared to freshly isolated T cells. Br J Pharmacol. 2003;138:1441–1450. doi: 10.1038/sj.bjp.0705193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kohm AP, Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev. 2001;53:487–525. [PubMed] [Google Scholar]
- 54.Loza MJ, Perussia B. Peripheral immature CD2-/low T cell development from type 2 to type 1 cytokine production. J Immunol. 2002;169:3061–3068. doi: 10.4049/jimmunol.169.6.3061. [DOI] [PubMed] [Google Scholar]
- 55.Baramki D, Koester J, Anderson AJ, Borish L. Modulation of T-cell function by (R)- and (S)-isomers of albuterol: anti-inflammatory influences of (R)-isomers are negated in the presence of the (S)-isomer. J Allergy Clin Immunol. 2002;109:449–454. doi: 10.1067/mai.2002.122159. [DOI] [PubMed] [Google Scholar]
- 56.Steinke JW, Baramki D, Borish L. Opposing actions of (R, R)-isomers and (S, S)-isomers of formoterol on T-cell function. J Allergy Clin Immunol. 2006;118:963–965. doi: 10.1016/j.jaci.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 57.Loza MJ, Peters SP, Foster S, Khan IU, Penn RB. beta-Agonist enhances type 2 T-cell survival and accumulation. J Allergy Clin Immunol. 2007;119:235–244. doi: 10.1016/j.jaci.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 58.Loza MJ, Foster S, Peters SP, Penn RB. Interactive effects of steroids and beta-agonists on accumulation of type 2 T cells. J Allergy Clin Immunol. 2008;121:750–755. doi: 10.1016/j.jaci.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 59.Penn RB. Embracing emerging pardigms of G protein-coupled receptor agonism and signaling to address airway smooth muscle pathobiology in asthma. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:149–169. doi: 10.1007/s00210-008-0263-1. [DOI] [PubMed] [Google Scholar]
- 60.Oostendorp J, Postma DS, Volders H, Jongepier H, Kauffman HF, Boezen HM, Meyers DA, Bleecker ER, Nelemans SA, Zaagsma J, Meurs H. Differential desensitization of homozygous haplotypes of the beta2-adrenergic receptor in lymphocytes. Am J Respir Crit Care Med. 2005;172:322–328. doi: 10.1164/rccm.200409-1162OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen LP, Lin R, Parra S, Omoluabi O, Hanania NA, Tuvim MJ, Knoll BJ, Dickey BF, Bond RA. Beta2-adrenoceptor signaling is required for the development of an asthma phenotype in a murine model. Proc Natl Acad Sci U S A. 2009;106:2435–2440. doi: 10.1073/pnas.0810902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Torgersen KM, Aandahl EM, Tasken K. Molecular architecture of signal complexes regulating immune cell function. Handb Exp Pharmacol. 2008:327–363. doi: 10.1007/978-3-540-72843-6_14. [DOI] [PubMed] [Google Scholar]
- 63.Skalhegg BS, Tasken K, Hansson V, Huitfeldt HS, Jahnsen T, Lea T. Location of cAMP-dependent protein kinase type I with the TCR-CD3 complex. Science. 1994;263:84–87. doi: 10.1126/science.8272870. [DOI] [PubMed] [Google Scholar]
- 64.Solheim SA, Petsalaki E, Stokka AJ, Russell RB, Tasken K, Berge T. Interactions between the Fyn SH3-domain and adaptor protein Cbp/PAG derived ligands, effects on kinase activity and affinity. FEBS J. 2008;275:4863–4874. doi: 10.1111/j.1742-4658.2008.06626.x. [DOI] [PubMed] [Google Scholar]
- 65.Tasken K, Ruppelt A. Negative regulation of T-cell receptor activation by the cAMP-PKA-Csk signalling pathway in T-cell lipid rafts. Front Biosci. 2006;11:2929–2939. doi: 10.2741/2022. [DOI] [PubMed] [Google Scholar]
- 66.Torgersen KM, Vaage JT, Rolstad B, Tasken K. A soluble LAT deletion mutant inhibits T-cell activation: reduced recruitment of signalling molecules to glycolipid-enriched microdomains. Cell Signal. 2001;13:213–220. doi: 10.1016/s0898-6568(01)00131-0. [DOI] [PubMed] [Google Scholar]
- 67.Torgersen KM, Vang T, Abrahamsen H, Yaqub S, Horejsi V, Schraven B, Rolstad B, Mustelin T, Tasken K. Release from tonic inhibition of T cell activation through transient displacement of C-terminal Src kinase (Csk) from lipid rafts. J Biol Chem. 2001;276:29313–29318. doi: 10.1074/jbc.C100014200. [DOI] [PubMed] [Google Scholar]